
Abstract
Eosinophilic esophagitis (EoE) is a leading cause of food bolus impaction in children and adults. The mechanism of dysphagia in EoE, particularly non-obstructive dysphagia, remains incompletely understood. While fibrostenotic processes appear to be critical in the development of dysphagia, somatosensory dysfunction and dysmotility also contribute. This review considers potential mechanisms of dysphagia and evaluates the utility of current and future treatment strategies in this context.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Eosinophilic esophagitis (EoE) is a chronic inflammatory condition affecting both children and adults, defined by the presence of ≥ 15 eosinophils per high-power field on esophageal biopsy [1]. This disorder has become increasingly common in the last two decades, causing significant morbidity for those afflicted as well as healthcare costs to society at large [2, 3]. Dysphagia and food bolus obstruction (FBO) events are the most frequent manifestations of EoE such that it is now a leading cause of FBO in older children and adults [4, 5]. FBO results in immediate distress, emergency department presentation, and often hospitalization for emergent endoscopy and bolus removal [6, 7]. Chronic dysphagia due to EoE causes lifestyle modifications that are inconvenient, expensive (changes in diet and medication), and associated with an increased likelihood of psychiatric comorbidity including anxiety and depression [8]. The need for a comprehensive understanding of the pathophysiology of dysphagia and FBO is thus obvious.
The pathophysiology of dysphagia in EoE is complex and multifactorial. Although acute inflammation and structural narrowing due to inflammatory infiltrate and edema contribute, chronic luminal narrowing and a decrement in compliance (distensibility) induced by basal zone hyperplasia, muscular hypertrophy, and subepithelial fibrosis appear to be pivotal to symptom development (Fig. 1). A dynamic element in the pathophysiology of dysphagia may also relate to the release of neuroactive mediators from mast cells and eosinophils causing impaired motility. Discerning which of these processes predominate is important to guide further management. A key consideration is the inconsistent relationship between a defined histological response to therapy (near disappearance of eosinophils at endoscopy and mucosal biopsy) and symptomatic and/or measured improvement in esophageal remodeling and function [9,10,11]. Data suggest that normalization of eosinophil count (the current treatment paradigm) may therefore be insufficient, highlighting the need to utilize and balance the results of a range of investigative modalities (as described below) in designing future therapeutic regimens.

available at https://smart.servier.com
Anatomical distortion predisposing to dysphagia in EoE. a: Normal esophageal layers. B Eosinophilic infiltration throughout all layers of the esophagus, with increased epithelial thickness, submucosal fibrosis, muscle hypertrophy and hyperplasia, and submucosal fibrosis. This figure was created using Servier Medical Art
This review explores the pathophysiology, complications, and treatment of these key contributors to dysphagia in EoE. While this is not a systematic review, it has been based largely on a structured interrogation of existing literature on EoE using Pubmed and Embase, including the search terms ‘eosinophilic esophagitis' or ‘EoE’ combined with ‘dysphagia,’ ‘endoscopy,’ ‘barium,’ ‘FLIP,’ ‘dysmotility,’ ‘achalasia,’ ‘manometry,’ and ‘sensitivity.’ All articles (including abstracts) that addressed the pathophysiology, investigations, manifestations, or treatment specific to the symptom of dysphagia in EoE were considered for inclusion. Reference lists from relevant review articles were also examined for additional suitable articles.
Mechanisms of Dysphagia
Anatomical Distortion
Tissue remodeling has been well documented in eosinophilic disorders, leading to long-term complications of disease. In asthma, mediators released by eosinophils lead to fibrosis and a progressive irreversible decline in lung function [12]. In hyper-eosinophilic syndrome, eosinophil products such as major basic protein (MBP) lead to extracellular fibrin deposition and eventually endomyocardial fibrosis [13]. Similarly, in EoE, chronic eosinophilic inflammation leads to fibrosis, angiogenesis, hypertrophy, and hyperplasia of multiple layers of the esophageal wall [13].
Basal Zone Hyperplasia
Basal zone hyperplasia (BZH), most frequently defined as a basal zone greater than 20% of the total epithelial thickness, has been demonstrated to correlate with the severity of mucosal eosinophilia in EoE [14, 15]. In addition, Whelan et al. [16] found that BZH correlated with both symptoms and endoscopic disease activity in patients with histologically ‘inactive’ EoE, proposing that BZH may therefore be used as a surrogate marker for active disease given the patchy nature of mucosal eosinophilia. Multiple factors contribute to the thickening of the epithelial layer seen in patients with EoE. In murine models, IL-5 or eosinophil-deficient mice do not undergo basal zone remodeling in response to an EoE-inducing antigen challenge, implying that eosinophilic mediators are key to this process [17]. MBP released by eosinophils leads to an increase in fibroblast growth factor 9 (FGF9) which drives basal cell proliferation [13]. In addition, plasminogen activator inhibitor-1 (PAI-1), induced by eosinophilic mediators, triggers BZH, while inhibition of PAI-1 has been demonstrated to reduce basal zone proliferation [18]. Williamson et al. [18] demonstrated that increased PAI-1 and BZH (seen more commonly in active EoE than controls) led to reduced esophageal distensibility (as a surrogate for dysphagia). BZH also likely contributes to esophageal narrowing which correlates with dysphagia and food bolus impaction [19, 20].
Smooth Muscle Hypertrophy and Hyperplasia
Smooth muscle changes have been depicted in studies using endoscopic ultrasound (EUS) in patients with EoE, with increased total esophageal wall and muscularis propria thickness compared to controls [21]. Degranulation of eosinophils releases toxic cationic proteins and histamine, resulting in continuous and repeated contraction of the muscularis propria, driving muscle hypertrophy [22]. In addition, degranulation of mast cells in parallel with eosinophils in EoE releases proteases that drive muscle cell hypertrophy, as supported by murine models demonstrating the absence of muscle hypertrophy in mast cell deficient mice [23]. EUS studies have highlighted the importance of muscle hypertrophy in the development of dysphagia in EoE, as dysphagic patients have greater muscularis propria thickness than asymptomatic patients [24]. Smooth muscle hypertrophy of the circular muscle layer is also hypothesized to underpin ‘trachealization’ which forms part of the endoscopic reference score (‘EREFS’), although no endoscopic feature has consistently correlated with dysphagia in EoE [22, 25].
Mucosal Vascular Proliferation
Vascular proliferation results in mucosal edema while concurrently facilitating additional eosinophil recruitment by increasing vascular permeability [26, 27]. Esophageal biopsies from patients with EoE exhibit higher levels of angiogenesis-promoting factors including VEGF, angiogenin, and IL-8: markers of activated vascular endothelium and increased vessel density [26,27,28]. Activated vessels and neovascularization result in dilated intercellular spaces and subsequent edema due to vascular leak [13]. Vascular cell adhesion molecule-1 (VCAM-1) is also more strongly expressed in vessels of active EoE patients, binding eosinophilic antigens and facilitating eosinophilic adhesion to the vessel wall [27].
Fibrostenotic Disease
Though EoE is often referred to as having ‘inflammatory’ and ‘fibrostenotic’ phenotypes; it is likely that the cumulative burden of eosinophilic inflammation over time leads to the development of progressive fibrostenotic disease [29]. A number of studies have demonstrated that younger patients have more inflammatory than fibrostenotic disease with less dysphagia, while untreated EoE eventually leads to fibrosis and stricture formation, correlating with dysphagia and food bolus impaction, in what is apparent as the endpoint of chronic inflammation [29,30,31,32,33]. As such, this fibrostenotic endpoint appears to be the key contributing factor to the development of dysphagia (Fig. 2).

available at https://smart.servier.com
Dysphagia due to hold-up of a food bolus resulting from fibrostenotic processes. This figure was created using Servier Medical Art
Subepithelial fibrosis, defined by abnormal collagen deposition in the lamina propria or submucosa seen on trichrome staining, has been demonstrated to be central to the development of dysphagia in children with EoE [34]. In their 2007 study, Chehade et al. reported dysphagia in 42% of patients with subepithelial fibrosis (accounting for 57–92% of children with EoE) while there were no patients with dysphagia in the absence of fibrosis [34, 35]. Fibrosis does not necessarily correlate with the degree of eosinophilia, but is more prominent in patients with markers of eosinophilic activation and degranulation [34]. The underlying mechanism of fibrosis likely relates to eosinophilic release of transforming growth factor beta-1 (TGF-β1) which is expressed at a higher rate in active eosinophilic esophagitis [27, 36]. TGF-β1 induces production of extracellular matrix (fibronectin and type I collagen) by esophageal fibroblasts and muscle cells [37]. Murine studies have confirmed the critical role of eosinophilic inflammation in driving fibrosis, demonstrating that IL-5 and eosinophil-deficient mice are protected from experimental pollen-induced esophageal subepithelial fibrosis [17].
Strictures
Esophageal strictures (focal fixed narrowings of the esophageal lumen) occur in almost 40% of patients with EoE, likely representing localized areas of subepithelial fibrosis in association with mucosal and muscular hyperplasia and hypertrophy [30]. The mechanism of stricture development is believed to be driven by chronic eosinophilic inflammation with release of pro-fibrotic mediators, as murine studies have again confirmed the absence of strictures in eosinophil-deficient mice exposed to EoE-inducing antigens [38]. Longer symptom duration prior to diagnosis in patients with EoE is associated with progressively lower likelihood of isolated inflammatory findings (edema, exudates, furrows) and increased risk of stricturing disease [30].
Narrow Caliber Esophagus (Diffuse Fibrosis)
Impaired esophageal dilation in EoE was recognized as early as 2002 and has been associated with higher incidence of dysphagia and food bolus obstruction [39, 40]. Although there has been no standardized definition of narrow caliber esophagus, it is characterized by a diffusely narrowed esophagus (rather than a focal stenosis or stricture) with a fixed internal diameter or impaired esophageal dilation, either extending the entire length of the esophagus or skipping segments [40]. Schoepfer et al. [41] highlighted the importance of esophageal caliber in the development of dysphagia in patients with EoE, demonstrating a significant improvement in dysphagia scores after endoscopic dilation, correlating with an increase in maximal esophageal diameter despite no change in eosinophilia. In support of fibrostenotic disease (including narrow caliber esophagus) as an endpoint of cumulative eosinophilic inflammation and subepithelial fibrosis, esophageal caliber does not change significantly in response to topical steroid therapy despite resolution of eosinophilia [40].
Investigation of Anatomical Distortion
Endoscopy
Upper gastrointestinal endoscopy (EGD) may detect luminal narrowing in severe cases due to the inability to pass the endoscope, or in causing mucosal or muscular tears (‘rents’) with scope passage [42]. However, while focal fibrostenotic changes including esophageal rings and strictures are detected with reasonable accuracy, EGD has poor sensitivity for the detection of more diffuse luminal narrowing [43,44,45]. Gentile et al. [45] notably demonstrated the sensitivity of EGD to be as low as 15% for detection of narrow caliber esophagus; therefore, alternative methods are required for accurate assessment of fixed luminal narrowing in EoE.
Barium Swallow
Barium swallow is useful in delineating fibrostenotic complications of EoE, including diffuse narrowing. Multiple studies have described rates of barium swallow abnormalities in patients with EoE (Table 1), with a reduction in maximal esophageal diameter (to less than 21 mm) in 38–59%, rings in 6–35%, strictures in 10–31%, and small caliber esophagus (> 8 cm long narrowed segment) in up to 52% of cases [19, 45,46,47,48,49,50,51]. Importantly, Al-Hussaini et al. [19] found that barium swallow findings predict symptom severity, with food bolus obstruction occurring in 69% of those with an abnormal barium swallow compared to 8% without. Podboy et al. [47] added further support to the fibrostenotic endpoint of long-term inflammation in their 2016 study that demonstrated an increased risk of strictures and small caliber esophagus in patients with longer symptom duration at the time of barium swallow.
Functional Lumen Imaging Probe (FLIP)
The functional lumen imaging probe (FLIP) uses high-resolution impedance planimetry to develop three-dimensional images of the esophageal lumen [52]. FLIP is comprised of a catheter with a distal overlying balloon which is inflated with saline and contains electrodes that measure diameter, volume, and pressure changes [52]. Using these measurements, FLIP is able to demonstrate the functional impact of esophageal remodeling and fibrosis, most often represented by the distensibility plateau (calculated by modeling the ‘pressure to cross-sectional area’ relationship at incremental volumes of saline in the balloon) [53, 54]. Patients with EoE have reduced esophageal distensibility which correlates with fibrostenotic features seen endoscopically (rings and strictures), increased risk of food bolus obstructions, and higher rates of future endoscopic dilation [43, 55,56,57,58,59,60,61]. A 2013 study by Nicodeme et al. [55, 62] found the distensibility plateau measured by FLIP to be the only factor on multivariate analysis that independently predicted future food bolus impaction. Importantly, distensibility on FLIP does not tend to correlate with mucosal eosinophilia, signifying that distensibility is a measure of remodeling more so than active inflammation [55, 57, 58].
Dysmotility
Pathogenesis
The dysphagia and resultant FBO that characterize EoE may relate not only to luminal narrowing and a lack of distensibility induced by inflammation and fibrosis, but also to incoordinate esophageal peristalsis. The abundant eosinophilic infiltration of the epithelium and lamina propria (Fig. 1) is demonstrable in deeper esophageal layers including muscularis mucosa and muscularis propria, where mast cells are also found in active EoE [23, 63]. Generally, routine esophageal biopsies in patients with EoE target the epithelium and thus document epithelial eosinophil counts. Although fragments of lamina propria are not infrequently seen incidentally in biopsies (sufficient for histological evaluation in up to 37–68% of biopsies), deeper esophageal muscle layers are rarely sampled [64, 65]. However, esophagectomy specimens from patients with EoE (though obtained in very few patients in exceptional circumstances) have demonstrated full thickness eosinophilic infiltration of the esophagus [37, 66,67,68].
Fibrosis, along with the direct impact of eosinophilic and mast cell inflammation, is hypothesized to be the predominant contributors to the dysfunction that is seen in longitudinal muscles in EoE, highlighted by EUS studies evaluating real-time muscle contraction during peristalsis [69]. While fibrosis of deeper muscle layers is difficult to assess histologically as described above, its role in dysmotility is extrapolated from other eosinophilic pathology such as endomyocardial fibrosis leading to end-stage heart failure in hypereosinophilic syndrome [70]. Additionally, eosinophils and mast cells have been demonstrated to infiltrate smooth muscle in EoE and are known to produce TGF-β1, a key driver of fibrosis [23, 63].
In addition to muscle fibrosis, eosinophils in EoE may directly impact dynamic esophageal muscle function. Both eosinophils and mast cells release mediators that influence muscle function, either by leading to enhanced contraction (e.g., leukotriene D4, prostaglandin F2 alpha) or relaxation (e.g., IL-6, IL-13) [37, 38, 71,72,73]. Eosinophil degranulation also results in the production of neurotoxic mediators including eosinophilic cationic protein and eosinophil-derived neurotoxin [74]. Given the established deep infiltration of eosinophils in EoE, including into the smooth muscle and myenteric plexus, these mediators are hypothesized to cause dysfunction and/or destruction of neurons which may contribute to dysmotility [73]. A theoretical framework is therefore plausible whereby a disturbance of esophageal motility results from esophageal neuromuscular dysfunction, in addition to primarily anatomical distortion [37, 68].
Achalasia in EoE?
The prevalence of dysmotility in EoE, as well as the demonstration of deep tissue eosinophilia facilitating myenteric plexus destruction, has led to conjecture regarding an association between EoE and achalasia [37, 68]. A recent large cohort study in the UK demonstrated an association between achalasia and a history of atopic disorders, supporting a potential role of eosinophilic disease in the development of achalasia [75]. A 2013 study of 96 patients undergoing laparoscopic Heller Myotomy for achalasia found increased mucosal eosinophilia in 34% (n = 17) of patients on endoscopic biopsy prior to surgery [76]. Savarino et al. [77] reported a case of type I achalasia in which > 50 eosinophils per high-power field were noted on esophageal biopsy, with complete resolution of symptoms and dysmotility after treatment with prednisolone. Ghisa et al. [78] described a group of 109 patients with EoE undergoing manometry in which 8 were diagnosed with achalasia. Further investigation of this association had previously been limited by difficulties accessing deep tissue specimens in patients with achalasia, although the increasing uptake of per-oral endoscopy myotomy (POEM) presents an opportunity for additional access for biopsies from muscle layers and myenteric plexus.
Despite this speculation, there is no convincing evidence of immune-related destruction of neurons in EoE. It is generally considered a disease amenable to medical treatment, where restorative remodeling occurs in response to appropriate therapy, unlike achalasia where changes are irreversible following destruction of the myenteric plexus [79, 80]. It is possible that there is a rare subgroup of patients with EoE in whom transmural eosinophilia leads to neuronal destruction similar to achalasia; however, it is more likely that these patients have concurrent pathologies (EoE and achalasia) given the population prevalence of EoE.
Manometry Abnormalities
Both conventional and now solid-state high-resolution manometry (HRM) platforms have demonstrated a range of abnormalities in patients with EoE, including abnormal peristalsis, esophagogastric junction outflow obstruction, and pan-esophageal pressurization (Table 2). Multiple controlled studies have reported higher mean lower esophageal sphincter resting pressure in patients with EoE compared to controls [81,82,83]. Abnormal peristalsis (either weak peristalsis or frequent failed peristalsis) has been reported in up to 45% of EoE patients, with a pooled prevalence of 26% across 12 studies including 449 patients (Table 2) [80,81,82,83,84,85,86,87,88,89,90,91]. The overall prevalence of dysmotility in EoE is difficult to ascertain due to heterogeneity between studies, but ranges between 21 and 76% [78, 80,81,82,83,84,85,86,87,88,89,90,91]. This heterogeneity may in part relate to the difficulties in interpretation of HRM in the context of the previously described anatomical distortion. Interestingly, as will be discussed in more detail below, abnormal motility resolves in many cases with medical therapy targeting eosinophilic inflammation, indicating that eosinophilic infiltration contributes to myenteric plexus and/or muscle dysfunction [78].
However, the relationship between abnormality manometry findings and dysphagia in EoE remains controversial. Multiple studies have been unable to demonstrate a difference in symptom scores between patients with or without abnormal motility on manometry in EoE, though these studies have been limited by small numbers of patients with abnormal motility [81, 85, 86]. On the contrary, in their 2009 prospective study utilizing prolonged manometry, Nurko et al. [83] recorded 21 episodes of dysphagia, all of which correlated with simultaneous abnormal motor function. Another prospective study in 2011 found that FBO is more common in patients with pan-esophageal pressurization (p < 0.05) [82].
Dysphagia as a Subjective Sensation
There has been inconsistent correlation between objective findings in EoE and symptom scores. In many studies, objective measures have correlated with food bolus obstructions rather than subjective symptom reporting. For example, Larsson et al. [9] found no correlation between eosinophilia and dysphagia symptom scores, but demonstrated correlation between upper esophageal eosinophil density and food bolus impactions (p < 0.05). In addition, multiple studies reporting the use of FLIP in EoE have demonstrated correlation between reduced distensibility and food bolus obstructions rather than subjective dysphagia [55, 62]. This discrepancy suggests the presence of an additional factor impacting the perception (or lack thereof) of dysphagia in EoE, which may in part relate to eosinophilia-induced sensory dysfunction.
Physiology of Esophageal Sensation
Noxious stimuli, either chemical, mechanical, or thermal, activate nociceptive receptors on esophageal neurons and are transmitted to the central nervous system via spinal and vagal nerves [92]. While numerous receptors have been identified, the transient receptor potential vanilloid 1 (TRPV1) receptor has been most extensively studied. In murine studies, vagal action potentials in response to mechanical stimulation of the esophagus were weaker in TRPV1-knockout than wild-type mice [93]. The role of TRPV1 in esophageal sensitivity has been demonstrated in gastresophageal reflux disease (GORD), where patients with symptomatic non-erosive reflux disease have increased expression of TRPV1 in their esophageal mucosa [94].
Impact of EoE on Esophageal Sensitivity
In EoE, as opposed to GORD, there is a paucity of data pertaining to sensory dysfunction and its relationship to symptoms. It is therefore not known whether these patients may have a similar pattern of hyposensitivity to that seen in silent GORD, or perhaps even hypersensitivity as is seen in reflux hypersensitivity and other non-gastrointestinal eosinophilic disorders. As clinicians managing many adult patients with EoE, we are struck by the frequency of FBO as the first presenting symptom, along with the lack of dysphagia often hampering enrolment of patients with severe fibrostenotic disease into clinical trials of promising pharmacotherapies.
An existing template for esophageal hyposensitivity exists in silent GORD and Barrett’s esophagus, where patients have reduced sensitivity to acid exposure and mechanical distention compared to controls [95, 96]. Although this has not been directly explored in EoE, hyposensitivity could in part explain the prevalence of FBO as the initial presentation of EoE in the majority of adults rather than non-obstructive dysphagia [5, 97,98,99,100,101]. Cumulative eosinophilic inflammation and the neurotoxic effects of released cytoplasmic granules may eventually lead to sensory neuronal loss or dysfunction, resulting in a reduction in esophageal sensation, particularly in those with a longer duration of disease [74]. Proof of concept for this hypothesis exists in the prevalence of peripheral neuropathy in up to 52% of those with hypereosinophilic syndrome [102, 103].
Alternatively, EoE may be associated with eosinophilia-induced neuronal hypersensitivity as is seen in other eosinophilic disorders including atopic dermatitis and eosinophilic asthma [104, 105]. Eosinophils have been demonstrated to increase neuronal TRPV1 expression in vitro, thereby priming esophageal neuronal sensitivity to mechanical and chemical stimulation [106]. Interestingly, in children with EoE the most common presenting symptoms are heartburn and dyspepsia, lending support to a hypothesis of eosinophil-induced esophageal hypersensitivity in the setting of predominantly inflammatory rather than fibrostenotic disease [107]. Accordingly, discordance between symptom severity and objective markers of inflammatory or fibrostenotic disease in EoE may relate to sensory dysfunction, including either sensory loss from longer term neurotoxicity or hypersensitivity related to eosinophilic inflammation. While this is currently merely speculation in the context of limited existing evidence, it is an area worthy of future study in order to guide targeted therapy for symptoms in EoE.
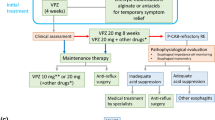
Management of Dysphagia in EOE
Currently, the treatment of dysphagia in EoE targets either disruption of the inflammatory process with diet or medications while anticipating an improvement in luminal narrowing due to positive remodeling, or alternatively treating fibrostenotic disease mechanically with dilation of the esophagus when immediate symptom relief is desired [108]. Specific treatments aimed at dysmotility and/or somatosensory dysfunction are yet to be investigated in EoE and thus are currently not recommended.
Inflammatory Processes
Dynamic inflammatory contributors to dysphagia generally respond to medical therapy in EoE. Proton-pump inhibitors (PPIs) are generally recommended as a first-line therapeutic option, leading to resolution of eosinophilia in up to 50% of patients [108, 109]. In those non-responsive to PPI, meta-analyses have established histological remission rates as high as 90% with topical corticosteroids [110,111,112]. Multiple modes of corticosteroid delivery have been described, including viscous slurries containing fluticasone or budesonide, while oral dispersible budesonide has recently become available and is associated with histological remission rates as high as 93% [113,114,115]. Alternatively, resolution of eosinophilia can be achieved in up to 70% of patients with the use of an elimination diet [110, 116]. Elimination diets in EoE involve varying degrees of restriction, generally including elimination of between 2 and 6 food groups (gluten and dairy, with or without a combination of eggs, soy, nuts and seafood) followed by gradual reintroduction of individual food groups to ascertain which food antigens are responsible for the eosinophilic inflammation [117].
In addition to resolution of eosinophilia, BZH improves with topical corticosteroids in treatment responders, with a reduction in esophageal wall thickness demonstrated in EUS studies [118, 119]. While data are limited beyond resolution of eosinophilia with elimination diets, BZH did improve in 36% of elimination diet responders in a study by Kagalwalla et al. [120]. Reduced vascularity, an endoscopic marker of mucosal edema, has also been demonstrated to respond to medical therapy [121].
Fibrostenotic Processes
While inflammatory processes respond to medical therapy, the response of fibrostenotic disease is variable. Early remodeling and subepithelial fibrosis may improve; however, a threshold appears to be reached after accumulation of an extended duration of fibrotic remodeling whereby the changes no longer respond to medical therapy. This may account for the persistence of dysphagia in many patients despite resolution of eosinophilia, highlighting the importance of fibrostenotic disease as a key driver of dysphagia in EoE.
Response to Topical Therapy and Elimination Diet
A reduction in profibrotic mediators including TGF-β1, FGF9, and IL-5 has been found in patients with EoE in response to topical corticosteroids [36]. In patients with histological remission as a result of topical corticosteroids, Straumann et al. [118] also found an improvement in fibrosis score (p = 0.026). In support of this, corticosteroids have also been shown to increase maximal esophageal diameter on barium swallow [49]. This reversal of remodeling correlates with a reduction in dysphagia symptom scores with topical corticosteroids [115, 118, 122, 123]. However, more advanced fibrostenotic disease may not respond to medical therapy. Murine studies have demonstrated persistence of strictures despite withdrawal of the EoE-inducing antigen, while human studies assessing the EREFS score before and after treatment have found that strictures and esophageal rings do not change with topical corticosteroids despite resolution of inflammatory features [38, 121].
There are minimal data regarding the response of remodeling to elimination diet; however, patients with resolution of eosinophilia would be expected to have a similar degree of regression of remodeling to those treated with topical therapy. Lieberman et al. [124] retrospectively analyzed biopsies from 17 children on elimination diets and found resolution of subepithelial fibrosis in 3 of 17. Carlson et al. [61] used EndoFLIP to assess for improvement in remodeling in 18 patients with EoE undergoing ‘medical management,’ of which 6 patients were on elimination diets only. In this cohort, the distensibility plateau improved from 13.9 to 16.8 mm (p = 0.007) although values were not reported specific to patients on elimination diets.
Response to Systemic Treatment
Systemic therapies for EoE, generally used in the clinical trial setting, include an expanding range of biologic agents [108]. Data on these agents (including anti-IL4, IL5 and IL13 monoclonal antibodies) are limited in the EoE setting beyond their impact on mucosal eosinophilia and symptoms [125,126,127]. However, Hirano et al. [126] demonstrated an improvement in the distensibility plateau on EndoFLIP following 12 weeks of dupilumab (an IL4 inhibitor). Of note, dupilumab does result in partial reversal of airway remodeling in eosinophilic asthma and therefore may have the capacity to similarly reverse fibrosis in EoE [128].
Systemic therapies targeting fibrosis are another area for further research. While there are no clinical trials in human subjects, pre-clinical studies have demonstrated that thiazolidinediones exert anti-fibrotic effects in EoE fibroblasts activated by TGF-β1, providing a mechanistic foundation for future studies [129].
Esophageal Dilation
Esophageal dilation targets fibrostenotic components of dysphagia in EoE without impacting mucosal eosinophilia [41]. This leads to an increase in esophageal diameter, correlating with symptomatic improvement [41, 130]. A significant advantage of dilation is that immediate symptom relief can be achieved while minimizing the potential for recurrent FBOs that may ensue as a result of critical stenoses. The efficacy of dilation for the relief of dysphagia supports fibrostenotic disease being the primary driver of dysphagia in EoE. For example, Schoepfer et al. [41] studied dilation in the absence of medical therapy and demonstrated a significant improvement in dysphagia scores for a median of 15 months. Further to this, Gentile et al. [45] reported symptomatic improvement in 7 out of 8 patients who had persistent dysphagia despite resolution of mucosal eosinophilia. Nevertheless, the interplay between inflammatory and fibrostenotic processes in EoE was highlighted by Schupack et al. [131], who demonstrated a reduction in need for recurrent dilation in patients on maintenance medical therapy for EoE by limiting recurrence of fibrostenotic disease.
Esophageal dilation was initially thought to be a high-risk procedure in EoE due to the fragility of the esophageal wall, with rates of perforation and hospitalization for chest pain reportedly as high as 5% and 7%, respectively [132]. However, esophageal dilation has subsequently been demonstrated to be a safe and effective procedure for the management of dysphagia in the context of fibrostenotic disease (Table 3). Multiple studies (including large meta-analyses) have now reported perforation rates as low as 0–1%, with all cases of perforation able to be conservatively managed without surgery [41, 130, 133,134,135,136,137,138,139,140,141,142,143,144]. While some degree of chest discomfort is common after dilation in EoE, significant chest pain requiring medical attention occurs in only 5–9.3% of patients according to meta-analyses [134, 136, 137, 142]. In regard to efficacy, recent studies have demonstrated an improvement in symptoms after dilation in 85–95% of patients [135,136,137].
Dysmotility
There are limited data available to guide the treatment of motility disorders in EoE. If one considers that dysmotility may result from neuroactive mediators released from inflammatory cells (eosinophils and/or mast cells), then reducing the inflammatory burden with diet and/or medications (see above) would be expected to improve motility to some degree. Pharmacotherapies that induce smooth muscle relaxation such as calcium channel blockers (e.g., nifedipine), nitrates (e.g., glyceryl trinitrate), or phosphodiesterase inhibitors (e.g., sildenafil) have not been trialed in EoE. Further research is therefore needed.
Motility disorders (even including achalasia) in patients with EoE have been demonstrated to respond to EoE-specific treatment in some cases [80, 145, 146]. Ghisa et al. [78] described 8 patients with EoE and achalasia, of which 3 had resolution of dysmotility after treatment with proton-pump inhibitors or topical corticosteroids. However, if dysmotility persists despite treatment of EoE, traditional achalasia therapies appear to remain effective. In the previously mentioned study by Ghisa et al. [78], the remaining 5 patients were successfully treated with pneumatic dilatation (3 cases) and/or laparoscopic Heller myotomy (3 cases). A 2018 case report also described successful per-oral endoscopic myotomy (POEM) in a patient with EoE and achalasia with ongoing dysmotility despite histological resolution of eosinophilia [147]. Of note, a 2013 study on Heller myotomy, in which 17 of 50 patients had esophageal eosinophilia prior to the procedure, found no impact of eosinophilia on clinical response to surgery [76]. However, caution should be applied when considering EoE as a cause of achalasia, as other potential explanations include coincidental concurrent existence of both disorders, or even the inaccuracies of manometry studies in the presence of a distorted esophagus in EoE.
Conclusion
The development of dysphagia in EoE is multifactorial; however, seems to be primarily a result of fibrostenotic disease rather than eosinophilic inflammation. Discerning if dysphagia (defined as a subjective sensation of difficulty swallowing) results from anatomical distortion, aberrant peristalsis or even hyper- or hyposensitivity to stimulus in a diseased esophagus is challenging. Accordingly, optimal management of dysphagia in EoE should include addressing inflammatory processes with medical therapy, while concurrently treating fibrostenotic disease with dilation when severe chronic fibrotic remodeling has occurred or immediate symptom relief is required. Dysmotility in EoE is less well understood, though appears responsive to medical therapies targeting EoE, or endoscopic and/or surgical interventions where medical therapy fails. Further investigation of the pathophysiology driving dysphagia in EoE may be the key to unlock targeted symptom-based therapies for this highly prevalent disorder.
References
Lucendo AJ, Molina-Infante J, Arias Á et al. Guidelines on eosinophilic esophagitis: evidence-based statements and recommendations for diagnosis and management in children and adults. United Eur Gastroenterol J 2017;5:335–358.
Dellon ES, Hirano I. Epidemiology and natural history of eosinophilic esophagitis. Gastroenterology 2018;154:319-332.e313.
Eke R, Dellon ES. Hospitalization trends and determinants of inpatient costs for eosinophilic esophagitis patients in the United States: results from the Nationwide Inpatient Sample analysis. Ann Gastroenterol 2021; 1–9.
Heerasing N, Lee SY, Alexander S, Dowling D. Prevalence of eosinophilic esophagitis in adults presenting with esophageal food bolus obstruction. World J Gastrointest Pharmacol Ther 2015;6:244–247.
Cook D, Zala A, Bollipo S, Potter MDE, Walker MM, Talley NJ. Esophageal food bolus obstruction and eosinophilic esophagitis. Internal Med J 2019;49:1032–1034.
Lucendo AJ, Arias-González L, Molina-Infante J, Arias Á. Determinant factors of quality of life in adult patients with eosinophilic esophagitis. United Eur Gastroenterol J 2018;6:38–45.
Aiolfi A, Ferrari D, Riva CG, Toti F, Bonitta G, Bonavina L. Esophageal foreign bodies in adults: systematic review of the literature. Scand J Gastroenterol 2018;53:1171–1178.
Taft TH, Guadagnoli L, Edlynn E. Anxiety and depression in eosinophilic esophagitis: a scoping review and recommendations for future research. J Asthma Allergy 2019;12:389–399.
Larsson H, Norder Grusell E, Tegtmeyer B, Ruth M, Bergquist H, Bove M. Grade of eosinophilia versus symptoms in patients with dysphagia and esophageal eosinophilia. Dis Esophagus 2016;29:971–976.
Pentiuk S, Putnam PE, Collins MH, Rothenberg ME. Dissociation between symptoms and histological severity in pediatric eosinophilic esophagitis. J Pediatr Gastroenterol Nutr 2009;48:152–160.
Alexander JA, Jung KW, Arora AS et al. Swallowed fluticasone improves histologic but not symptomatic response of adults with eosinophilic esophagitis. Clin Gastroenterol Hepatol 2012;10:742-749.e741.
Broide D. Immunologic and inflammatory mechanisms that drive asthma progression to remodelling. J Allergy Clin Immunol 2008;121:560–572.
Aceves SS. Remodeling and fibrosis in chronic eosinophil inflammation. Dig Dis 2014;32:15–21.
Blanchard C, Wang N, Stringer KF et al. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest 2006;116:536–547.
Furuta GT, Liacouras CA, Collins MH et al. Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology 2007;133:1342–1363.
Whelan KA, Godwin BC, Wilkins B et al. Persistent basal cell hyperplasia is associated with clinical and endoscopic findings in patients with histologically inactive eosinophilic esophagitis. Clin Gastroenterol Hepatol 2020;18:1475-1482.e1471.
Mishra A, Wang M, Pemmaraju VR et al. Esophageal remodeling develops as a consequence of tissue specific IL-5-induced eosinophilia. Gastroenterology 2008;134:204–214.
Williamson P, Proudfoot J, Gharibans A et al. Plasminogen activator inhibitor-1 as a marker of esophageal functional changes in pediatric eosinophilic esophagitis. Clin Gastroenterol Hepatol 2020.
Al-Hussaini A, AboZeid A, Hai A. How does esophagus look on barium esophagram in pediatric eosinophilic esophagitis? Abdom Radiol 2016;41:1466–1473.
Aceves SS, Ackerman SJ. Relationships between eosinophilic inflammation, tissue remodeling, and fibrosis in eosinophilic esophagitis. Immunol Allergy Clin N Am 2009;29:197–xiv.
Fox VL, Nurko S, Teitelbaum JE, Badizadegan K, Furuta GT. High-resolution EUS in children with eosinophilic “allergic” esophagitis. Gastrointest Endosc 2003;57:30–36.
Mann NS, Leung JW. Pathogenesis of esophageal rings in eosinophilic esophagitis. Med Hypotheses 2005;64:520–523.
Niranjan R, Mavi P, Rayapudi M, Dynda S, Mishra A. Pathogenic role of mast cells in experimental eosinophilic esophagitis. Am J Physiol Gastrointest Liver Physiol 2013;304:G1087-1094.
Muroi K, Kakushima N, Furukawa K et al. Subjective symptoms in patients with eosinophilic esophagitis are related to esophageal wall thickness and esophageal body pressure. Dig Dis Sci 2021;66:2291–2300. https://doi.org/10.1007/s10620-020-06527-5.
Rodríguez-Sánchez J, Barrio-Andrés J, Nantes Castillejo O et al. The endoscopic reference score shows modest accuracy to predict either clinical or histological activity in adult patients with eosinophilic esophagitis. Aliment Pharmacol Ther 2017;45:300–309.
Persad R, Huynh HQ, Hao L et al. Angiogenic remodeling in pediatric EoE is associated with increased levels of VEGF-A, angiogenin, IL-8, and activation of the TNF-α-NFκB pathway. J Pediatr Gastroenterol Nutr 2012;55:251–260.
Aceves SS, Newbury RO, Dohil R, Bastian JF, Broide DH. Esophageal remodeling in pediatric eosinophilic esophagitis. J Allergy Clin Immunol 2007;119:206–212.
McNamee EN, Biette KA, Hammer J et al. Targeting granulocyte-macrophage colony-stimulating factor in epithelial and vascular remodeling in experimental eosinophilic esophagitis. Allergy 2017;72:1232–1242.
Dellon ES, Kim HP, Sperry SL, Rybnicek DA, Woosley JT, Shaheen NJ. A phenotypic analysis shows that eosinophilic esophagitis is a progressive fibrostenotic disease. Gastrointest Endosc 2014;79:577-585.e574.
Schoepfer AM, Safroneeva E, Bussmann C et al. Delay in diagnosis of eosinophilic esophagitis increases risk for stricture formation in a time-dependent manner. Gastroenterology 2013;145:1230–1236.e1231–1232.
Singla MB, Chehade M, Brizuela D et al. Early comparison of inflammatory vs. fibrostenotic phenotype in eosinophilic esophagitis in a multicenter longitudinal study. Clin Transl Gastroenterol 2015;6:e132–e132.
Koutlas NT, Dellon ES. Progression from an Inflammatory to a fibrostenotic phenotype in eosinophilic esophagitis. Case Rep Gastroenterol 2017;11:382–388.
Warners MJ, Oude Nijhuis RAB, de Wijkerslooth LRH, Smout A, Bredenoord AJ. The natural course of eosinophilic esophagitis and long-term consequences of undiagnosed disease in a large cohort. Am J Gastroenterol 2018;113:836–844.
Chehade M, Sampson HA, Morotti RA, Magid MS. Esophageal subepithelial fibrosis in children with eosinophilic esophagitis. J Pediatr Gastroenterol Nutr 2007;45:319–328.
Li-Kim-Moy JP, Tobias V, Day AS, Leach S, Lemberg DA. Esophageal subepithelial fibrosis and hyalinization are features of eosinophilic esophagitis. J Pediatr Gastroenterol Nutr 2011;52:147–153.
Lucendo AJ, Arias A, De Rezende LC et al. Subepithelial collagen deposition, profibrogenic cytokine gene expression, and changes after prolonged fluticasone propionate treatment in adult eosinophilic esophagitis: a prospective study. J Allergy Clin Immunol 2011;128:1037–1046.
Rieder F, Nonevski I, Ma J et al. T-helper 2 cytokines, transforming growth factor β1, and eosinophil products induce fibrogenesis and alter muscle motility in patients with eosinophilic esophagitis. Gastroenterology 2014;146:1266–1277.e1261–1269.
Mavi P, Rajavelu P, Rayapudi M, Paul RJ, Mishra A. Esophageal functional impairments in experimental eosinophilic esophagitis. Am J Physiol Gastrointest Liver Physiol 2012;302:G1347-1355.
Vasilopoulos S, Murphy P, Auerbach A et al. The small-caliber esophagus: an unappreciated cause of dysphagia for solids in patients with eosinophilic esophagitis. Gastrointest Endosc 2002;55:99–106.
Eluri S, Runge TM, Cotton CC et al. The extremely narrow-caliber esophagus is a treatment-resistant subphenotype of eosinophilic esophagitis. Gastrointest Endosc 2016;83:1142–1148.
Schoepfer AM, Gonsalves N, Bussmann C et al. Esophageal dilation in eosinophilic esophagitis: effectiveness, safety, and impact on the underlying inflammation. Am J Gastroenterol 2010;105:1062–1070.
Rodriguez-Sanchez J, Barrio-Andres J, Nantes Castillejo O et al. The endoscopic reference score shows modest accuracy to predict either clinical or histological activity in adult patients with eosinophilic esophagitis. Aliment Pharmacol Ther 2017;45:300–309.
Menard-Katcher C, Swerdlow MP, Mehta P, Furuta GT, Fenton LZ. Contribution of esophagram to the evaluation of complicated pediatric eosinophilic esophagitis. J Pediatr Gastroenterol Nutr 2015;61:541–546.
Nelson MJ, Miller FH, Moy N et al. Comparison of endoscopy and radiographic imaging for detection of esophageal inflammation and remodeling in adults with eosinophilic esophagitis. Gastrointest Endosc 2018;87:962–968.
Gentile N, Katzka D, Ravi K et al. Esophageal narrowing is common and frequently under-appreciated at endoscopy in patients with esophageal eosinophilia. Aliment Pharmacol Ther 2014;40:1333–1340.
Muinuddin A, O’Brien PG, Hurlbut DJ, Paterson WG. Diffuse esophageal narrowing in eosinophilic esophagitis: a barium contrast study. J Can Assoc Gastroenterol 2019;2:1–5.
Podboy A, Katzka DA, Enders F et al. Esophageal narrowing on barium esophagram is more common in adult patients with eosinophilic esophagitis than PPI-responsive esophageal eosinophilia. Aliment Pharmacol Ther 2016;43:1168–1177.
Savarino E, Tolone S, Caccaro R et al. Clinical, endoscopic, histological and radiological characteristics of Italian patients with eosinophilic esophagitis. Dig Liver Dis 2015;47:1033–1038.
Lee J, Huprich J, Kujath C et al. Esophageal diameter is decreased in some patients with eosinophilic esophagitis and might increase with topical corticosteroid therapy. Clin Gastroenterol Hepatol 2012;10:481–486.
Diniz LO, Putnum PE, Towbin AJ. Fluoroscopic findings in pediatric eosinophilic esophagitis. Pediatr Radiol 2012;42:721–727.
White SB, Levine MS, Rubesin SE, Spencer GS, Katzka DA, Laufer I. The small-caliber esophagus: radiographic sign of idiopathic eosinophilic esophagitis. Radiology 2010;256:127–134.
Donnan EN, Pandolfino JE. EndoFLIP in the esophagus: assessing sphincter function, wall stiffness, and motility to guide treatment. Gastroenterol Clin N Am 2020;49:427–435.
Donnan EN, Pandolfino JE. Applying the functional luminal imaging probe to esophageal disorders. Curr Gastroenterol Rep 2020;22:10.
Carlson DA, Kou W, Lin Z et al. Normal values of esophageal distensibility and distension-induced contractility measured by functional luminal imaging probe panometry. Clin Gastroenterol Hepatol 2019;17:674-681.e671.
Nicodème F, Hirano I, Chen J et al. Esophageal distensibility as a measure of disease severity in patients with eosinophilic esophagitis. Clin Gastroenterol Hepatol 2013;11:1101-1107.e1101.
Horii Y, Dohi O, Naito Y et al. Efficacy of magnifying narrow band imaging for delineating horizontal margins of early gastric cancer. Digestion 2019;100:93–99.
Chen JW, Pandolfino JE, Lin Z et al. Severity of endoscopically identified esophageal rings correlates with reduced esophageal distensibility in eosinophilic esophagitis. Endoscopy 2016;48:794–801.
Hassan M, Aceves S, Dohil R et al. Esophageal compliance quantifies epithelial remodeling in pediatric patients with eosinophilic esophagitis. J Pediatr Gastroenterol Nutr 2019;68:559–565.
Kwiatek MA, Hirano I, Kahrilas PJ, Rothe J, Luger D, Pandolfino JE. Mechanical properties of the esophagus in eosinophilic esophagitis. Gastroenterology 2011;140:82–90.
Menard-Katcher C, Benitez AJ, Pan Z et al. Influence of age and eosinophilic esophagitis on esophageal distensibility in a pediatric cohort. Am J Gastroenterol 2017;112:1466–1473.
Carlson DA, Hirano I, Zalewski A, Gonsalves N, Lin Z, Pandolfino JE. Improvement in esophageal distensibility in response to medical and diet therapy in eosinophilic esophagitis. Clin Transl Gastroenterol 2017;8:e119.
Hirano I, Pandolfino JE, Boeckxstaens GE. Functional lumen imaging probe for the management of esophageal disorders: expert review from the clinical practice updates committee of the AGA institute. Clin Gastroenterol Hepatol 2017;15:325–334.
Aceves SS, Chen D, Newbury RO, Dohil R, Bastian JF, Broide DH. Mast cells infiltrate the esophageal smooth muscle in patients with eosinophilic esophagitis, express TGF-β1, and increase esophageal smooth muscle contraction. J Allergy Clin Immunol 2010;126:1198-1204.e1194.
Philpott H, Nandurkar S, Royce SG, Gibson PR. Ultrathin unsedated transnasal gastroscopy in monitoring eosinophilic esophagitis. J Gastroenterol Hepatol 2016;31:590–594.
Collins MH, Martin LJ, Alexander ES et al. Newly developed and validated eosinophilic esophagitis histology scoring system and evidence that it outperforms peak eosinophil count for disease diagnosis and monitoring. Dis Esophagus 2017;30:1–8.
Saffari H, Peterson KA, Fang JC, Teman C, Gleich GJ, Pease LF 3rd. Patchy eosinophil distributions in an esophagectomy specimen from a patient with eosinophilic esophagitis: implications for endoscopic biopsy. J Allergy Clin Immunol 2012;130:798–800.
Fontillón M, Lucendo AJ. Transmural eosinophilic infiltration and fibrosis in a patient with non-traumatic Boerhaave’s syndrome due to eosinophilic esophagitis. Am J Gastroenterol 2012;107:1762.
Nicholson AG, Li D, Pastorino U, Goldstraw P, Jeffery PK. Full thickness eosinophilia in esophageal leiomyomatosis and idiopathic eosinophilic esophagitis. A common allergic inflammatory profile? J Pathol 1997;183:233–236.
Korsapati H, Babaei A, Bhargava V, Dohil R, Quin A, Mittal RK. Dysfunction of the longitudinal muscles of the esophagus in eosinophilic esophagitis. Gut 2009;58:1056–1062.
Mankad R, Bonnichsen C, Mankad S. Hypereosinophilic syndrome: cardiac diagnosis and management. Heart 2016;102:100–106.
Rosenberg HF, Dyer KD, Foster PS. Eosinophils: changing perspectives in health and disease. Nat Rev Immunol 2013;13:9–22.
Daniel EE, Crankshaw J, Sarna S. Prostaglandins and myogenic control of tension in lower esophageal sphincter in vitro. Prostaglandins 1979;17:629–639.
Spechler SJ. Eosinophilic esophagitis: novel concepts regarding pathogenesis and clinical manifestations. J Gastroenterol 2019;54:837–844.
Gleich GJ, Adolphson CR, Leiferman KM. The biology of the eosinophilic leukocyte. Annu Rev Med 1993;44:85–101.
King D, Thomas T, Chandan JS et al. Achalasia is associated with atopy in patients younger than 40 years of age. Am J Gastroenterol 2021;116:416–419.
Cools-Lartigue J, Chang SY, McKendy K et al. Pattern of esophageal eosinophilic infiltration in patients with achalasia and response to Heller myotomy and Dor fundoplication. Dis Esophagus 2013;26:766–775.
Savarino E, Gemignani L, Zentilin P et al. Achalasia with dense eosinophilic infiltrate responds to steroid therapy. Clin Gastroenterol Hepatol 2011;9:1104–1106.
Ghisa M, Laserra G, Marabotto E et al. Achalasia and obstructive motor disorders are not uncommon in patients with eosinophilic esophagitis. Clin Gastroenterol Hepatol 2021;19:1554–1563.
Park W, Vaezi MF. Etiology and pathogenesis of achalasia: the current understanding. Am J Gastroenterol 2005;100:1404–1414.
Hejazi RA, Reddymasu SC, Sostarich S, McCallum RW. Disturbances of esophageal motility in eosinophilic esophagitis: a case series. Dysphagia 2010;25:231–237.
von Arnim U, Kandulski A, Weigt J, Malfertheiner P. Correlation of high-resolution manometric findings with symptoms of dysphagia and endoscopic features in adults with eosinophilic esophagitis. Dig Dis 2017;35:472–477.
Martín Martín L, Santander C, Lopez Martín MC et al. Esophageal motor abnormalities in eosinophilic esophagitis identified by high-resolution manometry. J Gastroenterol Hepatol 2011;26:1447–1450.
Nurko S, Rosen R, Furuta GT. Esophageal dysmotility in children with eosinophilic esophagitis: a study using prolonged esophageal manometry. Am J Gastroenterol 2009;104:3050–3057.
Colizzo JM, Clayton SB, Richter JE. Intrabolus pressure on high-resolution manometry distinguishes fibrostenotic and inflammatory phenotypes of eosinophilic esophagitis. Dis Esophagus 2016;29:551–557.
Nennstiel S, Bajbouj M, Becker V et al. High-resolution manometry in patients with eosinophilic esophagitis under topical steroid therapy-a prospective observational study (HIMEOS-study). Neurogastroenterol Motil 2016;28:599–607.
van Rhijn BD, Oors JM, Smout AJ, Bredenoord AJ. Prevalence of esophageal motility abnormalities increases with longer disease duration in adult patients with eosinophilic esophagitis. Neurogastroenterol Motil 2014;26:1349–1355.
Monnerat MM, Lemme EM. Eosinophilic esophagitis: manometric and pHmetric findings. Arq Gastroenterol 2012;49:113–117.
Moawad FJ, Maydonovitch CL, Veerappan GR, Bassett JT, Lake JM, Wong RK. Esophageal motor disorders in adults with eosinophilic esophagitis. Dig Dis Sci 2011;56:1427–1431. https://doi.org/10.1007/s10620-011-1655-5.
Roman S, Hirano I, Kwiatek MA et al. Manometric features of eosinophilic esophagitis in esophageal pressure topography. Neurogastroenterol Motil 2011;23:208–214, e111.
Bassett J, Maydonovitch C, Perry J, Sobin L, Osgard E, Wong R. Prevalence of esophageal dysmotility in a cohort of patients with esophageal biopsies consistent with eosinophilic esophagitis. Dis Esophagus 2009;22:543–548.
Martín Martín L, Santander Vaquero C, Sánchez Prudencio S, Cantero Perona J, Gisbert JP, Moreno-Otero R. Eosinophilic esophagitis in the adult–clinical, endoscopic, pH-metric, and manometric findings. Rev Esp Enferm Dig 2008;100:476–480.
Miwa H, Kondo T, Oshima T, Fukui H, Tomita T, Watari J. Esophageal sensation and esophageal hypersensitivity - overview from bench to bedside. J Neurogastroenterol Motil 2010;16:353–362.
Bielefeldt K, Davis BM. Differential effects of ASIC3 and TRPV1 deletion on gastresophageal sensation in mice. Am J Physiol Gastrointest Liver Physiol 2008;294:G130-138.
Guarino MP, Cheng L, Ma J et al. Increased TRPV1 gene expression in esophageal mucosa of patients with non-erosive and erosive reflux disease. Neurogastroenterol Motil 2010;22:746–751, e219.
Tack J, Pandolfino JE. Pathophysiology of gastresophageal reflux disease. Gastroenterology 2018;154:277–288.
Byrne PJ, Mulligan ED, O’Riordan J, Keeling PW, Reynolds JV. Impaired visceral sensitivity to acid reflux in patients with Barrett’s esophagus. The role of esophageal motility. Dis Esophagus 2003;16:199–203.
Wong S, Ellison S, Haj Ali S et al. Characteristics and progression of childhood-onset and adult-onset eosinophilic esophagitis. J Gastroenterol Hepatol 2021.
Mahesh VN, Holloway RH, Nguyen NQ. Changing epidemiology of food bolus impaction: is eosinophilic esophagitis to blame? J Gastroenterol Hepatol 2013;28:963–966.
Sperry SL, Crockett SD, Miller CB, Shaheen NJ, Dellon ES. Esophageal foreign-body impactions: epidemiology, time trends, and the impact of the increasing prevalence of eosinophilic esophagitis. Gastrointest Endosc 2011;74:985–991.
Katzka DA. Demographic data and symptoms of eosinophilic esophagitis in adults. Gastrointest Endosc Clin N Am 2008;18:25–32; viii.
Prasad GA, Talley NJ, Romero Y et al. Prevalence and predictive factors of eosinophilic esophagitis in patients presenting with dysphagia: a prospective study. Am J Gastroenterol 2007;102:2627–2632.
Lee KH, Kim JE, Park CJ. Peripheral neuropathy associated withhypereosinophilic syndrome. Ann Dermatol 2008;20:149–152.
Chao CC, Hsieh ST, Shun CT, Hsieh SC. Skin denervation and cutaneous vasculitis in eosinophilia-associated neuropathy. Arch Neurol 2007;64:959–965.
Foster EL, Simpson EL, Fredrikson LJ et al. Eosinophils increase neuron branching in human and murine skin and in vitro. PLoS One 2011;6:e22029.
Drake MG, Scott GD, Blum ED et al. Eosinophils increase airway sensory nerve density in mice and in human asthma. Sci Transl Med 2018;10.
Wai K. Eosinophils increase transient receptor potential V1 and substance P expression in Dorsal Root Ganglia in vivo. PSU McNair Scholars Online J 2019;13.
Kapel RC, Miller JK, Torres C, Aksoy S, Lash R, Katzka DA. Eosinophilic esophagitis: a prevalent disease in the United States that affects all age groups. Gastroenterology 2008;134:1316–1321.
Hirano I, Chan ES, Rank MA et al. AGA institute and the joint task force on allergy-immunology practice parameters clinical guidelines for the management of eosinophilic esophagitis. Ann Allergy Asthma Immunol 2020;124:416–423.
Lucendo AJ, Arias Á, Molina-Infante J. Efficacy of proton pump inhibitor drugs for inducing clinical and histologic remission in patients with symptomatic esophageal eosinophilia: a systematic review and meta-analysis. Clin Gastroenterol Hepatol 2016;14:13-22.e11.
Cotton CC, Eluri S, Wolf WA, Dellon ES. Six-food elimination diet and topical steroids are effective for eosinophilic esophagitis: a meta-regression. Dig Dis Sci 2017;62:2408–2420. https://doi.org/10.1007/s10620-017-4642-7.
de Heer J, Miehlke S, Rösch T et al. Histologic and clinical effects of different topical corticosteroids for eosinophilic esophagitis: lessons from an updated meta-analysis of placebo-controlled randomized trials. Digestion 2021;102:377–385.
Murali AR, Gupta A, Attar BM, Ravi V, Koduru P. Topical steroids in eosinophilic esophagitis: systematic review and meta-analysis of placebo-controlled randomized clinical trials. J Gastroenterol Hepatol 2016;31:1111–1119.
Dellon ES, Katzka DA, Collins MH et al. Budesonide oral suspension improves symptomatic, endoscopic, and histologic parameters compared with placebo in patients with eosinophilic esophagitis. Gastroenterology 2017;152:776-786.e775.
Dellon ES, Woosley JT, Arrington A et al. Efficacy of budesonide vs fluticasone for initial treatment of eosinophilic esophagitis in a randomized controlled trial. Gastroenterology 2019;157:65-73.e65.
Lucendo AJ, Miehlke S, Schlag C et al. Efficacy of budesonide orodispersible tablets as induction therapy for eosinophilic esophagitis in a randomized placebo-controlled trial. Gastroenterology 2019;157:74-86.e15.
Arias Á, González-Cervera J, Tenias JM, Lucendo AJ. Efficacy of dietary interventions for inducing histologic remission in patients with eosinophilic esophagitis: a systematic review and meta-analysis. Gastroenterology 2014;146:1639–1648.
Chehade M, Brown S. Elimination diets for eosinophilic esophagitis: making the best choice. Expert Rev Clin Immunol 2020;16:679–687.
Straumann A, Conus S, Degen L et al. Budesonide is effective in adolescent and adult patients with active eosinophilic esophagitis. Gastroenterology 2010;139:1526–1537, 1537.e1521.
Yamabe A, Irisawa A, Shibukawa G et al. Clinical effects of eosinophilic esophagitis observed using endoscopic ultrasound. Clin J Gastroenterol 2014;7:305–309.
Kagalwalla AF, Wechsler JB, Amsden K et al. Efficacy of a 4-food elimination diet for children with eosinophilic esophagitis. Clin Gastroenterol Hepatol 2017;15:1698-1707.e1697.
Dellon ES, Cotton CC, Gebhart JH et al. Accuracy of the eosinophilic esophagitis endoscopic reference score in diagnosis and determining response to treatment. Clin Gastroenterol Hepatol 2016;14:31–39.
Kia L, Nelson M, Zalewski A et al. Oral delivery of fluticasone powder improves esophageal eosinophilic inflammation and symptoms in adults with eosinophilic esophagitis. Dis Esophagus 2018;31.
Konikoff MR, Noel RJ, Blanchard C et al. A randomized, double-blind, placebo-controlled trial of fluticasone propionate for pediatric eosinophilic esophagitis. Gastroenterology 2006;131:1381–1391.
Lieberman JA, Morotti RA, Konstantinou GN, Yershov O, Chehade M. Dietary therapy can reverse esophageal subepithelial fibrosis in patients with eosinophilic esophagitis: a historical cohort. Allergy 2012;67:1299–1307.
Hirano I, Collins MH, Assouline-Dayan Y et al. RPC4046, a monoclonal antibody against IL13, reduces histologic and endoscopic activity in patients with eosinophilic esophagitis. Gastroenterology 2019;156:592-603.e510.
Hirano I, Dellon ES, Hamilton JD et al. Efficacy of dupilumab in a phase 2 randomized trial of adults with active eosinophilic esophagitis. Gastroenterology 2020;158:111-122.e110.
Assa’ad AH, Gupta SK, Collins MH et al. An antibody against IL-5 reduces numbers of esophageal intraepithelial eosinophils in children with eosinophilic esophagitis. Gastroenterology 2011;141:1593–1604.
Xiong XF, Zhu M, Wu HX, Fan LL, Cheng DY. Efficacy and safety of dupilumab for the treatment of uncontrolled asthma: a meta-analysis of randomized clinical trials. Respir Res 2019;20:108.
Nhu QM, Hsieh L, Dohil L et al. Antifibrotic effects of the thiazolidinediones in eosinophilic esophagitis pathologic remodeling: a preclinical evaluation. Clin Transl Gastroenterol 2020;11:e00164.
Dellon ES, Gibbs WB, Rubinas TC et al. Esophageal dilation in eosinophilic esophagitis: safety and predictors of clinical response and complications. Gastrointest Endosc 2010;71:706–712.
Schupack DA, Ravi K, Geno DM et al. Effect of maintenance therapy for eosinophilic esophagitis on need for recurrent dilation. Dig Dis Sci 2021;66:503–510. https://doi.org/10.1007/s10620-020-06192-8.
Hirano I. Dilation in eosinophilic esophagitis: to do or not to do? Gastrointest Endosc 2010;71:713–714.
Greenberg S, Chang NC, Corder SR, Reed CC, Eluri S, Dellon ES. Dilation-predominant approach versus routine care in patients with difficult-to-treat eosinophilic esophagitis: a retrospective comparison. Endoscopy 2021.
Dougherty M, Runge TM, Eluri S, Dellon ES. Esophageal dilation with either bougie or balloon technique as a treatment for eosinophilic esophagitis: a systematic review and meta-analysis. Gastrointest Endosc 2017;86:581-591.e583.
Menard-Katcher C, Furuta GT, Kramer RE. Dilation of pediatric eosinophilic esophagitis: adverse events and short-term outcomes. J Pediatr Gastroenterol Nutr 2017;64:701–706.
Moawad FJ, Molina-Infante J, Lucendo AJ, Cantrell SE, Tmanova L, Douglas KM. Systematic review with meta-analysis: endoscopic dilation is highly effective and safe in children and adults with eosinophilic esophagitis. Aliment Pharmacol Ther 2017;46:96–105.
Moole H, Jacob K, Duvvuri A et al. Role of endoscopic esophageal dilation in managing eosinophilic esophagitis: a systematic review and meta-analysis. Medicine 2017;96:e5877.
Al-Hussaini A. Savary dilation is safe and effective treatment for esophageal narrowing related to pediatric eosinophilic esophagitis. J Pediatr Gastroenterol Nutr 2016;63:474–480.
Runge TM, Eluri S, Cotton CC et al. Outcomes of esophageal dilation in eosinophilic esophagitis: safety, efficacy, and persistence of the fibrostenotic phenotype. Am J Gastroenterol 2016;111:206–213.
Lipka S, Keshishian J, Boyce HW, Estores D, Richter JE. The natural history of steroid-naïve eosinophilic esophagitis in adults treated with endoscopic dilation and proton pump inhibitor therapy over a mean duration of nearly 14 years. Gastrointest Endosc 2014;80:592–598.
Ukleja A, Shiroky J, Agarwal A, Allende D. Esophageal dilations in eosinophilic esophagitis: a single center experience. World J Gastroenterol 2014;20:9549–9555.
Moawad FJ, Cheatham JG, DeZee KJ. Meta-analysis: the safety and efficacy of dilation in eosinophilic esophagitis. Aliment Pharmacol Ther 2013;38:713–720.
Jung KW, Gundersen N, Kopacova J et al. Occurrence of and risk factors for complications after endoscopic dilation in eosinophilic esophagitis. Gastrointest Endosc 2011;73:15–21.
Jacobs JW Jr, Spechler SJ. A systematic review of the risk of perforation during esophageal dilation for patients with eosinophilic esophagitis. Dig Dis Sci 2010;55:1512–1515. https://doi.org/10.1007/s10620-010-1165-x.
Karapiperis D, Malmstrom C, Vrakas S et al. Eosinophilic esophagitis and gastresophageal reflux disease: an overlapping of clinical, endoscopic and manometric features. Cureus 2021;13:e15774.
Amieva-Balmori M, Cano-Contreras AD, Remes-Troche JM. “Jackhammer esophagus” and eosinophilic esophagitis. Rev Gastroenterol Mex 2015;80:217–219.
Tanaka S, Toyonaga T, Kawara F et al. A case of Jackhammer esophagus caused by eosinophilic esophagitis in which per-oral endoscopic myotomy resulted in symptom improvement. Clin J Gastroenterol 2018;11:377–381.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No authors have any conflicts of interest to disclose. No grant support has been provided for this review.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Young, E., Philpott, H. Pathophysiology of Dysphagia in Eosinophilic Esophagitis: Causes, Consequences, and Management. Dig Dis Sci 67, 1101–1115 (2022). https://doi.org/10.1007/s10620-022-07419-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-022-07419-6