
Abstract
Purpose To examine the presence and functional significance of annular fundus autofluorescence abnormalities in patients with different retinal dystrophies. Methods Eighty one patients were ascertained who had a parafoveal ring of high density on fundus autofluorescence imaging. Sixty two had had a clinical diagnosis of retinitis pigmentosa (RP) or Usher syndrome with normal visual acuity. Others included a case of Leber congenital amaurosis and genetically confirmed cases of cone or cone-rod dystrophy (GUCA1A, RPGR, RIMS1), “cone dystrophy with supernormal rod ERG” (KCNV2) and X-linked retinoschisis (RS1). International-standard full-field and pattern electroretinography (ERG; PERG) were performed. Some patients with rod-cone or cone-rod dystrophy underwent multifocal ERG (mfERG) testing and photopic and scotopic fine matrix mapping (FMM). Results In patients with RP, the radius of the parafoveal ring of high density correlated with PERG P50 (R = 0.83, P < 0.0005, N = 62) and encircled areas of preserved photopic function. In the other patients, AF rings either resembled those seen in RP or encircled an area of central atrophy. Ring radius was inversely related to the PERG P50 component in 4 of 18 cases with a detectable response. FMM showed that arcs of high density were associated with a gradient of sensitivity change. Conclusions Parafoveal rings of high density autofluorescence are a non-specific manifestation of retinal dysfunction that can occur in different retinal dystrophies. Electrophysiology remains essential for accurate diagnosis. The high correlation of autofluorescence with PERG, mfERG and FMM demonstrates that AF abnormalities have functional significance and may help identify suitable patients and retinal areas amenable to future therapeutic intervention.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lipofuscin is derived from the shed and degraded photoreceptor outer segments and normally accumulates in the retinal pigment epithelium (RPE) with age [1–4]. An abnormal increase or depletion of lipofuscin manifests as high or low density areas respectively in fundus autofluorescence (AF) images [5] and such changes may aid in the detection and characterisation of a wide range of inherited retinal disorders, either by accentuating the visibility of fundus abnormalities or by revealing changes not accessible by routine examination or fluorescein angiography [5–13]. Lipofuscin has been shown to fragment when exposed to light [14] and its presence suggests continuing metabolic demand [5]. Absence of autofluorescence suggests either blocking of the incident radiation, photoreceptor cell death and RPE atrophy [5, 6] or disruption of the vitamin A cycle [15, 16].
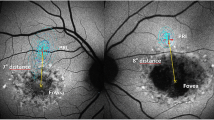
Some patients with genetically different forms of retinitis pigmentosa manifest a ring of high density AF representing abnormal parafoveal accumulation of lipofuscin which usually encircles preserved foveal areas [17–20]. Similar parafoveal rings have recently been documented in other retinal dystrophies including some patients with Leber congenital amaurosis [21], Best disease [13], X-linked retinoschisis [22] and cone-rod dystrophy consequent upon mutation in GUCA1A [23], GUCY2D [24], RIMS1 [25, 26], or RPGR ORF15 [26, 27]. Abnormal annular AF has also been described in some cases of “cone dystrophy with supernormal rod ERG” consequent upon mutation in KCNV2 [28, 29]. Unlike RP, these disorders often result in atrophic macular changes, manifesting as low density AF within the ring.
The main aims of the current study were to review the electrophysiological phenotypes associated with annular AF abnormalities, and to examine two heterogeneous groups of patients in more detail by comparing indices of macular function associated with abnormal macular AF in rod-cone and cone-rod dystrophies.
Materials and methods
Autofluorescence imaging was performed using a scanning laser ophthalmoscope according to previously described techniques [5, 30]. Eighty one patients with abnormal AF of the posterior pole in the form of a high density parafoveal ring were reviewed. Sixty two had a clinical diagnosis of retinitis pigmentosa or Usher syndrome with a visual acuity of 6/9 or better and included 30 cases described previously [17]. Nineteen other individuals were ascertained from previous studies [21–23, 25–29] including 14 with cone-rod or cone dystrophy consequent upon mutation in RPGR ORF15 (4 cases), RIMS1 (8 cases) or GUCA1A (2 cases). Two subjects had “cone dystrophy with supernormal rod ERG” consequent upon mutation in KCNV2, two had RS1mutations and manifested rings in one or both eyes. One patient had Leber congenital amaurosis.
Full-field ERGs were performed according to extended testing protocols incorporating the ISCEV minimum standard [31] in order to assess generalised retinal function. A stimulus 0.6 log units greater than the ISCEV maximum was also used, to demonstrate better the a-wave under conditions of dark adaptation. Pattern ERGs evoked by high contrast checkerboard reversal were recorded according to ISCEV recommendations [32] using standard parameters; checkerboard size 12 × 15 degrees, check size 45′, Michelson contrast 0.98. The PERG P50 component was used as an index of macular function. Additional PERG testing was performed using a range of circular checkerboard fields ranging from 3 to 18 degrees in diameter presented in a random order [17]. Check size was constant at 45′. International-standard mfERGs [33, 34] and fine matrix mapping were performed in some cases. Fine matrix mapping measures rod and cone system sensitivity and has been described previously [18, 35, 36]. In brief, detection thresholds for a spot of light were determined at 1-degree intervals over selected 9 × 9-degree retinal areas. Data were presented both as sensitivity contours, illustrating the position and orientation of tested retinal locations and as three-dimensional threshold profiles, plotted using interpolated values at 0.25-degree intervals, obtained by Gaussian filtering.
Results
All patients were selected according to fundus autofluorescence that was characterised by an abnormal high density parafoveal ring (Figs. 1, 2, 4–8). Sixty two with a clinical diagnosis of retinitis pigmentosa or Usher syndrome and a visual acuity of 6/9 or better had evidence of preserved RPE within the ring. Nineteen others had either cone or cone-rod dystrophy, “cone-dystrophy with supernormal rod ERG”, X-linked retinoschisis or Leber congenital amaurosis. Many of the older non-RP cases had evidence of macular RPE atrophy within the ring (Figs. 2b–d, 7c, 8b–c). Conversely, AF imaging in some RP patients showed mild to moderate atrophic RPE changes within the vascular arcades but external to the ring (Figs. 4d and 5e).

Full-field ERGs, PERGs and AF in 3 patients with rod-cone dystrophy (a–c) including a patient with RP18 (a) and Usher syndrome (b). Row D shows full-field ERGs and AF in a case of Leber congenital amaurosis; ERGs and the AF image were obtained in the presence of nystagmus and are consequently noisy. Normal examples are shown for comparison (e). LU indicates log units greater (+) or less (−) than the ISCEV standard flash

Full-field ERGs, PERGs and AF in cone rod-dystrophy consequent upon mutation in RPGR (a), RIMS1 (b), “cone dystrophy with supernormal rod ERG” (c) and in a case of X-linked retinoschisis (RS1; d). Normal examples are shown for comparison (e). LU indicates log units greater (+) or less (−) than the ISCEV standard flash
Figures 1 and 2 show representative full-field ERGs and PERGs in eight individuals with different retinal dystrophies. Figure 1 shows data from 3 subjects with rod-cone dystrophy and normal visual acuity including a case of autosomal dominant RP18 (Fig. 1a) and Usher syndrome (Fig. 1b). Figure 1d shows undetectable ERGs in a patient with Leber congenital amaurosis; ERGs are contaminated by the effects of nystagmus but no ERG is detectable, in keeping with severe generalised photoreceptor dysfunction. Figure 2 shows representative examples of cone-rod dystrophy consequent upon mutations in RPGR (Fig. 2a), RIMS1 (Fig. 2b) and KCNV2 (“cone dystrophy with supernormal rod ERG”; Fig. 2c). Figure 2d shows the ERG and AF findings in a patient with X-linked retinoschisis.
In the 62 RP cases PERG P50 components varied between normal (>2 uV) and undetectable, consistent with varying degrees of macular involvement. There was no significant correlation with the bright flash ERG a-wave amplitude or 30 Hz flicker ERG amplitude. Figure 3 shows high positive correlation between PERG P50 and mean ring radius (r = 0.83, P < 0.0005). Data are shown from one RP patient that was tested comprehensively (Fig. 4); PERGs were normal to the smallest diameter checkerboard but minimal enlargement was seen as the stimulus field size was increased (Fig. 4b). Multifocal ERGs showed widespread reduction with relative preservation of the response associated with the central stimulus element (Fig. 4a), consistent with a central island of visual field preservation (Fig. 4c). The internal edge of visual field constriction corresponded closely with the ring of high density, as shown by photopic FMM (Fig. 4d). Scotopic fine matrix mapping revealed rod sensitivity losses that were severe and that encroached upon the central macula within the ring. Additional examples of mfERGs, visual fields and fine matrix mapping in RP patients are shown in Figs. 5 and 6.

Comparison of mean ring radius with PERG P50 in 62 patients with rod-cone dystrophy (RP; broken linear regression line) and normal visual acuity and in 19 patients with other retinal dystrophies, including 4 cone or cone-rod dystrophy cases in which there was a detectable PERG (solid regression line)

Multifocal ERGs (a), small field PERGs (b), Humphrey visual field (c) and photopic (d) and scotopic (e) fine matrix mapping in a patient with a clinical diagnosis of RP. Diamonds and error bars in (b) show mean values and standard deviations for 8 normal subjects; triangles and squares show patient data from right and left eyes. Contour plots (d and e) show sensitivity gradients over tested retinal locations; corresponding 3-D plots show retinal location (abscissa, degrees) and thresholds (ordinate, log units). Labelling (x) shows correspondence between the orientation of contour and threshold plots. Normal photopic and scotopic values are plotted in Fig. 5a and d

Contour sensitivity plots (rows 1 and 3) and 3-D threshold plots (rows 2 and 4) obtained in representative normal subjects (a, d) and in 3 RP patients (b, e and c, f). Subjects were tested under photopic (a–c) and/or scotopic conditions (d–f). Labelling (x) shows correspondence between the orientation of contour and threshold plots. Abscissa shows retinal location (degrees), ordinate axes show threshold (log units). Corresponding photopic FMM in individual (e) has been published elsewhere [18]. Normal 3-D plots show averaged data from 14 (a) or 12 (d) normal subjects

AF images, mfERGs and corresponding Humphrey visual fields in 3 patients with RP and normal visual acuity
Pattern ERGs were detectable in only 4 patients with cone or cone-rod dystrophy (GUCA1A, RPGR or RIMS1, Fig. 3). Pattern ERG P50 was inversely related to ring size in these subjects (Fig. 3). Data from one subject with cone-rod dystrophy (RIMS1) are shown in Fig. 7. Multifocal ERG showed widespread reduction with only relative preservation of the central response (Fig. 7a). Standard Humphrey visual fields showed a central scotoma and some superior field loss (Fig. 7b). Fine matrix mapping revealed severe threshold elevation across the macula but with relatively preserved photopic sensitivity over a central island of preserved RPE AF (Fig. 7c). Threshold values inside the ring are maximally elevated over a concentric atrophic area and show a gradient of increasing sensitivity over the arc of high density (Fig. 7c and e). The PERGs in this patient were undetectable (data not shown). Additional examples of fine matrix mapping in RIMS1 and RPGR patients are illustrated in Fig. 8. Central RPE atrophy was not always present (Figs. 2a, 8a).

Multifocal ERGs (a), Humphrey visual field (b) and photopic (c, d) and scotopic (e, f) fine matrix mapping in a patient with cone-rod dystrophy consequent upon RIMS1 mutation. Labelling (x) shows correspondence between the orientation of contour and 3-D threshold plots. Abscissa shows retinal location (degrees), ordinate axes show threshold (log units). Threshold values for half the tested area have been removed from c and e, to expose the foveal values that would otherwise be obscured

Contour sensitivity plots (rows 1 and 3) and 3-D threshold plots (rows 2 and 4) obtained in 3 patients with cone-rod dystrophy consequent upon RPGR (column 1) or RIMS1 mutations (columns 2 and 3). Subjects were tested under photopic (a–c) and scotopic conditions (d–f). Labelling (x) shows correspondence between the orientation of contour and threshold plots. Abscissa shows retinal location (degrees), ordinate axes show threshold (log units)
Discussion
This study reviews a heterogeneous group of 81 patients with genetically-determined retinal diseases that manifest a common feature on fundus autofluorescence imaging in the form of a parafoveal ring of high density. Patients with RP and normal visual acuity had rings that encircled preserved central AF. Mild to moderate atrophic changes were occasionally detected within the vascular arcades but eccentric to the ring (Figs. 4d, 5e). In the other retinal dystrophies that were examined in this and in previous studies [25–27], the ring could also encircle preserved central AF but in older subjects there was often central RPE atrophy within the annulus and preserved AF at more eccentric locations.
As patients with RP or different forms of retinal dystrophy can have indistinguishable AF abnormalities, the AF appearance cannot be used to establish a diagnosis in such cases. Non-specific annular increases in AF may be associated with a wide variety of distinctive or pathognomonic full-field ERG changes that are essential for accurate diagnosis and functional phenotyping (see below).
The data from 62 patients with RP and normal visual acuity show a high positive correlation between ring size and the PERG P50 component extending and confirming findings in a cohort of 30 of these cases [17], demonstrating the robust nature of this relationship. The findings are corroborated by high spatial resolution fine matrix mapping, visual field data and mfERG testing that shows preserved photopic sensitivity within central areas bordered by the ring, consistent with previous reports [18–20, 37]. Scotopic sensitivity losses encroach upon central macular areas suggesting that rod-system dysfunction precedes abnormal parafoveal accumulation of lipofuscin and progressive visual field loss [18]. Serial data indicate that the rings may vary greatly in terms of their stability; to date only 3 cases have been reported in which progressive ring constriction has occurred [37]. The rate of AF ring constriction may prove to be of prognostic value in predicting retention of visual acuity and visual field preservation, but further monitoring is required.
In addition to RP, the parafoveal ring of high density may occur in cone or cone-rod dystrophy consequent upon mutation in GUCA1A [23], GUCY2D [24], RPGR [25, 26], RIMS1 [26, 27], in “cone dystrophy with supernormal rod ERG” (KCNV2) [28], in X-linked retinoschisis (RS1) [22] and in Leber congenital amaurosis [21]. Similar AF findings have also been documented in Best Disease [13] and other maculopathies [10]. In young patients with cone-rod dystrophy, small rings may have preserved central AF and are similar to those seen in RP cases [26]. Older individuals tend to manifest central atrophic changes that are encircled by the ring and there may be a central island of RPE preservation. Fine matrix mapping suggests that the abnormal accumulation of lipofuscin in cases of cone-rod dystrophy represents a transitional stage between relatively preserved parafoveal function and severe central dysfunction that is likely to precede central atrophy [26, 27]. In patients with cone-rod dystrophy consequent upon RPGR or RIMS1 mutations, serial studies have recently demonstrated ring expansion [26], suggesting an expanding front of macular photoreceptor dysfunction. This contrasts with RP where the opposite occurs [37]. It is possible that rings associated with other causes of maculopathy may also expand with time as lesions become larger with age. It is noted that RPGR mutations are more commonly associated with X-linked retinitis pigmentosa [38] with visual acuity reduction [39]; in the current study not all patients underwent genetic screening but none were known to have X-linked RP.
Lipofuscin accumulation in the RPE is likely to reflect metabolic activity which is largely determined by outer segment renewal. There is evidence of light-induced degradation of lipofuscin [14], so that its presence would depend upon continuing metabolic demand. Evidence from RCS rats indicates that failure of RPE cells to phagocytose outer segments results in reduced lipofuscin levels [40]. Lipofuscin formation is almost completely dependent on a normal visual cycle and on the availability of dietary vitamin A [41]. In the RPE65 knockout mouse, the visual cycle is impaired and fluorescence is reduced [42]. Similarly, patients with RPE65 mutations can have reduced autofluorescence in the presence of near-normal fundi and OCT findings that do not suggest reduction in the photoreceptor layer thickness [15, 16]. It is widely accepted that photoreceptor degeneration results in atrophy and reduced rather than stable levels of autofluorescence and it is likely that autofluorescence is lost within a few weeks of photoreceptor loss. The presence of foveal or parafoveal AF in the photoreceptor dystrophies outlined in the current study, suggest that photoreceptors are likely to be intact and thus may be amenable to functional rescue. Recent developments, for example in the field of gene therapy, make early identification of candidate patients increasingly important.
Patients with RP classically present with impaired night vision and visual field constriction, consistent with generalised retinal dysfunction involving rod more than cone photoreceptors. In the early stages the fundi may be near normal and rod-cone dystrophy is established by full-field ERG testing (Fig. 1). In patients with normal visual acuity, the degree of macular sparing cannot be predicted from the severity of peripheral dysfunction. The cone and cone-rod dystrophies are typically characterised by progressive worsening of visual acuity, dyschromatopsia, and photophobia with eventual central scotomata and peripheral field abnormalities. Ophthalmoscopic abnormalities, when present, are generally confined to the macula. Photopic full-field ERGs are typically delayed and subnormal with milder scotopic ERG abnormalities (Fig. 2a and b). A notable exception is the autosomal dominant cone dystrophy consequent upon mutation in GUCA1A; cone-mediated ERGs show amplitude reduction without significant implicit time delay [23] and although not diagnostic, may enable focussed mutational screening. Patients with “cone dystrophy with supernormal rod ERG” exhibit a wide range of fundus AF abnormalities including ring-like or bull’s eye changes, central atrophy or increased foveal AF [28]. Full field ERGs are pathognomonic in this disorder (Fig. 2c), have recently been shown to be consequent upon mutation in KCNV2 and have been described in detail [28, 29]. X-linked retinoschisis is usually associated with an electronegative bright flash ERG in keeping with inner retinal dysfunction (Fig. 2d); two cases (aged 47 years and 49 years) with annular AF abnormalities have been documented within a series of seven atypical but genetically confirmed cases [22]. The rings of high density in these individuals surround areas of central atrophy and may represent an outer boundary of macular photoreceptor dysfunction in addition to generalised inner rather than outer retinal disease. Commonly, younger individuals with X-linked retinoschisis manifest stellate macular lesions that are also visible in AF images [43].
Conclusions
A parafoveal ring of high density autofluorescence is a non-specific manifestation seen in different retinal dystrophies. Electrophysiology remains essential for accurate diagnosis. The high correlation of autofluorescence with PERG, mfERG and FMM demonstrates that AF abnormalities have functional significance and may be an important parameter in the monitoring of these patients. Autofluorescence may be of prognostic value and may help identify suitable patients and retinal areas amenable to future therapeutic intervention.
References
Wing GL, Blanchard GC, Weiter JJ (1978) The topography and age relationship of lipofuscin concentration in the retinal pigment epithelium. Invest Ophthalmol Vis Sci 17:601–607
Feeney-Burns L, Eldred GE (1983) The fate of the phagosome: conversion to “Age Pigment” and impact in human retinal pigment epithelium. Trans Ophthal Soc UK 103:416–421
Kennedy CJ, Rakoczy PE (1995) Constable IJ. Lipofuscin of the retinal pigment epithelium: a review. Eye 9:763–771
Delori FC, Goger DG, Dorey CK (2001) Age-related accumulation and spatial distribution of lipofuscin in RPE of normal subjects. Invest Ophthalmol Vis Sci. 42:1855–1866
von Rückmann A, Fitzke FW, Bird AC (1995) Distribution of fundus autofluorescence with a scanning laser ophthalmoscope. Br J Ophthalmol 79:407–412
von Rückmann A, Fitzke FW, Bird AC (1999) Distribution of pigment epithelium autofluorescence in retinal disease state recorded in vivo and its change over time. Graefes Arch Clin Exp Ophthalmol 237:1–9
Lois N, Holder GE, Fitzke FW, Plant C, Bird AC (1999) Intrafamilial variation of phenotype in Stargardt macular dystrophy-fundus flavimaculatus. Invest Ophthalmol Vis Sci 40:2668–2675
Lois N, Halfyard AS, Bird AC, Holder GE, Fitzke FW (2000) Quantitative evaluation of fundus autofluorescence imaged “in vivo” in eyes with retinal disease. Br J Ophthalmol 84:741–745
Lois N, Holder GE, Bunce C, Fitzke FW, Bird AC (2001) Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch Ophthalmol 119:359–369
Kurz-Levin MM, Halfyard AS, Bunce C, Bird AC, Holder GE (2002) Clinical variations in assessment of bull’s-eye maculopathy. Arch Ophthalmol 120:567–575
Holz FG, Bellman C, Staudt S, Schütt F, Völcker HE (2001) Fundus autofluorescence and development of geographic atrophy in age-related macular degeneration. Invest Ophthalmol Vis Sci 42:1051–1056
Holder GE, Robson AG, Hogg CR, Kurz-Levin M, Lois N, Bird AC (2003) Pattern ERG: clinical overview, and some observations on associated fundus autofluorescence imaging in inherited maculopathy. Doc Ophthalmol 106:17–23
Jarc-Vidmar M, Kraut A, Hawlina M (2003) Fundus autofluorescence imaging in Best’s vitelliform dystrophy. Klin Monatsbl Augenheilkd 220:861–867
Zhou J, Jang YP, Kim SR, Sparrow JR (2006) Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci USA 103:16182–16187
Lorenz B, Wabbels B, Wegscheider E, Hamel CP, Drexler W, Preising MN (2004) Lack of fundus autofluorescence to 488 nanometers from childhood on in patients with early-onset severe retinal dystrophy associated with mutations in RPE65. Ophthalmology 111:1585–1594
Henderson R, Lorenz B, Moore AT (2006) Clinical and molecular genetic aspects of Leber’s congenital amaurosis. In: Lorenz B (Eds) Essentials in ophthalmology. Springer-Verlag, Berlin, pp 133–155
Robson AG, El-Amir A, Bailey C, Egan CA, Fitzke FW, Webster AR, Bird AC, Holder GE (2003) Pattern ERG correlates of abnormal fundus autofluorescence in patients with retinitis pigmentosa and normal visual acuity. Invest Ophthalmol Vis Sci 44:3544–3550
Robson AG, Egan CA, Luong VA, Bird AC, Holder GE, Fitzke FW (2004) Comparison of fundus autofluorescence with photopic and scotopic fine-matrix mapping in patients with retinitis pigmentosa and normal visual acuity. Invest Ophthalmol Vis Sci 45:4119–4125
Robson AG, Egan C, Holder GE, Bird AC, Fitzke FW (2003) Comparing rod and cone function with fundus autofluorescence images in retinitis pigmentosa. Adv Exp Med Biol 533:41–47
Popovic P, Jarc-Vidmar M, Hawlina M (2005) Abnormal fundus autofluorescence in relation to retinal function in patients with retinitis pigmentosa. Graefes Arch Clin Exp Ophthalmol 243:1018–1027
Scholl HP, Chong NH, Robson AG, Holder GE, Moore AT, Bird AC (2004) Fundus autofluorescence in patients with leber congenital amaurosis. Invest Ophthalmol Vis Sci 45:2747–2752
Tsang SH, Vaclavik V, Bird AC, Robson AG, Holder GE (2007) Novel phenotypic and genotypic findings in X-linked retinoschisis. Arch Ophthalmol 125:259–267
Downes SM, Holder GE, Fitzke FW, Payne AM, Warren MJ, Bhattacharya SS, Bird AC (2001) Autosomal dominant cone and cone-rod dystrophy with mutations in the guanylate cyclase activator 1A gene-encoding guanylate cyclase activating protein-1. Arch Ophthalmol 119:96–105
Downes SM, Payne AM, Kelsell RE, Fitzke FW, Holder GE, Hunt DM, Moore AT, Bird AC (2001) Autosomal dominant cone-rod dystrophy with mutations in the guanylate cyclase 2D gene encoding retinal guanylate cyclase-1. Arch Ophthalmol 119:1667–1673
Ebenezer ND, Michaelides M, Jenkins SA, Audo I, Webster AR, Cheetham ME, Stockman A, Maher ER, Ainsworth JR, Yates JR, Bradshaw K, Holder GE, Moore AT, Hardcastle AJ (2005) Identification of novel RPGR ORF15 mutations in X-linked progressive cone-rod dystrophy (XLCORD) families. Invest Ophthalmol Vis Sci 46:1891–1898
Robson AG, Michaelides M, Luong VA, Holder GE, Bird AC, Webster AR, Moore AT, Fitzke FW (2007) Functional correlates of fundus autofluorescence abnormalities in patients with RPGR or RIMS1 mutations causing cone or cone-rod dystrophy. Br J Ophthalmol. Online First: 25 Oct 2007
Michaelides M, Holder GE, Hunt DM, Fitzke FW, Bird AC, Moore AT (2005) A detailed study of the phenotype of an autosomal dominant cone-rod dystrophy (CORD7) associated with mutation in the gene for RIM1. Br J Ophthalmol 89:198–206
Michaelides M, Holder GE, Webster AR, Hunt DM, Bird AC, Fitzke FW, Mollon JD, Moore AT (2005) A detailed phenotypic study of “cone dystrophy with supernormal rod ERG”. Br J Ophthalmol 89:332–339
Wu H, Cowing JA, Michaelides M, Wilkie SE, Jeffery G, Jenkins SA, Mester V, Bird AC, Robson AG, Holder GE, Moore AT, Hunt DM, Webster AR (2006) Mutations in the gene KCNV2 encoding a voltage-gated potassium channel subunit cause “cone dystrophy with supernormal rod electroretinogram” in humans. Am J Hum Genet 79:574–579
Robson AG, Moreland JD, Pauleikhoff D, Morrissey T, Holder GE, Fitzke FW, Bird AC, van Kuijk FJ (2003) Macular pigment density and distribution: comparison of fundus autofluorescence with minimum motion photometry. Vision Res 43:1765–1775
Marmor MF, Holder GE, Seeliger MW, Yamamoto S (2004) Standard for clinical electroretinography (2004 update) Doc Ophthalmol 108:107–114
Holder GE, Brigell MG, Hawlina M, Meigen T, Vaegan, Bach M (2007) ISCEV standard for clinical pattern electroretinography—2007 update. Doc Ophthalmol 114:111–116
Marmor MF, Hood DC, Keating D, Kondo M, Seeliger MW, Miyake Y (2003) Guidelines for basic multifocal electroretinography (mfERG). Doc Ophthalmol 106:105–115
Bellmann C, Neveu MM, Scholl HP, Hogg CR, Rath PP, Jenkins S, Bird AC, Holder GE (2004) Localized retinal electrophysiological and fundus autofluorescence imaging abnormalities in maternal inherited diabetes and deafness. Invest Ophthalmol Vis Sci 45:2355–2360
Chen JC, Fitzke FW, Pauleikhoff D, Bird AC (1992) Functional loss in age-related Bruch’s membrane change with choroidal perfusion defect. Invest Ophthalmol Vis Sci 33:334–340
Westcott MC, Garway-Heath DF, Fitzke FW, Kamal D, Hitchings RA (2002) Use of high spatial resolution perimetry to identify scotomata not apparent with conventional perimetry in the nasal field of glaucomatous subjects. Br J Ophthalmol 86:761–766
Robson AG, Saihan Z, Jenkins SA, Fitzke FW, Bird AC, Webster AR, Holder GE (2006) Functional characterisation and serial imaging of abnormal fundus autofluorescence in patients with retinitis pigmentosa and normal visual acuity. Br J Ophthalmol 90:472–479
Shu X, Black GC, Rice JM, Hart-Holden N, Jones A, O’Grady A, Ramsden S, Wright AF (2007) RPGR mutation analysis and disease: an update. Hum Mutat 28:322–328
Sandberg MA, Rosner B, Weigel-DiFranco C, Dryja TP, Berson EL (2007) Disease course of patients with X-linked retinitis pigmentosa due to RPGR gene mutations. Invest Ophthalmol Vis Sci 48:1298–1304
Katz ML, Drea CM, Eldred GE, Hess HH, Robison WG (1986) Influence of early photoreceptor degeneration on lipofuscin in the retinal pigment epithelium. Exp Eye Res 43:561–573
Radu RA et al (2005): Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseases. Invest Ophthalmol Vis Sci 46:4393–4401
Katz ML, Redmond MT (2001) Effect of Rpe65 knockout on accumulation of lipofuscin fluorophores in the retinal pigment epithelium. Invest Ophthalmol Vis Sci 42:3023–3030
Wabbels B, Demmler A, Paunescu K, Wegscheider E, Preising MN, Lorenz B (2006) Fundus autofluorescence in children and teenagers with hereditary retinal diseases. Graefes Arch Clin Exp Ophthalmol 244:36–45
Acknowledgements
The Foundation Fighting Blindness (AGR). We are grateful to Vy Luong at the Institute of Ophthalmology and to the electrophysiology technicians at Moorfields Eye Hospital.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Robson, A.G., Michaelides, M., Saihan, Z. et al. Functional characteristics of patients with retinal dystrophy that manifest abnormal parafoveal annuli of high density fundus autofluorescence; a review and update. Doc Ophthalmol 116, 79–89 (2008). https://doi.org/10.1007/s10633-007-9087-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10633-007-9087-4