
Abstract
Purpose
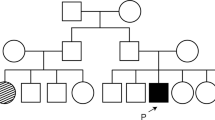
The c.1430A > G (Asp477Gly) variant in RPE65 has been reported in Irish and Scottish families with either an autosomal dominant retinal dystrophy (adRD) that resembles choroideremia, a vitelliform macular dystrophy or an isolated macular atrophy. We report novel features on multimodal imaging and the natural history of a family harbouring this variant in combination with the BEST1 c.37C > T (Arg13Cys) variant.
Methods
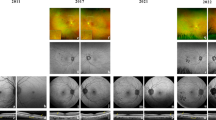
Members of a family with an adRD were examined clinically to ascertain phenotype and underwent genetic testing. Multimodal imaging included widefield colour fundus photography, quantitative autofluorescence (qAF) and spectral domain optical coherence tomography. Electrophysiology and microperimetry were also performed.
Results
Vision loss was attributed to foveal atrophy in the proband and choroidal neovascularisation and a vitello-eruptive lesion in one affected son. Peripheral retinal white dots corresponding to subretinal deposits were seen in three patients. The median qAF8 values in the proband (I:1) were low (40 and 101 in OD and OS) at age 79. Similarly, the qAF8 values for the middle son (II:2) were also low (100 and 87 in ODS and OS) at age 60. Electrophysiology showed disproportionate reduction in Arden ratio prior to the gradual loss of full-field responses. Microperimetry demonstrated an enlarging scotoma in the proband.
Conclusions
The coexistence of the pathogenic BEST1 c.37C > T variant may modify clinical features observed in RPE65 adRD. This study expands our understanding of RPE65 adRD as a retinoid cycle disorder supported by the reduced qAF, fine white retinal dots and corresponding subretinal deposits on OCT in affected members.






Similar content being viewed by others

Data availability
Data sharing is not applicable to this article as no datasets were generated or analysed during this study.
References
Zahid S, Branham K, Schlegel D, Pennesi ME, Michaelides M, Heckenlively J, Jayasundera T (2018) RPE65. In: Zahid S (ed) Retinal dystrophy gene atlas. Springer International Publishing, New York, p 251
Moiseyev G, Chen Y, Takahashi Y, Wu BX, Ma JX (2005) RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc Natl Acad Sci U S A 102(35):12413–12418
Jin M, Li S, Moghrabi WN, Sun H, Travis GH (2005) RPE65 is the retinoid isomerase in bovine retinal pigment epithelium. Cell 122(3):449–459
Redmond TM, Poliakov E, Yu S, Tsai JY, Lu Z, Gentleman S (2005) Mutation of key residues of RPE65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc Natl Acad Sci U S A 102(38):13658–13663
Morimura H, Fishman GA, Grover SA, Fulton AB, Berson EL, Dryja TP (1998) Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or Leber congenital amaurosis. Proc Natl Acad Sci U S A 95(6):3088–3093
Weleber RG, Michaelides M, Trzupek KM, Stover NB, Stone EM (2011) The phenotype of severe early childhood onset retinal dystrophy (SECORD) from mutation of RPE65 and differentiation from Leber congenital amaurosis. Invest Ophthalmol Vis Sci 51(1):292–302
Bowne SJ, Humpries MM, Sullivan LS, Kenna PF, Tam LCS, Kiang AS, Campbell M, Weinstock GM, Koboldt DC, Ding L et al (2011) A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur J Hum Genet 19:1074–1081
Hull S, Mukherjee R, Holder GE, Moore AT, Webster AR (2016) The clinical features of retinal disease due to a dominant mutation in RPE65. Mol Vis 22:626–635
Jauregui R, Park KS, Tsang SH (2018) Two-year progression analysis of RPE65 autosomal dominant retinitis pigmentosa. Ophthalmic Genet 39:544–549
Choi EH, Suh S, Sandler CL, Ortiz Hernandez CJ, Bulman ER, Khadka N, Dong Z, Shi W, Palczewski K, Kiser PD (2018) Insights into the pathogenesis of dominant retinitis pigmentosa associated with a D477G mutation in RPE65. Hum Mol Genet 17(13):2225–2243
Li Y, Furhang R, Ray A, Duncan T, Soucy J et al (2019) Aberrant RNA splicing is the major pathogenic effect in a knock-in mouse model of the dominantly inherited c.1430A>G human RPE65 mutation. Hum Mutat 40(4):426–443
Wells J, Wroblewski J, Keen J, Inglehearn C, Jubb C et al (1993) Mutations in the human retinal degeneration slow (RDS) gene can cause either retinitis pigmentosa or macular dystrophy. Nat Genet 3(3):213–218
Jauregui R, Cho A, Oh JK, Tanaka AJ et al. Phenotypic expansion of autosomal dominant retinitis pigmentosa associated with the D477G mutation in RPE65. Cold Spring Harb Mol Case Stud 2020 3;6(1): 004952.
Kenna PF, Humphries MM, Kiang AS et al (2020) Advanced late-onset retinitis pigmentosa with dominant-acting D477G RPE65 mutation is responsive to oral synthetic retinoid therapy. BMJ Open Ophthalmol 5(5):e000462
Li Y, Zhang Y, Xu Y, Kittredge A et al (2017) Patient-specific mutations impair BESTROPHIN1’s essential role in mediating Ca 2+-dependent Cl - currents in human RPE. Elife 24(6):e29914
Burke TR, Duncker T, Woods RL et al (2014) Quantitative fundus autofluorescence in recessive stargardt disease. Invest Ophthalmol Vis Sci 55:2841–2852
Greenberg JP, Duncker T, Woods RL, Smith TR, Sparrow JR et al (2013) Quantitative fundus autofluorescence in healthy eyes. Invest Ophthalmol Vis Sci 54:5684–5693
Marmor MF, Arden GB, Nilsson SE, Zrenner E (1989) Standard for clinical electroretinography. Arch Ophthalmol 107:816–819
De Roach JN, McLaren TL, Paterson RL, O’Brien EC, Hoffmann L, Mackey DA et al (2013) Establishment and evolution of the Australian inherited retinal disease register and DNA bank. Clin Exp Ophthalmol 41(5):476–483
Chiang JP, Lamey T, McLaren T, Thompson JA, Montgomery H, De Roach J (2015) Progress and prospects of next-generation sequencing testing for inherited retinal dystrophy. Expert Rev Mol Diagn 15(10):1269–1275
den Dunnen JT, Dalgleish R, Maglott DR et al (2016) HGVS Recommendations for the description of sequence variants: 2016 update. Hum Mutat 37(6):564–569
Thompson JA, De Roach JN, McLaren TL et al (2017) The genetic profile of Leber congenital amaurosis in an Australian cohort. Mol Genet Genomic Med 5(6):652–667
Richards S, Nazneen A, Bale S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med 17(5):405–424
Jarvik GP, Browning BL (2016) Consideration of Cosegregation in the Pathogenicity Classification of Genomic Variants. Am J Hum Genet 98(6):1077–1081
ACGS, Best Practice Guidelines for Variant Classification 2019, S Ellard et al, Editors, Association for Clinical Genomic Science: UK: p.32
Group, C.S.V.I.W. ClinGen Sequence Variant Interpretation Work Group recommendations for ACMG/AMP guideline criteria code modifications nomenclature 2017 [cited 2020 15–09–2020]; Available from: https://www.clinicalgenome.org/site/assets/files/3459/svi_criteria_nomenclature_recommendation_v1.pdf
Shin Y et al (2017) A Dominant Mutation in Rpe 65, D477G, Delays Dark Adaptation and Disturbs the Visual Cycle in the Mutant Knock-In Mice. Am J Pathol 187(3):517–527
Karczewski KJ. (2019) Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes, in bioRxiv. Cold Spring Harbor Laboratory
Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (URL: http://evs.gs.washington.edu/EVS/) [15 September 2020 accessed]
Kinnick TR, Mullins RF, Dev S et al (2011) Autosomal recessive vitelliform macular dystrophy in a large cohort of vitelliform macular dystrophy patients. Retina 31:581–595
Abdalla YF, De Salvo G, Elsahn A, Self JE (2017) Novel presenting phenotype in a child with autosomal dominant Best’s Vitelliform Macular Dystrophy. Ophthalmic Surg Laser Imaging Retina 48:580–585
Shyam R et al (2017) RPE65 has an additional function as the lutein to meso-zeaxanthin isomerase in the vertebrate eye. PNAS 114:10882–10887
Schuerch K, Woods RL, Lee W et al (2017) Quantifying fundus autofluorescence in patients with retinitis pigmentosa. Invest Ophthalmol Vis Sci 58(3):1843–1855
de Carvalho JRL, Kim HJ, Ueda KX et al (2020) Effects of deficiency in the RLBP1-encoded visual cycle protein CRALBP on visual dysfunction in humans and mice. J Biol Chem 295(19):6767–6780
Felius J, Thompson DA, Khan NW (2002) Clinical course and visual function in a family with mutations in the RPE65 gene. Arch Ophthalmol 120:55–61
El Matri L, Ambresin A, Schorderet DF (2006) Phenotype of three consanguineous Tunisian families with early-onset retinal degeneration caused by an R91W homozygous mutation in the RPE65 gene. Graefes Arch Clin Exp Ophthalmol 244:1104–1112
Dessalces E, Bocquet B, Bourien J, Zanlonghi X, Verdet R, Meunier I, Hamel CP (2013) Early-onset foveal involvement in retinitis punctata albescens with mutations in RLBP1. JAMA Ophthalmol 131:1314–1323
Genead MA, Fishman GA, Lindeman M (2010) Spectral-domain optical coherence tomography and fundus autofluorescence characteristics in patients with fundus albipunctatus and retinitis punctata albescens. Ophthalmic Genet 31:66–72
Caldwell GM, Kakuk LE, Griesinger IB et al (1999) Bestrophin gene mutations in patients with best vitelliform macular dystrophy. Genomics 58:98–101
Wong RL, Hou P, Choy KW et al (2010) Novel and homozygous BEST1 mutations in Chinese patients with Best vitelliform macular dystrophy. Retina 30:820–827
Tian R, Yang G, Wang J, Chen Y (2014) Screening for BEST1 gene mutations in Chinese patients with bestrophinopathy. Mol Vis 20:1594–1604
Gao F-J, Qi YH, Hu FY et al. (2020) Mutation spectrum of the bestrophin-1 gene in a large Chinese cohort with bestrophinopathy Br J Ophthalmol. 104,846–851
Levi SR, Oh JY, de Carvalho JR et al (2020) Quantitative Autofluorescence Following Gene Therapy With Voretigene Neparvovec. JAMA Ophthalmol 138:919–921
Acknowledgements
The authors would like to thank Amanda Scurry and Jayme Glynn for their assistance in organising the patient appointments for the clinical assessments. The AIRDR acknowledges the assistance of Ling Hoffman and Isabella Urwin from the Department of Medical Technology and Physics at Sir Charles Gairdner Hospital.
Funding
This work was supported by the Australian National Health and Medical Research Council under GNT116360 (FKC), GNT1188694 (FKC), GNT1054712 (FKC) and MRF1142962 (FKC), McCusker Foundation (FKC), Miocevich Retina Fellowship (RCHJ) and Retina Australia (JAT, TL, JNDR, TLM). The sponsor or funding organisation had no role in the design or conduct of this research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare they have no conflict of interest or competing interests.
Consent to participate
Written informed consent was obtained from all participants included in this case series.
Consent for publication
Written informed consent for publication of clinical information, test results and images was obtained from all participants included in this case series.
Ethical approval
The WARD study and Australian Inherited Retinal Disease Registry and DNA bank study protocols adhered to the tenets of the Declaration of Helsinki, and ethics approval was obtained from the Human Ethics Office of Research Enterprise, The University of Western Australia (RA/4/1/8932 and RA/4/1/7916) and Sir Charles Gairdner Hospital Human Research Ethics Committee (approval number 2001–053).
Statement of human rights
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Statement on the welfare of animals
This article does not contain any studies with animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pappalardo, J., Heath Jeffery, R.C., Thompson, J.A. et al. A novel phenotype in a family with autosomal dominant retinal dystrophy due to c.1430A > G in retinoid isomerohydrolase (RPE65) and c.37C > T in bestrophin 1 (BEST1). Doc Ophthalmol 143, 61–73 (2021). https://doi.org/10.1007/s10633-021-09819-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10633-021-09819-x