
Abstract
Visceral myopathy is a rare, life-threatening disease linked to identified genetic mutations in 60% of cases. Mostly due to the dearth of knowledge regarding its pathogenesis, effective treatments are lacking. The disease is most commonly diagnosed in children with recurrent or persistent disabling episodes of functional intestinal obstruction, which can be life threatening, often requiring long-term parenteral or specialized enteral nutritional support. Although these interventions are undisputedly life-saving as they allow affected individuals to avoid malnutrition and related complications, they also seriously compromise their quality of life and can carry the risk of sepsis and thrombosis. Animal models for visceral myopathy, which could be crucial for advancing the scientific knowledge of this condition, are scarce. Clearly, a collaborative network is needed to develop research plans to clarify genotype–phenotype correlations and unravel molecular mechanisms to provide targeted therapeutic strategies. This paper represents a summary report of the first ‘European Forum on Visceral Myopathy’. This forum was attended by an international interdisciplinary working group that met to better understand visceral myopathy and foster interaction among scientists actively involved in the field and clinicians who specialize in care of people with visceral myopathy.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The European Forum on Visceral Myopathy 2022 (EFVM2022) was held from April 27th to April 29th, 2022 in Camogli, Genova, Italy (https://poic-e-dintorni.org/efvm-2022/). This was the first international meeting focused on visceral myopathy (VSCM), a life-threatening disease characterized by profound impairment of the gastrointestinal tract, urinary system, and uterine muscles. VSCM is an extremely rare disease currently without mechanism-based therapy. Speakers and attendees included clinicians and researchers from around the world, representatives from support organizations, as well as families affected by this disease. The goals of the conference were to appraise what is known about VSCM, share research data, and raise awareness on this poorly understood condition. The present review represents a position paper about VSCM and is organized into six sections, which recapitulate the evidence-based data presented by the speakers: ‘Clinical manifestations, diagnosis, and initial management,’ ‘Genetics and disease mechanisms,’ ‘Smooth muscle cell biology,’ ‘Model systems,’ ‘Outcomes, current therapy and future innovative treatments,’ and ‘Tools and activities to support VSCM research.’
Clinical Manifestations, Diagnosis, and Initial Management
Clinical Manifestations
‘Visceral myopathy’ refers to altered integrity of the smooth muscle of the whole gastrointestinal tract and two specific extra-digestive organs (urinary tract and uterus). Muscle disease restricted to these organs occurs because visceral (bowel, bladder, ureter, and uterus) smooth muscle differs from vascular or airway smooth muscle, as well as from cardiac or skeletal muscle. Each muscle type is uniquely adapted to its role by virtue of remarkably divergent cell shapes and expression of muscle subtype-specific proteins. For example, vascular smooth muscle differs from visceral smooth muscle in relative abundance of specific actin isoforms expressed in each cell type. For this reason, people with the actin-based forms of visceral myopathy have impaired bowel, bladder, and uterine smooth muscle function, typically without vascular, cardiac or skeletal muscle manifestations [1]. Visceral myopathy symptoms often present during infancy but can also begin at later ages [1, 2]. Symptoms include a massively dilated bowel full of air, fluid, and often partially digested food. Contents move very slowly through the bowel. Nausea, vomiting, constipation or diarrhea, abdominal distension, and pain are common, often with extremely severe symptoms. Most people with visceral myopathy rely at least intermittently on parenteral nutrition for survival. In addition to parenteral nutrition, feeding tubes in the stomach (gastrostomy) or jejunum (jejunostomy) are commonly required. These tubes may be used for slow drip feeding, but also to decompress the bowel by draining air and fluid. These intestinal manifestations of visceral myopathy are also called myopathic chronic intestinal pseudo-obstruction—mCIPO. In people with mCIPO, urinary bladder is frequently dilated and empties poorly. Thus, bladder catheter drainage may be needed to reduce the risk of infection and chronic kidney disease due to obstructive uropathy. Symptom onset and severity vary considerably for people with VSCM, even in family members with the same disease-causing mutation [3, 4]. For example, in some cases, children experiencing severe disease from infancy can have parents that are only mildly affected, suggesting that second-site genetic modifiers or non-genetic factors (e.g., diet and gut microbes) are likely to impact disease manifestations. Waxing and waning disease severity also occurs in single affected individuals, with rapidly deteriorating bowel dysmotility in the setting of acute infections (e.g., upper respiratory infection). In the most severe forms of VSCM, the bladder (and ureter) can be dilated at birth (megacystis/megaureter) and the colon has a reduced diameter (microcolon), presumably because movement of intraluminal contents is required for normal prenatal colon development. This combination of neonatal symptoms is called Megacystis Microcolon Intestinal Hypoperistalsis Syndrome (MMIHS). MMIHS is often fatal in childhood, even with optimal care with studies reporting up to 80% mortality before 20 years of age [5]. Death may result from sepsis or volvulus of massively dilated bowel loops, even with state-of-the-art treatment.
Diagnosis and Initial Management
Imaging tests are crucial at the time of diagnosis to define anatomy and exclude mechanical obstruction. Although symptoms of mechanical obstruction overlap those of VSCM, management is distinct. Mechanical obstruction requires prompt surgical intervention to prevent death. In contrast, surgery is not helpful when symptoms are from myopathy and recovery from surgery is often prolonged in people with VSCM. Even in cases with established mCIPO, distinguishing mechanical from non-mechanical obstruction can be difficult, since dilated bowel is prone to volvulus and adhesions may trap distended bowel. Common diagnostic methods to define visceral anatomy and exclude mechanical obstruction include abdominal computerized tomography (CT with/without oral contrast medium), magnetic resonance imaging (MRI) enterography (MRE), and upper gastrointestinal radiology (Fig. 1) and small bowel follow through. CT and MRE are often used because of their high diagnostic accuracy. Gastrointestinal (GI) motility can be investigated via gastric emptying tests, cine-MRI of the whole gut, and by colonic and antroduodenal manometry (ADM). Gastric emptying may predict the likelihood that feeding into the stomach will be beneficial, while ADM may predict success of enteral feeding (via jejunostomy), or response to medical treatment. Muscle contractility and pharmacological responses to various drugs can be tested in vitro in oxygenated organ baths. However, this approach is usually restricted to research and only applicable in few centers with translational (basic and clinical) laboratory expertise.

Radiological (abdomen X-ray) images of a VSCM patient with chronic intestinal pseudo-obstruction (mCIPO). A Single supine view of the abdomen shows diffuse bowel distention. An ostomy projects over the right lower quadrant, near a cluster of surgical staples. Multiple gallstones are demonstrated in the right upper quadrant measuring up to 16 mm maximum dimension. The external portion of the central venous catheter is partially imaged at the lower right chest. B The duodenum is distended, and peristalsis is inefficient. Transit through the duodenal sweep was aided by patient positioning. Beyond the duodenojejunal junction, the contrast medium progressed slowly through very dilated segments of bowel. Bowel caliber measures up to 6–7 cm. No stricture was identified
When mCIPO is suspected, full thickness bowel biopsy may be recommended using minimally invasive approaches, such as laparoscopic or robot-assisted surgery. Alternatively, interventional endoscopic techniques can be used to obtain full thickness biopsies, but may be less commonly available. Histopathology reveals intestinal myopathic changes (e.g., abnormal smooth muscle layering) in ~ 50% of cases [6, 7], a finding that corroborates the clinical features (e.g., a myopathic pattern at ADM) and supports a diagnosis of VSCM. However, histopathological findings do not impact management unless muscle layer immune cell infiltration (hence inflammatory or immune-mediated leiomyopathy) is demonstrated. If immune cell infiltration of muscle is seen, immunosuppressive treatment may be started [8], but may not help if there is already extensive smooth muscle fibrosis. Venting gastrostomy and diverting ileostomy are almost universally beneficial, whereas caution should be exercised before proceeding with any other surgical intervention, especially in forms of CIPO characterized by diffuse involvement of the smooth muscle. Even minimally invasive surgery carries risks of post-surgical sequelae, and recovery of bowel function after surgery is often prolonged in people with VSCM. Stoma reversal might be considered in a minority of people with mCIPO who have long-standing stable disease but minimal symptoms (e.g., at least 2 years without pseudo-obstruction episodes, no enteral/parenteral feeding requirement, minimal bowel dilatation). A Duhamel pull-through [9] seems to be a preferred option to reestablish intestinal continuity and close ostomies.
Genetics and Disease Mechanisms
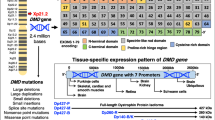
ACTG2, the gene most commonly mutated in people with VSCM, encodes actin G2 (actin gamma 2, smooth muscle), one of the 6 actin isoforms in humans [10]. ACTG2 is the primary smooth muscle actin in bowel, bladder, and uterus. However, visceral smooth muscle cells also express ACTA2 (actin alpha 2, smooth muscle), and all cells express the cytoplasmic acting ACTB (actin beta) and ACTG1 (actin gamma 1). In contrast to visceral smooth muscle, vascular (blood vessel) smooth muscle cells produce more ACTA2 than ACTG2. Cardiac smooth muscle expresses primarily ACTC1 (actin alpha cardiac muscle 1) and skeletal muscle ACTA1 (actin alpha 1, skeletal muscle) [10]. These differences in gene expression explain why ACTG2 variants cause VSCM, whereas similar variants in ACTA2 cause aneurysms and aortic dissection [11]. ACTC1 variants cause cardiomyopathies (heart disease) [12] and ACTA1 variants cause skeletal muscle weakness (e.g., nemaline rod myopathy) [13]. So far (April 2022), 132 ACTG2 variants have been reported to produce VSCM, with most reported in fewer than 5 families. Almost all ACTG2 variants that cause VSCM are due to changes in a single amino acid and cause disease in heterozygotes (i.e., one mutant allele is enough to cause VSCM). For this reason, VSCM may show a dominant inheritance pattern or result from de novo ACTG2 mutation, especially in the most severely affected individuals [3, 4]. In 80% of the cases, the affected amino acid is an arginine residue [14], due to the fact that C to T variation occurs readily and the first codon for arginine starts with C. A severe clinical phenotype is observed for 60% of known ACTG2 variants, requiring parenteral nutrition, or drastic therapeutic measures (e.g., small bowel transplant), and even with current state-of-the-art therapy outcomes remain poor (i.e., early death). ACTG2 R257C is the most commonly identified cause of VSCM and, along with ACTG2 R178 variants, it causes a very disabling form of disease [3]. ACTG2 R40 variants have variable severity myopathy, whereas ACTG2 R38 variants are usually less severe. Second site modifiers (genetic or non-genetic) impact disease course and are responsible for the considerable variety in symptom onset and severity even within families [15, 16]. An example is represented by a family of eleven people with a broad spectrum of visceral manifestations, where eight affected members showed severe complications due to biliary and/or urinary tracts dysfunction in addition to CIPO. All affected mothers had a history of assisted deliveries owing to poor progress during labor and weak uterine contractions. The genetic tests performed in this family revealed an ACTG2 G269E variant [16]. If we could identify second site modifiers, they might provide additional therapeutic targets to reduce disease severity without the need to correct the ACTG2 mutation.
In addition to ACTG2, variants in several other genes may cause severe gut dysmotility due to VSCM including myosin heavy chain 11 (MYH11) [17], myosin light chain kinase (MYLK) [18], and myosin light chain 9 (MYL9) [19] (essential components of the contractile apparatus in smooth muscle cells), leiomodin 1 (LMOD1, a protein that nucleates formation of actin filaments in smooth muscle) [18], and filamin A (FLNA, an actin crosslinking protein that anchors membrane proteins to the cytoskeleton) [20]. Table 1 summarizes the most commonly identified genes associated with VSCM. The encoded proteins primarily function as part of the actin–myosin contractile apparatus are needed for actin polymerization, or regulate actin–myosin and actin–actin interactions. The reasons that mutations in these genes cause visceral myopathy remain incompletely understood, but defects are thought to reduce muscle strength or the ability of smooth muscle to resist passive stretch (Fig. 2).

Visceral myopathy causes profound weakness of bowel, bladder, and uterine smooth muscle resulting in very dilated bowel and bladder. At a cellular level, smooth muscle expressing visceral myopathy-associated gene mutations appears weak and may be unable to resist passive stretch, but there is still very limited data about how specific disease-causing mutations affect smooth muscle cell biology. Many of the proteins encoded by VSCM-associated genes have central roles in actin or actin–myosin-dependent biochemical functions. More detailed information about how specific mutations affect biochemical functions of VSCM-associated proteins could lead to mechanism-based therapy. Created with BioRender.com
Mutations in other genes including LIG3 [21], POLG [22], RAD21 [23], TFAPB2 [24], TYMP [25], SGO1 [26], and SOX10 [27] can cause severe CIPO, likely due to an underlying neuropathy (e.g., RAD21, SOX10, TFAPB2) or neuromyopathy (POLG, TYMP, LIG3, SGO1), although characterization studies are still awaited.
ACTG2 Variants and Bowel Physiology
The bowel has a complex ensemble of tasks, sensing intraluminal nutrients and generating mechanical forces to determine motility patterns that enhance nutrient digestion and absorption, and then expel waste (undigested residues). Smooth muscle provides the force (via active contraction and relaxation) for bowel motility [28]. Specifically, circular muscle contracts to narrow the intestinal diameter, whereas longitudinal muscle shortens the intestine along the longitudinal axis. Smooth muscle cells work in concert with pacemaker cells [i.e., interstitial cells of Cajal (ICC)], and platelet-derived growth factor receptor alpha + (PDGFRA +) cells. These cells are coupled by gap junctions forming the SMC-ICC-PDGFRA “SIP syncytium,” which is controlled by the enteric nervous system (ENS) and influenced by extrinsic sympathetic and parasympathetic innervation as well as hormonal signals [29]. ACTG2 encodes a globular actin monomer (G-actin) that can polymerize to form actin filaments (F-actin). Actin interacts with dozens of other proteins to control polymerization, depolymerization, bundling, and branching of filaments. F-actin interactions with smooth muscle myosin (MYH11) generate the force needed for cell contraction or changes in cell shape [30]. Actin is also required for maintenance of cell polarity, endocytosis, exocytosis, cell division, chromatin remodeling, and regulation of transcription. While some of these processes are thought to be primary functions for cytoplasmic actins ACTB and ACTG1, the roles played by each actin isoform remain incompletely understood. Disease-causing actin variants may interfere with some or all these processes. One key issue is that we need to define normal ACTG2 cellular functions to understand which processes are affected by the ACTG2 disease-causing variants. As an example, whereas ACTG2 variants may reduce actin filament formation or actin–myosin interactions, G-actin also binds and sequesters myocardin-related transcription factor (MRTF) in the cytoplasm, preventing MRTF-SRF (serum response factor) interactions that drive expression of muscle contractile apparatus genes (e.g., ACTG2 and MYH11) [1]. When not bound to MRTF, SRF can induce the expression of extracellular matrix (ECM) and pro-mitogenic genes in concert with ELK1 (ETS transcription factor ELK1). This is a potentially important disease mechanism since smooth muscle cells can undergo phenotypic switching from contractile to “synthetic” or “proliferative” state [31]. This phenotypic switch might also be triggered by mechanical forces [tissue stiffness via Yes1 associated transcriptional regulator (YAP1)] or inflammatory mediators [via nuclear factor kappa B (NFκB)] since YAP1 and NFκB bind to and sequester myocardin (MYOCD), a key transcription factor needed to evoke contractile gene expression in the smooth muscle. Bowel dilatation, which is a common feature in people with VSCM, may change the contractile phenotype as tension on muscle releases YAP1 thereby facilitating its entrance into the nucleus. Furthermore, infections might induce phenotypic switching via nuclear translocation of NFκB.
Smooth Muscle Cell Biology
Visceral smooth muscle differentiates from highly proliferative mesenchymal precursors that migrate and produce extracellular matrix to enable intestinal growth. As mesenchymal precursors differentiate into contractile smooth muscle, they produce myocardin, ACTA2, and ACTG2. Further maturation toward a contractile phenotype leads to MYLK, MYH11 and calponin 1 (CNN1) expression. Remarkably, throughout life, the contractile phenotype of smooth muscle cells is not permanent. Smooth muscle can undergo “phenotypic class switching,” with loss of contractile apparatus genes, increased expression of extracellular matrix proteins, increased proliferation, and increased cell mobility [32, 33]. Phenotypic class switching is controlled in part by the putative RNA binding mitochondrial protein limb and CNS expressed 1 (LIX1), which is highly expressed in mesenchymal precursor cells able to differentiate into contractile smooth muscle [34]. LIX1 levels decline during the transition from precursor to contractile smooth muscle cells. In chick embryo, the sustained expression of LIX1 increases alpha smooth muscle actin expression and prevents CNN1 expression, while maintaining the expression of the intermediate filament protein DESMIN. In contrast, LIX1 silencing in chick splanchnic mesoderm reduces cell proliferation and decreases alpha smooth muscle actin and MYOCD expression [35]. Interestingly, LIX1 was abnormally expressed in 80% of the mCIPO bowel muscles examined (N = 20), suggesting that phenotypic switching from contractile to synthetic smooth muscle cells occurs in vivo in people with mCIPO, causing muscle weakness (Pascal De Santa Barbara unpublished data). Consistent with this hypothesis, many pediatric mCIPO colon biopsies had low levels of alpha smooth muscle actin and a 50% reduction in ACTG2 mRNAs [31]. Furthermore, PDGFRA was expressed at high levels in smooth muscle cells from people with mCIPO. While PGDFRA is low in contractile smooth muscle, its level is high in smooth muscle precursor cells. This again suggests that a phenotypic switch away from the contractile smooth muscle phenotype occurs in vivo in human mCIPO. Interestingly, silencing LIX1 dramatically reduces YAP1/TAZ [34, 35]. Since YAP1 competes with SRF for myocardin [36], reduced YAP1 could enhance the formation of SRF-myocardin complexes that directly induce transcription of smooth muscle contractile genes. These observations suggest that silencing LIX1 could facilitate maintenance of contractile smooth muscle cells in culture or in vivo, potentially representing a novel therapy.
Human intestinal mesenchymal precursors differentiate into circular muscle beginning in the rostral bowel at 8 weeks of gestation [37]. At this stage, MYH11 is present in the region of the circular smooth muscle, but not in the region where longitudinal muscle will eventually form. By 10 weeks of gestation, MYH11 is also present in the longitudinal muscle indicating differentiated contractile smooth muscle cells. Smooth muscle differentiation proceeds in a rostral to caudal progression through the bowel during the 1st trimester of fetal development [37]. It is hypothesized that a common mesenchymal precursor gives rise to smooth muscle, ICC, and PDGFRA cells of the SIP syncytium [38,39,40,41]. In support of this hypothesis, in human bowel at week 8 of gestation, there are cells in the presumptive circular muscle region that co-express the KIT and PDGFRA receptors (Silvia Perin unpublished data). By 9 weeks of human gestation, KIT and PDGFRA are expressed in non-overlapping cell populations (presumptive ICC and PDGFRA cells, respectively). In mouse embryos, presence of KIT and PDGFRA cells in the developing gut was reported at day 12 of gestation [41]. Over time, these genes are expressed in separate cell populations in bowel muscle layers [29]. Until now, the exact origin of gut smooth muscle remains unclear with evidence that smooth muscle can differentiate from gut mesenchyme [42, 43], serosal mesothelium [44], and from pericytes of the vasculature (Silvia Perin unpublished). In support of these hypotheses, it has been shown that bowel pericyte-like cells grown in serum-depleted media generate smooth muscle-like cells over 7 days in culture and that repopulation of decellularized human bowel scaffolds generates tissue with well-oriented smooth muscle layers (Silvia Perin unpublished data).
Model Systems
Traditional Cell Culture
Current VSCM studies rely on a limited number of models. Mostly, they mimic ACTG2-based VSCM, often focusing on the ACTG2 R257C variant, one of the most severe and common forms of the disease. A cellular model of ACTG2 R257C VSCM was produced by Hashmi et al. [45] mimicking human heterozygous disease by overexpressing either wild type (WT) or R257C ACTG2 proteins in cultured human intestinal smooth muscle cells (HISMC). In the presence of the ACTG2 R257C protein, actin filament bundles visualized with phalloidin (which stains all actin filaments) or via electron microscopy were indistinguishable from cells overexpressing WT ACTG2, despite the severe symptoms in people carrying the R257C variant. In contrast, staining for only the tagged ACTG2 protein demonstrated actin filament bundles containing ACTG2 R257C were shorter, thinner, and less branched than those containing tagged WT ACTG2. These results suggest compensation for the ACTG2 R257C variant by other cellular actins (ACTB, ACTG1, or endogenous WT ACTG2). Still, the severity of the human disease indicates ongoing cellular dysfunction caused by mutant ACTG2. Consistent with this hypothesis, human smooth muscle expressing ACTG2 R257C migrate faster and spread more quickly on plastic culture dishes than cells that express WT ACTG2 protein, suggesting that ACTG2 R257C induces phenotypic switching in smooth muscle from the contractile to the synthetic state. Unfortunately, phenotype switching is incompletely modeled when cells are cultured on plastic because hard surfaces sensed by smooth muscle rapidly reduce cellular levels of many contractile apparatus proteins (e.g., 100–1000 fold reductions in ACTG2 and MYH11 mRNA in 24 h).
Another cellular model of VSCM is represented, somewhat surprisingly, by fibroblasts from people with VSCM [46]. Three fibroblast cell lines carrying the ACTG2 R257C variant and one carrying the milder ACTG2 R38H variant were analyzed (at early and uniform cell passages). Compared to controls, represented by fibroblasts from people with Hirschsprung disease (HSCR) or normal bowel function (non-CIPO), ACTG2 R257C fibroblasts are thicker than non-CIPO controls and present higher cytoskeletal anisotropy, migrate faster, and are softer. Traction force microscopy (TFM) using 12 kPa stiffness substrates revealed the most robust differences between groups with dramatically reduced force generation by ACTG2 mutant fibroblasts, intermediate force from HSCR-derived fibroblasts, and much more force from healthy control-derived fibroblasts. Cell migration differences correlate well with human intestinal smooth muscle cells transfected with WT or mutant ACTG2 [45] but are still surprising, since fibroblasts were not previously appreciated to express ACTG2. Nonetheless, the observations suggest a readily accessible patient-derived cell type (skin-derived fibroblasts) can be used to gain insight into visceral myopathy-inducing human variants.
3D In Vitro Models
Due to the complex biology and structure of human bowel, 2D cell cultures only sometimes recapitulate normal physiology or development. 3D in vitro models of human bowel could be generated from human-induced pluripotent stem cells (iPSC) or embryonic stem cells (ESC), using developmental pathways needed for the intestinal development in vivo human intestinal organoids (HIO) from stem cells present several features of natural human bowel. However, they resemble fetal bowel until implanted in vivo (e.g., into mesentery or kidney capsule) in immunodeficient mice [47]. Although the implanted HIO receives signals, such as nutrients and hormones, from the mouse host via vascular and lymphatic networks, 90% of its cells remain of human origin. Adding vagal neural crest-like cells (vNCC, derived from iPSC or ESC) produces a complex organoid, containing neurons and glia [48] that enhance HIO cell diversity and improve smooth muscle maturation. Neural crest-lineage cells can also increase bowel motility, as they differentiate into a functional neuroglial network resembling the enteric nervous system. This HIO-derived bowel can be expanded in vivo, by including a Nitinol memory foam spring within the organoid lumen [49]. Mechanical force of the spring leads to thicker circular and longitudinal muscle layers and more robust responses to acetylcholine, a major pro-contraction neurotransmitter. The described HIOs could be generated from people affected with CIPO using iPSC, as previously demonstrated in the case of a Hirschsprung’s disease caused by PHOX2B mutation [48]. The mature organoids provide muscle layering and enteric nervous system organization, but their development status is approximate second trimester of gestation rather than normal adult human bowel. Furthermore, while generated HIO can contract and relax, the complex motility patterns typical of normal human bowel have not yet been demonstrated in HIO. Forthcoming engineering strategies will be needed to capture the normally intricate motility patterns needed for survival, and these strategies might include bioprinting tubular structures or other engineering approaches.
Another method to generate human bowel in vitro is to repopulate decellularized bowel scaffolds that provide extracellular matrix, orient cells, and promote differentiation. Repopulating the decellularized scaffold requires precursors that can be induced to differentiate into bowel smooth muscle (see ‘Future therapies’ section).
Innovative Cell Culture Systems
Traditional cell culture systems are suboptimal for studying smooth muscle cell biology and VSCM, because smooth muscles are acutely responsive to mechanical forces and change over time in response to rhythmic stretching. Innovative culture methods including gut-on-a-chip approaches can allow uniaxial stretching, while bioreactors such as those commercialized by FlexCell can provide static or cyclical strain to cultured cells. Recently, a new bioreactor has been developed [50], equipped with electrically curved membranes that can be dynamically tuned to create radial and circumferential strains. Cells grown in such bioreactors had more actin fiber bundles which aligned along the direction of the radial strain, a very different phenotype from that obtained when the same cells experience planar stretching. Another approach to repeatedly induce cyclic strain with little hysteresis is represented by the innovative system for bioprinting called the Core–Shell Microbeads Creator (COSMIC). The beads present a core of Pluronic® and alginate, surrounded by paramagnetic nanoparticles that allow the generation of force and motion through magnets [51]. These magneto-responsive gels permit wireless muscle-like actuation, facilitating cell growth and organization in layers. Although applications in vivo are yet unexplored, in theory Core–Shell paramagnetic microbeads might be also adapted to apply controlled force to the bowel as an in vivo “gut assist” device for people with intestinal dysmotility, including VSCM-related CIPO.
Animal Models
A new Drosophila model of visceral myopathy was generated by knocking down Drosophila Act79B (92% identical to ACTG2) in fly visceral mesoderm using Mef2-Act79B RNAi (Antonio Galeone unpublished data). These flies have markedly delayed transit of food through the bowel and fewer gut contractions than WT. To more closely model human VSCM, inducible upstream-activated sequence fly lines were generated producing Drosophila Act79B on one allele and the human WT (UAS-ACTG2) or mutant (UAS-ACTG2 R257C) from the other allele. This model could be used for drug screening since testing involved feeding flies with colored food and observing motility and transit through larval midgut. Candidate drugs could then be evaluated in cell culture, tissue-engineered intestine, or mutant animal models currently under development by other investigators.
Recently, a mouse model carrying the most common heterozygous variant in ACTG2 (R257C), was generated by CRISPR/Cas9 genome-editing [52]. This model showed dilated intestine and bladder reminiscent of the MMIHS phenotype identified in patients. Interestingly, no microscopic morphological changes were observed in the intestine, bladder, stomach, and uterus of these mice by hematoxylin–eosin staining. However, primary intestinal smooth muscle cells isolated from the ACTG2 R257C mouse showed reduced cellular contractility, possibly due to impaired ACTG2 polymerization.
Outcomes, Current Therapies, and Future Innovative Treatments
Outcomes
The most severe form of VSCM, MMIHS, had death rates of 80% during childhood (1976 to 2011 literature review) [53] and 40% mortality before 10 years of age (28 children) [54]. A more recent study reported 86% survival to 20 years of age in patients with MMIHS (N = 25, median age for involved subjects = 9.2 years), but 50% of survivors (11/22) had an intestinal transplant [55]. Neuropathic CIPO has similar symptoms to the myopathic form (Table 2), but a recent study reported 55% death in childhood for mCIPO compared to 8.5% death in childhood for neuropathic CIPO [56]. Even in the case of a “good” outcome, it is common for children affected with mCIPO to spend months in hospitals, with a median length of stay of 6 days and a median cost of $ 52,000 in the United States [57]. Ileostomy and gastrostomy reduced the median cost of hospitalization by about 20%, consistent with the hypothesis that low pressure routes for release of gas and stool are valuable when bowel muscles are weak. Many people with VSCM require total parenteral nutrition (TPN) for survival, at least intermittently. Infection and liver injury represent potentially fatal complications of TPN: survival on TPN is reported to be 90% at 1 year, 70% at 5 years and 60% at 10 years [58]. Electrolyte imbalance, malnutrition, and small intestinal bacterial overgrowth are common problems that dramatically affect health [59, 60]. Over time, people with mCIPO may have replacement of bowel muscle by collagen. These factors, coupled with complex care (ostomy management, enteral feeding, TPN, repeated school and work absence), and daily debilitating symptoms (abdominal pain, abdominal distension, vomiting, nausea, constipation, fatigue) can dramatically reduce the quality of life.
Current therapies
Current therapies primarily address complications of mCIPO. Small intestinal bacterial overgrowth is treated with antibiotics. Fluid and electrolyte imbalances and malnutrition are treated by combinations of intravenous and enteral therapy. All these therapies improve the quality of life and can be life-saving [61]. Narcotics should be avoided, if possible, since opioids exert an anti-motility effect that worsens symptoms and complicates the clinical picture. Furthermore, long-term (weeks) opioid use increases pain sensitivity, causing “narcotic bowel syndrome,” requiring tapering down of opioids to avoid severe, life-threatening complications [56]. Prokinetic agents may be helpful by preventing acetylcholine degradation at the synapse (neostigmine, pyridostigmine), blocking dopamine D2 receptors (domperidone, metoclopramide), activating somatostatin receptors (octreotide), or activating serotonin/5-hydroxytryptamine (5-HT4) receptors (cisapride, prucalopride, velusetrag). 5-HT4 receptors are found pre-synaptically in ascending interneurons and excitatory motor neurons in the bowel as well as in substance P-expressing extrinsic nerve fibers important for visceral pain sensitivity. The primary pro-motility effect of 5-HT4 agonists is via an increased acetylcholine release at the synapse cleft. Acetylcholine induces smooth muscle contraction by activating muscarinic receptors (M2, M3) on smooth muscle cells and on ICC. Therefore, avoiding anti-cholinergic drugs is important [59, 60].
Diet and gut microbes can also be manipulated to enhance bowel motility but optimal therapy is not yet established. Animal models show gut microbes and metabolites impact bowel motility, alter enteric neuron number, influence epithelial cell function, and change bowel immune system function in ways that may contribute to VSCM symptoms [62]. Poor bowel motility and the lack of regular diet predisposes to accumulation of abnormal gut microbial communities, causing dysbiosis, which is common in people with VSCM. Consistent with these observations, a small study of nine people with CIPO showed that fecal transplant significantly reduced abdominal pain and bloating and improved bacterial overgrowth (in 5 out of 7 cases) at eight weeks after treatment [63]. Mechanistically, gut microbes and their metabolites can alter neuronal subtypes in the enteric nervous system via the arylhydrocarbon receptor, increase serotonin synthesis in enteroendocrine cells by inducing tryptophan hydroxylase transcription, increase serotonin in enteric neurons, impact enteric neuron numbers and antimicrobial peptide production via toll like receptors, increase motilin secretion by M-cells (via the bile acid receptor TGR5, long chain fatty acid receptor FFA1, and monoacylglycerol receptor GPR119), and deconjugate cholic acid (a bile acid) to produce deoxycholic acid, which increases bowel motility via TGR5 receptors [64,65,66]. Overall, limited data are available about fecal transplant, the effectiveness of probiotics, or the modulation of metabolites to improve the life of people with VSCM.
Nutrition remains a challenge for people with visceral myopathy. When oral feeding is poorly tolerated, nutrients may be provided via gastrostomy or jejunostomy, often as slow continuous drip feeding. When the enteral feedings are poorly tolerated, partial or total parenteral (intravenous) nutrition are provided, often in combination with small amounts of enteral feeding. In infants, breast milk is considered the best choice, providing nearly complete nutrition with little residual waste, and providing a variety of trophic and antimicrobial molecules. When breast milk is not available, feeding should start with whole protein-based formulas before trying partially hydrolyzed or amino acid-based formulas. Since dysmotility impairs mixing of complex nutrients with digestive enzymes, people with VSCM may benefit from medium chain triglycerides (MCT) that are absorbed without need for hydrolysis. Electrolytes and micronutrients need to be carefully monitored and abnormalities are corrected when identified since vitamin deficiencies and electrolyte abnormalities may affect bowel motility. For example, deficiencies in thiamine (vitamin B1), riboflavin (vitamin B2), niacin (vitamin B3), pyridoxine, cobalamin (vitamin B12), tocopherol (vitamin E), biotin, retinol (vitamin A), folate, iron, and copper cause nervous system disease, while vitamin D deficiency and selenium deficiencies cause muscle weakness.
As reported in the initial section, surgical interventions useful in the VSCM treatment include venting gastrostomy, jejunostomy, and ileostomy, because bowel distension increases the effort necessary to contract the bowel, facilitates bacterial overgrowth, and increases the risk of bacterial translocation, sepsis, and volvulus. Unfortunately, ostomy prolapse is common in people with VSCM and skin irritation around the ostomy can also be a problem. Performing the least invasive procedures is strongly recommended, avoiding open abdominal surgery when possible (e.g., cystostomy is preferred to Mitrofanoff procedure for bladder emptying).
When the strategies described above fail, an alternative therapy is intestinal transplant. The Necker-Enfants Malades in Paris, the largest transplant center in Europe, performed the world’s first small bowel transplant in 1987 [67]. Data from 2021 [68] indicated 5-year post-bowel transplant survival of around 60% in the United States and 50% globally, suggesting chronic TPN may be safer than intestinal transplant. Primary post-transplant problems include early acute rejection, late rejection, graft failure, and complications of immunosuppression including late infection, renal failure, and cancer. The presence of megacystis (large bladder) increases the risk of post-transplant infection. Immunosuppression doses required for bowel transplant are twice as high as for liver transplant, which contributes to adverse outcomes. While outcomes are slowly improving, bowel transplants remain uncommon (~ 4000 bowel transplants since 1988 worldwide, compared to more than 1000 liver transplants each year in France), limiting experience needed to optimize post-transplant management. Because of these issues, the indications for bowel transplant are total intestinal failure with no hope of weaning off TPN, plus complications that make continued TPN impossible or very risky (e.g., repeated line infections, extensive vascular thrombosis, inability to manage TPN at home, progressive TPN-associated liver injury, growth failure, and psychological intolerance of TPN). Although in VSCM, small intestine is the target organ for transplant, stomach should be transplanted along with small bowel, since delayed gastric emptying is common in people with VSCM. Moreover, it is important to transplant right colon and connect it to the rectum, leaving an open ostomy. In the past 10 years, the Necker-Enfants Malades has performed 14 intestinal transplants (3 were second transplants), 4 of them to treat VSCM with ongoing pain, repeated vomiting, limited vascular accesses, and recurrent infections. Of the 14 transplanted, 7 are alive, including 3 with VSCM, with improved quality of life, even though severe complications (including autoimmune hemolytic anemia) arose in the first-year post-transplant in 2 transplant recipients. Three of the transplant recipients died and 4 had the intestinal graft removed.
Future therapies
Future therapies for VSCM might be based on regenerative medicine, the process of creating artificial human bowel for transplantation. The feasibility of generating new organs is demonstrated by artificial trachea created by seeding a decellularized trachea scaffold with human cells. The engineered trachea was then implanted into a child with tracheal stenosis, allowing the child to live [69]. To create new small bowel, decellularized intestine can be seeded with human-derived intestinal organoids. Alternatively, stem cell-based strategies allow bowel like-organoids to be generated without decellularized scaffolds. While these advances are remarkable, it is not yet known how long regions of small bowel can be created that contain the machinery needed for normal bowel motility. Whole organs might also be grown using cells from affected individuals, to learn more about disease mechanisms or to avoid transplant rejection and need for immunosuppression. For people with VSCM, pathogenic variants would need to be corrected in the stem cells before populating the scaffold. However, adverse outcomes may occur when working with engineered organs [70], so a cautious approach to this treatment is recommended. While many challenges need to be overcome before this approach is useful, regenerative medicine (organogenesis) remains an exciting potential future therapy.
Tools and Activities to Support VSCM Research
Patient support groups and societies are of great values in severe and rare diseases such as VSCM. An example of such organization is ‘POIC e dintorni’ an Italian patient advocacy organization (PAO). A video shown on their website (https://poic-e-dintorni.org/this-is-pediatric-cipo) provides a window into the life of a child with VSCM, emphasizing that the disease affects specifically bowel, bladder, and uterus. In this context, if nutritional needs are met, infections are avoided, and abdominal distension is managed by ostomy, venting, and careful attention to diet and medicines, children can live at home most of the time, attend school, and pursue most of the activities of childhood. ‘POIC e dintorni’ (https://poic-e-dintorni.org/) supports families affected by VSCM by sharing experiences, building a community of families and providing them with guidance and financial assistance, increasing public awareness about VSCM, and funding research. In Italy, ‘Uniti per la P.I.P.O.’ (www.unitiperlapipo.it) PAO is another association with a similar aim to improve the lives of children with pediatric CIPO (PIPO). In France, the ‘Association de POIC’ (https://www.association-poic.fr/) PAO was born to support research and help French-speaking families affected by CIPO. Finally, in Europe, the ‘European Reference Network for Rare Diseases’ (ERN-RND) defines best practices for treatment for gastrointestinal congenital anomalies (e.g., Hirschsprung disease, esophageal atresia, etc.), aimed at examining the transition to adulthood, supporting patient registries, and providing affected people with access to expert care.
Although known as one of the most recent and innovative strategies to support data collection on rare diseases, ‘Citizen Science’ has not yet been applied to VSCM. This approach relies on public volunteers to help science by collecting information and collaborating with scientists to analyze and classify data. Another helpful tool that could support VSCM research is ‘biobanking,’ an organized method of storing human biospecimens (blood, urine, tissue, cell lines, DNA/RNA, iPSC, and other specimen types) and associated metadata, often indispensable to foster research in the context of rare diseases. The organization and support of Biobanks is not trivial. It is crucial to clearly define a priori how samples must be collected and retrieved from the biobank, the governance rules, the plans for operational continuity (long-term management), how to disseminate results, how to manage sample and data protection compliantly to ELSI (Ethical, Legal, and Social Issues), access policies, informed consent documents, material transfer agreements, and standard operating procedures. A biobank must rely on a dedicated IT-infrastructure (database) to manage samples and associated data, equipped with a user-friendly interface to make them available to the scientific community. Since the amount of data linked to individual specimens determines the value of the biological samples for future research, biobanks gain value if sample information is connected to the medical records of the participants and their updated clinical data [71]. Carefully annotated human biospecimens stored in a biobank are invaluable not only for human genetic research, but also to support a wide range of new scientific approaches to advances in the field of such rare diseases. A virtuous example of biobanking is represented by the Telethon Network of Genetic Biobank (TNGB), which interconnects 11 Italian Rare Diseases Biobanks, hosting ~ 127,000 biospecimens from people affected with 950 genetic diseases, available for distribution. More than 50,000 samples have been distributed to national and international research groups resulting in > 700 scientific manuscripts [72].
Development of patient registries represents another crucial initiative to support research in rare diseases. A registry is a collection of standardized information about a group of people who share a condition [73]. Crucial issues in registry development include managing privacy and protection of collected data, establishing governance rules for the registry, securing the ongoing support from patient associations and from the medical community (e.g., hospital administration). Patient involvement is recommended when developing registries to allow building services based on real needs of affected individuals and to implement a totally transparent path for the dissemination of information. The value returned to subjects by collaborative network-based observational registries includes improved connections with their network of specialists, and awareness of contributing to knowledge. The value of the registry increases over time, although few registries are successful long term.
Finally, building alliances among medical providers, researchers, and families is crucial for carrying out research of orphan diseases, improving outcomes, advocating for care and research, and building partnerships to find better treatments. These alliances require clear communication, defined roles, time dedicated to understand the culture of the partners, and full disclosure of goals, methods, and the value added by partnerships.
Conclusions
VSCM is an understudied orphan disease that severely compromises the life of affected individuals and their families. This report is based on proceeding of the first European Forum on Visceral Myopathy (2022), attended by scientists and clinicians with expertise in this disease. The meeting spearheaded the birth of a community and fostered new collaborations. Over the past decade, we have learned a lot about VSCM genetics and can identify variants in around 60% of affected individuals. These discoveries position us to build new model systems and to find mechanism-based cures and treatments for these devastating diseases. Dedicated VSCM biobanks or registries could have a tremendous impact on the study of this rare disease. Collaboration among dedicated scientists across several different disciplines who are willing to partner in international and multidisciplinary networks will likely yield significant improvements over the next decade.
References
Hashmi SK, Ceron RH, Heuckeroth RO. Visceral myopathy: clinical syndromes, genetics, pathophysiology, and fall of the cytoskeleton. Am J Physiol Gastrointest Liver Physiol 2021;320:G919–G935.
Fournier N, Fabre A. Smooth muscle motility disorder phenotypes: A systematic review of cases associated with seven pathogenic genes (ACTG2, MYH11, FLNA, MYLK, RAD21, MYL9 and LMOD1). Intractable Rare Dis Res 2022;11:113–119.
Wangler MF, Gonzaga-Jauregui C, Gambin T et al. Heterozygous de novo and inherited mutations in the smooth muscle actin (ACTG2) gene underlie megacystis-microcolon-intestinal hypoperistalsis syndrome. PLoS Genet 2014;10:e1004258.
Matera I, Rusmini M, Guo Y et al. Variants of the ACTG2 gene correlate with degree of severity and presence of megacystis in chronic intestinal pseudo-obstruction. Eur J Hum Genet 2016;24:1211–1215.
Thorson W, Diaz-Horta O, Foster J 2nd et al. De novo ACTG2 mutations cause congenital distended bladder, microcolon, and intestinal hypoperistalsis. Hum Genet 2014;133:737–742.
Kapur RP. Histopathological, Ultrastructural, and Immunohistochemical Findings in MYH11-Variant Visceral Myopathy. Pediatr Dev Pathol 2023;26:39–51.
Kapur RP, Goldstein AM, Loeff DS, Myers CT, Paschal CR. Intestinal Pathology in Patients With Pathogenic ACTG2-Variant Visceral Myopathy: 16 Patients From 12 Families and Review of the Literature. Pediatr Dev Pathol 2022;25:581–597.
De Giorgio R, Camilleri M. Human enteric neuropathies: morphology and molecular pathology. Neurogastroenterol Motil 2004;16:515–531.
Thapar N, Saliakellis E, Benninga MA et al. Paediatric Intestinal Pseudo-obstruction: Evidence and Consensus-based Recommendations From an ESPGHAN-Led Expert Group. J Pediatr Gastroenterol Nutr 2018;66:991–1019.
Kashina AS. Regulation of actin isoforms in cellular and developmental processes. Semin Cell Dev Biol 2020;102:113–121.
Guo DC, Pannu H, Tran-Fadulu V et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet 2007;39:1488–1493.
Despond EA, Dawson JF. Classifying Cardiac Actin Mutations Associated With Hypertrophic Cardiomyopathy. Front. Physiol. 2018;9:405.
Labasse C, Brochier G, Taratuto AL et al. Severe ACTA1-related nemaline myopathy: intranuclear rods, cytoplasmic bodies, and enlarged perinuclear space as characteristic pathological features on muscle biopsies. Acta Neuropathol Commun. 2022;10:101.
Assia Batzir N, Kishor Bhagwat P, Larson A et al. Recurrent arginine substitutions in the ACTG2 gene are the primary driver of disease burden and severity in visceral myopathy. Hum Mutat 2020;41:641–654.
Sandy NS, Huysentruyt K, Mulder DJ et al. The Diverse Phenotype of Intestinal Dysmotility Secondary to ACTG2-related Disorders. J Pediatr Gastroenterol Nutr 2022;74:575–581.
Klar J, Raykova D, Gustafson E et al. Phenotypic expansion of visceral myopathy associated with ACTG2 tandem base substitution. Eur J Hum Genet 2015;23:1679–1683.
Dong W, Baldwin C, Choi J et al. Identification of a dominant MYH11 causal variant in chronic intestinal pseudo-obstruction: results of whole-exome sequencing. Clin Genet 2019;96:473–477.
Halim D, Brosens E, Muller F et al. Loss-of-function variants in MYLK cause recessive megacystis microcolon intestinal hypoperistalsis syndrome. Am J Hum Genet 2017;101:123–129.
Moreno CA, Sobreira N, Pugh E et al. Homozygous deletion in MYL9 expands the molecular basis of megacystis-microcolon-intestinal hypoperistalsis syndrome. Eur J Hum Genet 2018;26:669–675.
Kapur RP, Robertson SP, Hannibal MC et al. Diffuse abnormal layering of small intestinal smooth muscle is present in patients with FLNA mutations and x-linked intestinal pseudo-obstruction. Am J Surg Pathol 2010;34:1528–1543.
Bonora E, Chakrabarty S, Kellaris G et al. Biallelic variants in LIG3 cause a novel mitochondrial neurogastrointestinal encephalomyopathy. Brain 2021;144:1451–1466.
Tang S, Dimberg EL, Milone M, Wong LJ. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)-like phenotype: an expanded clinical spectrum of POLG1 mutations. J Neurol 2012;259:862–868.
Bonora E, Bianco F, Cordeddu L et al. Mutations in RAD21 disrupt regulation of APOB in patients with chronic intestinal pseudo-obstruction. Gastroenterology 2015;148:771–782.
Zada A, Kuil LE, de Graaf BM et al. TFAP2B Haploinsufficiency Impacts Gastrointestinal Function and Leads to Pediatric Intestinal Pseudo-obstruction. Front Cell Dev Biol 2022;10:901824.
Boschetti E, D’Angelo R, Tardio ML et al. Evidence of enteric angiopathy and neuromuscular hypoxia in patients with mitochondrial neurogastrointestinal encephalomyopathy. Am J Physiol Gastrointest Liver Physiol. 2021;320:G768–G779.
Chetaille P, Preuss C, Burkhard S et al. Mutations in SGOL1 cause a novel cohesinopathy affecting heart and gut rhythm. Nat Genet 2014;46:1245–1249.
Bianco F, Lattanzio G, Lorenzini L et al. Novel understanding on genetic mechanisms of enteric neuropathies leading to severe gut dysmotility. Eur J Histochem 2021;65:3289.
Fish EM, Burns B. Physiology, Small Bowel. [Updated 2022 Oct 14]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. 2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532263/
Schneider S, Hashmi SK, Thrasher AJ, Kothakapa DR, Wright CM, Heuckeroth RO. Single Nucleus Sequencing of Human Colon Myenteric Plexus-Associated Visceral Smooth Muscle Cells, Platelet Derived Growth Factor Receptor Alpha Cells, and Interstitial Cells of Cajal. Gastro Hep Adv 2023;2:380–394. https://doi.org/10.1016/j.gastha.2022.12.004.
Murthy KS. Signaling for contraction and relaxation in smooth muscle of the gut. Annu Rev Physiol 2006;68:345–374.
Martire D, Garnier S, Sagnol S et al. Phenotypic switch of smooth muscle cells in paediatric chronic intestinal pseudo-obstruction syndrome. J Cell Mol Med 2021;25:4028–4039.
Le Guen L, Marchal S, Faure S et al. Mesenchymal-epithelial interactions during digestive tract development and epithelial stem cell regeneration. Cell Mol Life Sci 2015;72:3883–3896.
Scirocco A, Matarrese P, Carabotti M et al. Cellular and Molecular Mechanisms of Phenotypic Switch in Gastrointestinal Smooth Muscle. J Cell Physiol 2016;231:295–302.
Guérin A, Angebault C, Kinet S et al. LIX1-mediated changes in mitochondrial metabolism control the fate of digestive mesenchyme-derived cells. Redox Biol 2022;56:102431.
McKey J, Martire D, de Santa Barbara P et al. LIX1 regulates YAP1 activity and controls the proliferation and differentiation of stomach mesenchymal progenitors. BMC Biol 2016;14:34.
Xie C, Guo Y, Zhu T et al. Yap1 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. J Biol Chem 2012;287:14598–14605.
Wallace AS, Burns AJ. Development of the enteric nervous system, smooth muscle and interstitial cells of Cajal in the human gastrointestinal tract. Cell Tissue Res 2005;319:367–382.
Torihashi S, Ward SM, Sanders KM. Development of c-Kit-positive cells and the onset of electrical rhythmicity in murine small intestine. Gastroenterology 1997;112:144–155.
Klüppel M, Huizinga JD, Malysz J et al. Developmental origin and Kit-dependent development of the interstitial cells of cajal in the mammalian small intestine. Dev Dyn 1998;211:60–71.
Torihashi S, Nishi K, Tokutomi Y et al. Blockade of kit signaling induces transdifferentiation of interstitial cells of cajal to a smooth muscle phenotype. Gastroenterology 1999;117:140–148.
Kurahashi M, Niwa Y, Cheng J et al. Platelet-derived growth factor signals play critical roles in differentiation of longitudinal smooth muscle cells in mouse embryonic gut. Neurogastroenterol Motil 2008;20:521–531.
De Santa Barbara P, Williams J, Goldstein AM et al. Bone morphogenetic protein signaling pathway plays multiple roles during gastrointestinal tract development. Dev Dyn 2005;234:312–322.
Torihashi S, Hattori T, Hasegawa H et al. The expression and crucial roles of BMP signaling in development of smooth muscle progenitor cells in the mouse embryonic gut. Differentiation 2009;77:277–289.
Rinkevich Y, Mori T, Sahoo D et al. Identification and prospective isolation of a mesothelial precursor lineage giving rise to smooth muscle cells and fibroblasts for mammalian internal organs, and their vasculature. Nat Cell Biol 2012;14:1251–1260.
Hashmi SK, Barka V, Yang C et al. Pseudo-obstruction-inducing ACTG2R257C alters actin organization and function. JCI Insight 2020;5:e140604.
Viti F, Pramotton FM, Martufi M et al. Patient’s dermal fibroblasts as disease markers for visceral myopathy. Biomaterials Advances 2023;148:213355.
Watson CL, Mahe MM, Múnera J et al. An in vivo model of human small intestine using pluripotent stem cells. Nat Med 2014;20:1310–1314.
Workman MJ, Mahe MM, Trisno S et al. Engineered human pluripotent-stem-cell-derived intestinal tissues with a functional enteric nervous system. Nat Med 2017;23:49–59.
Poling HM, Wu D, Brown N et al. Mechanically induced development and maturation of human intestinal organoids in vivo. Nat Biomed Eng 2018;2:429–442.
Costa J, Ghilardi M, Mamone V et al. Bioreactor With Electrically Deformable Curved Membranes for Mechanical Stimulation of Cell Cultures. Front Bioeng Biotechnol 2020;8:22.
Guazzelli N, Cacopardo L, Ahluwalia A. Engineering magneto-responsive core-shell microbeads for mimicking peristalsis and alveolar breathing in vitro. Biomedical Science and Engineering. 2021. https://doi.org/10.4081/bse.2021.173.
Cai Hui, Xiao Yongtao, Chen Shanshan et al. Heterozygous Actg2R257C mice mimic the phenotype of megacystis microcolon intestinal hypoperistalsis syndrome. Neurogastroenterol Motil 2023;35:e14472.
Gosemann JH, Puri P. Megacystis microcolon intestinal hypoperistalsis syndrome: systematic review of outcome. Pediatr Surg Int 2011;27:1041–1046.
Soh H, Fukuzawa M, Kubota A, Kawahara H, Ueno T, Taguchi T. Megacystis microcolon intestinal hypoperistalsis syndrome: A report of a nationwide survey in Japan. J Pediatr Surg 2015;50:2048–2050.
Prathapan KM, King DE, Raghu VK et al. Megacystis Microcolon Intestinal Hypoperistalsis Syndrome: A Case Series With Long-term Follow-up and Prolonged Survival. J Pediatr Gastroenterol Nutr 2021;72:e81–e85.
Ko D, Yang HB, Youn J, Kim HY. Clinical Outcomes of Pediatric Chronic Intestinal Pseudo-Obstruction. J Clin Med 2021;10:2376.
Batra S, Rahman S, Rana MS, Matta S, Darbari A. Epidemiology and healthcare utilization of inpatient admissions in children with pediatric intestinal pseudo-obstruction. Neurogastroenterol Motil 2020;32:e13781.
Pironi L, Goulet O, Buchman A et al. Outcome on home parenteral nutrition for benign intestinal failure: a review of the literature and benchmarking with the European prospective survey of ESPEN. Clin Nutr 2012;31:831–845.
Di Nardo G, Karunaratne TB, Frediani S et al. Chronic intestinal pseudo-obstruction: Progress in management? Neurogastroenterol Motil 2017;29:e13231.
Di Nardo G, Di Lorenzo C, Lauro A et al. Chronic intestinal pseudo-obstruction in children and adults: diagnosis and therapeutic options. Neurogastroenterol Motil 2017;29:e12945.
Billiauws L, Corcos O, Joly F. Dysmotility disorders: a nutritional approach. Curr Opin Clin Nutr Metab Care 2014;17:483–488.
Gershon MD. 5-Hydroxytryptamine (serotonin) in the gastrointestinal tract. Curr Opin Endocrinol Diabetes Obes. 2013;20:14–21.
Gu L, Ding C, Tian H et al. Serial Frozen Fecal Microbiota Transplantation in the Treatment of Chronic Intestinal Pseudo-obstruction: A Preliminary Study. J Neurogastroenterol Motil 2017;23:289–297.
Yano JM, Yu K, Donaldson GP et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 2015;161:264–276.
De Vadder F, Grasset E, Mannerås Holm L et al. Gut microbiota regulates maturation of the adult enteric nervous system via enteric serotonin networks. Proc Natl Acad Sci USA 2018;115:6458–6463.
Guo C, Chen WD, Wang YD. TGR5, Not Only a Metabolic Regulator. Front Physiol 2016;26:646.
Lacaille F. Thirty years after the first intestinal transplantation in 1987: which indications are left in 2018? Curr Opin Organ Transplant. 2018;23:196–198.
Proli F, Metou-Lopes A, Ayachi A et al. Quality of life in long term survivors of pediatric intestinal transplantation compared with liver transplantation and home parenteral nutrition: A prospective single-center pilot study. Pediatr Transplant 2021;25:e13982.
Hamilton NJ, Kanani M, Roebuck DJ et al. Tissue-Engineered Tracheal Replacement in a Child: A 4-Year Follow-Up Study. Am J Transplant 2015;15:2750–2757.
Elliott MJ, Butler CR, Varanou-Jenkins A et al. Tracheal Replacement Therapy with a Stem Cell-Seeded Graft: Lessons from Compassionate Use Application of a GMP-Compliant Tissue-Engineered Medicine. Stem Cells Transl Med 2017;6:1458–1464.
Filocamo M, Casareto L, Baldo C. Biobanking for Genetic Diseases. eLS. Chichester: John Wiley & Sons Ltd; 2017.
Filocamo M, Baldo C, Goldwurm S et al. Telethon Network of Genetic Biobanks: a key service for diagnosis and research on rare diseases. Orphanet J Rare Dis 2013;8:129.
Workman TA. Engaging Patients in Information Sharing and Data Collection: The Role of Patient-Powered Registries and Research Networks [Internet]. Rockville (MD): Agency for Healthcare Research and Quality (US); 2013 Sep. Report No.: AHRQ 13-EHC124-EF.
Acknowledgments
The European Forum on Visceral Myopathy (EFVM 2022) was funded by the European Joint Programme for Rare Diseases—Networking Support Scheme—Round 3, and by ‘POIC e dintorni’ patient advocacy organization (https://poic-e-dintorni.org/). ROH is supported by the Irma and Norman Braman Endowment, the Suzi and Scott Lustgarten Center Endowment, NIH R01 DK128282, and The Children’s Hospital of Philadelphia Frontier Program Center for Precision Diagnosis and Therapy for Pediatric Motility Disorders.
Funding
Open access funding provided by Consiglio Nazionale Delle Ricerche (CNR) within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
The idea for the article came from the final round table during EFVM2022 conference. Each author provided contributions related to his/her field of expertise. ROH initially drafted the work. All authors critically revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Viti, F., De Giorgio, R., Ceccherini, I. et al. Multi-disciplinary Insights from the First European Forum on Visceral Myopathy 2022 Meeting. Dig Dis Sci 68, 3857–3871 (2023). https://doi.org/10.1007/s10620-023-08066-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-023-08066-1