
Abstract
Purpose
To identify the genetic defect, and to phenotype, three consanguineous Tunisian families presenting with early-onset retinal degeneration (EORD).
Methods
All accessible family members were included. They underwent blood sampling and ophthalmological examination including, when possible, full-field ERG and pupillometry. A genome-wide linkage analysis was initiated. Mutation analysis of the RPE65 gene within the linked interval was performed by bi-directional sequencing.
Results
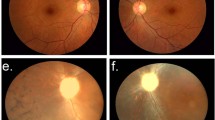
Eleven out of 53 examined members were clinically affected with an EORD. Linkage analysis revealed a maximal lod score of 4.02 (θ=0.1) for the marker D1S207 on 1p31. Mutational screening of the RPE65 gene identified a homozygous R91W mutation co-segregating with the disease in all affected individuals. Eleven homozygotes had nystagmus and acuities ranging from CF to NLP. Two retinal patterns were identified: pattern 1 presented mid-peripheral deep white dot deposits and virtually no clumped pigmentation, whereas pattern 2 showed mid-peripheral pigmented clumps without any white deposits. Homozygotes had no detectable full-field ERG and an abnormal pupillary light reflex. Eleven heterozygotes had normal visual function.
Conclusion
We identified and characterised an endemic form of early onset rod-cone dystrophy in a consanguineous population from northeastern Tunisia, due to the prevalence of a single RPE65 mutation. Two funduscopic patterns were identified: white dot deposits in earlier stages and clumped pigment in later stages.





Similar content being viewed by others


Notes
Moiseyev et al identified the RPE65 protein as the isomerohydrolase transforming all‐trans retinyl‐ester into 11‐cis retinol. Moiseyev G, Chen Y, Takahashi Y, Wu BX, Ma JX. RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc Natl Acad Sci U S A. 2005 Aug 30;102(35):12412–12418
References
Aleman TS, Jacobson SG, Chico JD et al (2004) Impairment of the transient pupillary light reflex in Rpe65(−/−) mice and humans in Leber congenital amaurosis. Invest Ophthalmol Vis Sci 45(4):1259–1271
Al–Khayer K, Hagstrom S, Pauer G et al (2004) Thirty-year follow-up of a patient with Leber congenital amaurosis and novel RPE65 mutations. Am J Ophthalmol 137(2):375–377
Cremers FP, van den Hurk JA, den Hollander AI (2002) Molecular genetics of Leber congenital amaurosis. Hum Mol Genet 11(10):1169–1176
Fazzi E, Signorini SG, Scelsa B et al (2003) Leber's congenital amaurosis: an update. Eur J Paediatr Neurol 7(1):13–22
Felius J, Thompson DA, Khan NW et al (2002) Clinical course and visual function in a family with mutations in the RPE65 gene. Arch Ophthalmol 120(1):55–61
Gu SM, Thompson DA, Srikumari CR et al (1997) Mutations in RPE65 cause autosomal recessive childhood onset severe dystrophy. NatGenet 17(2):194–197
Hamel CP, Jenkins N, Gilbert DJ et al (1994) The gene for the retinal pigment epithelium-specific protein RPE65 is localized to human 1p31 and mouse 3. Genomics 20:509–512
Hamel CP, Jenkins N, Gilbert DJ et al (2001) Retinal dystrophies caused by mutations in RPE65: assessment of visual functions. Br J Ophthalmol 85(4):424–427
Hanein S, Perrault I, Gerber S et al (2004) Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum Mutat 23(4):306–317
Heckenlively JR, Foxmann SG (1988) Congenital and early onset forms of retinitis pigmentosa. In: Retinitis pigmentosa. Lippincott, Philadelphia, pp 107–149
Katz ML, Redmond TM (2001) Effect of Rpe65 knockout on accumulation of lipofuscin fluorophores in the retinal pigment epithelium. Invest Ophthalmol Vis Sci 42(12):3023–3030
Lorenz B, Gyurus P, Preising M et al (2000) Early-onset severe rod-cone dystrophy in young children with RPE65 mutations. Invest Ophthalmol Vis Sci 41(9):2735–2742
Lorenz B, Wabbels B, Wegscheider E et al (2004) Lack of fundus autofluorescence to 488 nanometers from childhood on in patients with early-onset severe retinal dystrophy associated with mutations in RPE65. Ophthalmology 111(8):1585–1594
Lotery AJ, Namperumalsamy P, Jacobson SG et al (2000) Mutation analysis of 3 genes in patients with Leber congenital amaurosis. Arch Ophthalmol 118(4):538–543
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215
Moiseyev G, Crouch RK, Goletz P et al (2003) Retinyl esters are the substrate for isomerohydrolase. Biochemistry 42(7):2229–2238
Morimura H, Berson EL, Dryja TP (1999) Recessive mutations in the RLBP1 gene encoding cellular retinaldehyde-binding protein in a form of retinitis punctata albescens. Invest Ophthalmol Vis Sci 40(5):1000–1004
Morimura H, Fishman GA, Grover SA et al (1998) Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or Leber congenital amaurosis. Proc Natl Acad Sci U S A 17;95(6):3088–3093
Narfstrom K, Katz ML, Bragadottir R et al (2003) Functional and structural recovery of the retina after gene therapy in the RPE65 null mutation dog. Invest Ophthalmol Vis Sci 44(4):1663–1672
Paunescu K, Wabbels B, Preising MN et al (2005) Longitudinal and cross-sectional study of patients with early-onset severe retinal dystrophy associated with RPE65 mutations. Graefes Arch Clin Exp Ophthalmol 243 (5):417–426
Poehner WJ, Fossarello M, Rapport AL et al (2000) A homozygous deletion in RPE65 in a small Sardinian family with autosomal recessive retinal dystrophy. Mol Vis 6:192–198
Redmond TM, Yu S, Lee E et al (1998) RPE65 is necessary for production of 11 cis vitamin A in the retinal cycle. Nat Genet 20:344–351
Seeliger MW, Grimm C, Stahlberg F et al (2001) New views on RPE65 deficiency: the rod system is the source of vision in a mouse model of Leber congenital amaurosis. Nat Genet 29(1):70–74
Sharon D, Sandberg MA, Caruso RC et al (2003) Shared mutations in NR2E3 in enhanced S-cone syndrome, Goldmann-Favre syndrome, and many cases of clumped pigmentary retinal degeneration. Arch Ophthalmol 121(9):1316–1323
Simovich MJ, Miller B, Ezzeldin H et al (2001) Four novel mutations in the RPE65 gene in patients with Leber congenital amaurosis. Hum Mutat 18(2):164
Thompson DA, Gyurus P, Fleisher LL et al (2000) Genetics and phenotypes of RPE65 mutations in inherited retinal degeneration. Invest Ophthalmol Vis Sci 41(13):4293–4299
To KW, Adamian M, Jakobiec FA et al (1996) Clinical and histopathologic findings in clumped pigmentary retinal degeneration. Arch Ophthalmol 114(8):950–955
Veske A, Nillson SE, Narfstrom K et al (1999) Retinal dystrophy of Swedish briard/briard-beagle dogs is due to 4-bp deletion in the RPE65. Genomics 57:57–61
Yamamoto H, Simon A, Eriksson U et al (1999) Mutations in the gene encoding 11-cis retinol dehydrogenase cause delayed dark adaptation and fundus albipunctatus. Nat Genet 22(2):188–191
Yzer S, van den Born LI, Schuil J et al (2003) A Tyr368His RPE65 founder mutation is associated with variable expression and progression of early onset retinal dystrophy in 10 families of a genetically isolated population. J Med Genet 40:709–713
Acknowledgements
RPE65 Study Group: Olfa Charfi, Corinne Kostic, Karim Baglouti, Leonidas Zografos. O.C. and K.B. are from the Hedi Rais Institute of Ophthalmology, Tunis, Tunisia; Y.A. , C.K. and L.Z. from the Department of Ophthalmology, Jules Gonin Eye Hospital, University of Lausanne, Lausanne, Switzerland.
Author information
Authors and Affiliations
Corresponding authors
Additional information
L. El Matri and A. Ambresin contributed equally to this work.
Rights and permissions
About this article
Cite this article
El Matri, L., Ambresin, A., Schorderet, D.F. et al. Phenotype of three consanguineous Tunisian families with early-onset retinal degeneration caused by an R91W homozygous mutation in the RPE65 gene. Graefe's Arch Clin Exp Ophthalmo 244, 1104–1112 (2006). https://doi.org/10.1007/s00417-005-0096-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-005-0096-2