
Abstract
Background
Long-term tenofovir disoproxil fumarate (TDF) treatment for chronic hepatitis B (CHB) is associated with sustained viral suppression and regression of fibrosis and cirrhosis at year 5 (240 weeks) and no TDF resistance through 6 years (288 weeks).
Aim
We assessed the efficacy, safety, and resistance of TDF for up to 7 years (336 weeks) in HBeAg-positive and HBeAg-negative CHB patients.
Methods
Patients who completed 1 year (48 weeks) of randomized treatment with TDF or adefovir dipivoxil were eligible to receive open-label TDF for a total duration of 8 years (384 weeks).
Results
Of 641 patients initially randomized, 585 (91.3 %) entered the open-label phase; 437/585 (74.7 %) remained on study at year 7. For patients on treatment at year 7, 99.3 % maintained viral suppression (HBV DNA < 69 IU/mL), 80.0 % achieved serum alanine aminotransferase normalization, and in HBeAg-positive patients, 84/154 (54.5 %) and 25/154 (11.8 %) achieved HBeAg and HBsAg loss, respectively. One/375 (0.3 %) HBeAg-negative patients achieved HBsAg loss. No resistance to TDF was detected through 7 years. During the open-label phase, grade 3/4 drug-related adverse events were uncommon (1.0 %); ten (1.7 %) patients had elevation of serum creatinine ≥0.5 mg/dL above baseline. No significant change in bone mineral density was observed from year 4 to year 7 (week 192 to week 336).
Conclusions
Long-term TDF treatment was associated with sustained virologic, biochemical, and serologic responses, without resistance. TDF treatment was well tolerated, with a low incidence of renal and bone events. These data confirm the safety and efficacy of long-term TDF for CHB.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Disease progression to cirrhosis, liver failure, or hepatocellular carcinoma (HCC) occurs in up to 40 % of patients with chronic hepatitis B (CHB) [1]. Elevated hepatitis B virus (HBV) DNA viral load correlates with increased risk of HCC and cirrhosis [2], and conversely, treatments that suppress viral replication without breakthrough can delay disease progression [3]. Several antiviral agents are approved for treatment of CHB [4], although prospective long-term clinical data beyond 5 years of treatment with oral antiviral agents for CHB are limited.
TDF is an orally available prodrug of the nucleotide analogue tenofovir, a potent and selective inhibitor of HBV DNA polymerase/reverse transcriptase (pol/RT) in vitro [5]. TDF is currently approved for treatment of CHB in patients 12 years of age and older. In two international, multicenter, randomized, double-blind phase 3 studies of once-daily TDF versus once-daily adefovir dipivoxil (ADV) for 1 year (48 weeks) in 641 patients, TDF was more effective than ADV in viral suppression and improving histologic inflammation [6]. In addition, at year 5 (week 240), TDF therapy led to histological improvement (defined as a ≥2-point reduction in Knodell necroinflammatory score with no worsening of fibrosis) in 87.4 % (304/348) of patients, and 74.0 % (71/96) had reversal of baseline cirrhosis [7]. No evidence of resistance to TDF has been observed through year 6 (288 weeks) of treatment [8]. Here, we report the efficacy, safety, and resistance results of patients remaining on study at year 7 (week 336) of follow-up.
Methods
Study Design
The designs of the two randomized, controlled studies, GS-US-174-0102 (NCT00117676; Study 102) and GS-US-174-0103 (NCT00116805; Study 103), have been described previously [6]. Briefly, hepatitis B e antigen (HBeAg)-positive or (HBeAg)-negative CHB patients (18–69 years of age) with compensated liver disease and Knodell necroinflammatory score ≥3 were randomized 2:1 to TDF 300 mg once daily or ADV 10 mg once daily for 1 year, after which all patients switched to or continued on TDF during the open-label phase for up to 7 years, for a total study duration of up to 8 years (384 weeks). Emtricitabine (FTC) could be added to the treatment regimen, at the discretion of the investigator, for confirmed viremia on or after 1.5 years (week 72).
Efficacy assessments included virologic response, defined as plasma HBV DNA levels <69 IU/mL (<400 copies/mL). The proportion of patients with plasma HBV DNA <29 IU/mL (<169 copies/mL, the lower limit of quantification of the COBAS Taqman assay) was also analyzed. Biochemical response based on normalized alanine aminotransferase (ALT) levels was assessed as described previously [6, 7]. Serologic endpoints included serum HBeAg loss and seroconversion to anti-HBe (HBeAg-positive patients), and serum hepatitis B surface antigen (HBsAg) loss and seroconversion to anti-HBs. Patients with confirmed HBsAg loss or seroconversion could stop treatment at the investigator’s discretion, provided they remained on follow-up. Viral resistance testing for genotypic changes within the HBV reverse transcriptase (HBV pol/RT) was performed annually for patients with HBV DNA ≥69 IU/mL (≥400 copies/mL) who experienced virologic breakthrough or persistent viremia, and those who discontinued from the study with HBV DNA ≥69 IU/mL (≥400 copies/mL) [8]. Conserved-site changes or polymorphic-site changes detected in > 1 patient by genotypic analysis were confirmed by phenotyping, as described previously [8, 9].
Safety and tolerability assessments, including adverse events (AEs), treatment discontinuations, and patient deaths, were conducted on an ongoing basis. Predefined renal endpoints included creatinine clearance <50 mL/min, serum creatinine ≥0.5 mg/dL above baseline, and serum phosphate <2 mg/dL. Bone mineral density (BMD) was assessed annually by dual-energy X-ray absorptiometry scan from year 4 (week 192) through year 7 (week 336).
Virologic and biochemical response were assessed using a modified, long-term evaluation-TDF (LTE-TDF) only analysis, in which (a) patients with missing data or with FTC added were counted as failures at all time points following addition, and (b) patients with HBsAg loss who discontinued study drug and met endpoint criteria at the last on-study visit were counted as successes and had the last value carried forward. An on-treatment analysis was also conducted, in which patients with non-missing data, regardless of the treatment received, were included in the analysis, and patients with missing data were excluded.
Results
Patient Disposition
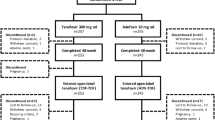
At year 7, a total of 437/641 (68.2 %) of HBeAg-positive and HBeAg-negative patients initially randomized and 437/585 (74.7 %) of patients entering the open-label phase remained on study (Fig. 1). In total, 148 of 585 (25.3 %) patients discontinued during the open-label phase for withdrawal of consent (n = 51); loss to follow-up (n = 37); investigator discretion (n = 27); safety, tolerability, or efficacy reason (n = 17); HBsAg or HBeAg seroconversion (n = 8); protocol violation (n = 7); and sponsor decision (n = 1) (Fig. 1).

Patient disposition at year 7
Virologic Response
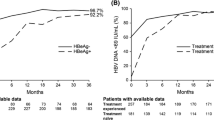
Suppression of HBV DNA levels at both < 69 and < 29 IU/mL was observed in nearly all patients (99.3 % for both measures) who remained on study at year 7, including 99.3 % (both measures) of HBeAg-negative patients and 99.4 % (both measures) of HBeAg-positive patients (Table 1).
Biochemical and Serologic Response
Biochemical responses at year 7 are summarized in Table 1. Of patients on study at year 7 who had abnormal ALT at baseline, 80.0 % experienced ALT normalization, including 83.5 % of HBeAg-negative patients and 74.2 % of HBeAg-positive patients. Of the 42 HBeAg-negative and 40 HBeAg-positive patients on study who did not achieve ALT normalization, the median [mean] ALT (U/L) levels were 49 [55] and 51 [57], respectively. Among 154 observed HBeAg-positive patients on study at year 7, 84 (54.5 %) experienced HBeAg loss and 61 (39.6 %) experienced seroconversion to anti-HBe. In a Kaplan–Meier analysis of the intent-to-treat population of HBeAg-positive patients, 27 (11.8 %) experienced HBsAg loss and 21 (9.7 %) experienced seroconversion to anti-HBs. Among 375 HBeAg-negative initially randomized patients, one experienced HBsAg loss at year 5. Of the 28 HBeAg-positive and HBeAg-negative patients who experienced HBsAg loss during the study, three remained on study drug through year 7, and 25 subsequently discontinued study drug and entered treatment-free follow-up (TFFU). Fourteen of these discontinued study participation before year 7, while 10 remained on study under TFFU, and one restarted treatment (Fig. 2).

Disposition at year 7 (week 336) of patients who experienced confirmed HBsAg loss. aCompleted through year 7. bOne patient experienced seroreversion, restarted on treatment, and seroconverted again
Resistance Surveillance
Overall, the majority of patients (533/585, 91.1 %) who entered open-label treatment had HBV DNA < 69 IU/mL at their last time point on TDF through year 7. Fifty-two patients (8.8 %) qualified for genotypic analysis at their last time point on TDF, with three patients qualifying during year 7. Thirty-nine of the 585 patients (6.7 %) who entered open-label treatment switched to FTC/TDF through year 6 of open-label treatment. Seven patients qualified for genotypic analysis at their last time point on FTC/TDF, with one patient qualifying during year 7. The 48 patients that qualified for genotypic analysis prior to year 7 were previously evaluated [9]. For the four patients who qualified for genotypic analysis during year 7, two had no changes in HBV pol/RT from baseline and two were unable to be genotyped. Three of the patients who qualified for genotypic analysis were non-adherent to study treatment, as confirmed by undetectable levels of study drug in plasma. All four patients achieved HBV DNA < 69 IU/ml prior to year 7, and at their qualifying time point during year 7, the mean HBV DNA level was 100.1 IU/ml (range 89-113 IU/ml). One of the four subjects discontinued from the study prior to week 336 and is not included in the on-treatment HBV DNA suppression analysis.
Safety
Safety findings during the open-label period are summarized in Table 2. Of 585 patients who entered the open-label phase, 13 (2.2 %) patients discontinued study drug because of AEs, including three (0.5 %) that were considered study drug related. Reasons for discontinuation (in some cases multiple) for these 13 patients included hepatic neoplasm (n = 3), endometrial cancer (n = 1), lung neoplasm (n = 1), fatigue (n = 1), abdominal pain (n = 1), septic shock (n = 1), non-specified injury (n = 1), road traffic accident (n = 1), disturbance in attention (n = 1), dizziness (n = 1), increased blood creatinine (i = 1), osteoporosis (n = 1), breast cancer (n = 1), and drug dependence (n = 1). Seven (1.2 %) patients had study drug-related serious AEs during the open-label period, in some cases multiple events, although none had new onset during year 7. These serious AEs included acute pancreatitis (n = 1), osteopenia (n = 1), osteoporosis (n = 2), renal failure (n = 1), increased ALT (n = 2), and facial spasm (n = 1). To date, 12 deaths have occurred during the open-label phase, none of which were study drug related.
Overall, 21/585 (3.6 %) patients had 25 confirmed AEs under renal impairment (defined as serum creatinine increase ≥0.5 mg/dL above baseline, serum phosphate <2 mg/dL, and creatinine clearance [CrCl] <50 mL/min [Cockcroft–Gault]) during the open-label phase. Up to year 5, there were seven patients who experienced serum creatinine increase ≥0.5 mg/dL above baseline, seven patients with serum phosphate <2 mg/dL, and two patients with CrCl < 50 mL/min. Between year 5 and year 7, an additional three patients experienced an increase in serum creatinine ≥ 0.5 mg/dL above baseline, two patients with serum phosphate <2 mg/dL, and four patients with CrCl < 50 mL/min. Comparing baseline characteristics of patients who did and did not have renal AEs, mean age (47 vs. 40 years; P = 0.003), mean CrCl (98.5 vs. 117.4 mL/min; P = 0.003), and mean serum phosphate (2.8 vs. 3.3 mg/dL; P = 0.002) were statistically different between groups. Of the 21 patients who developed renal insufficiency, seven patients had hypertension, two patients had diabetes mellitus, two patients had underlying renal disease (HBV-associated glomerulonephritis, IgA nephropathy), two patients had nephrolithiasis, three patients were older than 60 years of age, and 12 patients had no known risk factors listed in their medical history. Additionally, no patients experienced concurrent decrease in CrCl < 50 mL/min and serum phosphate <2 mg/dL.
Annual BMD assessments were undertaken at year 4 through year 7 in patients enrolled in the open-label phase. Overall, no significant changes in BMD were observed from year 4 (325 hip and spine scans) to year 7 (288 hip scans and 294 spine scans), and no trends in T or Z scores were apparent over the 3 years (Supplemental Table 1).
Discussion
The results described in this report demonstrate that long-term treatment with TDF for chronic HBV infection provides potent and durable viral suppression and normalization of serum ALT levels. In addition, the proportion of patients with HBsAg loss was 11.8 % (Kaplan–Meier estimation) at up to 7 years of TDF treatment. Of particular importance, no resistance to TDF has been detected with up to 7 years of treatment. These results also demonstrate that TDF treatment is safe and well tolerated, with few discontinuations related to AEs and no new safety signals identified through 7 years. Renal AEs occurred infrequently, were generally mild, and improved with dose modification. Although introduced only at year 4, serial assessments of BMD suggest no evidence of bone loss over 3 years of evaluation.
The lack of resistance to TDF after prolonged exposure is striking, considering the high rates of resistance observed with other nucleotide/nucleoside analogues, ranging from 20 % at 5 years of ADV treatment [10] to 65 % at 5 years of lamivudine treatment [11]. Entecavir treatment is associated with a low rate of resistance in treatment-naive patients (1.2 % of patients treated for up to 5 years) [12], although rates of entecavir resistance are considerably higher in those who are ADV nonresponders (4 % of patients treated for a median of 20 months) [13], lamivudine resistant (51 % of patients treated for up to 5 years) [12], and lamivudine and ADV dual resistant (91 % of patients with virologic breakthrough after treatment for a median of 24 months) [14]. In this study, we report the lack of resistance to TDF through 7 years in mostly treatment-naive patients. Additionally, lack of resistance to TDF has been reported through 3.5 years (168 weeks) in prior ADV-treated patients [15] and through 2 years (96 weeks) in lamivudine-resistant patients [16]. The absence of viral resistance to TDF carries important implications for clinical outcomes, because patients without resistance development may have a lower risk of liver disease progression [17].
The proportion of patients with HBsAg loss in HBeAg-positive patients through year 7 was 11.8 % by Kaplan–Meier analysis. This is consistent with rates of HBsAg loss determined by different methodologies reported in the literature for other oral antiviral agents: a retrospective study in HBeAg-positive patients reported HBsAg loss in 18/354 (5.1 %) patients after 96 weeks of entecavir treatment and 10/355 (2.8 %) patients after 96 weeks of lamivudine treatment [18], and an open-label extension of a randomized study of entecavir in HBeAg-positive patients reported HBsAg loss in 2/145 (1.4 %) patients after up to 5 years of treatment [19]. Although this is higher than the rate of spontaneous HBsAg loss (0.5–0.8 % per year) [20], HBsAg loss rates remain low with current CHB nucleos(t)ide therapy, indicating that achievement of HBsAg loss is a relatively rare but dependable clinical end point. Achieving higher rates of HBsAg loss might require the addition of immune-based therapies, with an approach currently under investigation in an ongoing TDF and pegylated interferon combination study (NCT01277601).
In summary, our 7-year follow-up is the first CHB study to show that long-term use of an oral nucleos(t)ide agent—TDF monotherapy for chronic HBV infection—is able to suppress viral replication without development of resistance and is well tolerated.
References
Lok AS. Chronic hepatitis B. N Engl J Med. 2002;346:1682–1683.
Chen CJ, Yang HI, Su J, et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA. 2006;295:65–73.
Iloeje UH, Yang HI, Su J, et al. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology. 2006;130:678–686.
Di Marco V, Di Stefano R, Ferraro D, et al. HBV-DNA suppression and disease course in HBV cirrhosis patients on long-term lamivudine therapy. Antivir Ther.. 2005;10:431–439.
De Clercq E, Ferir G, Kaptein S, Neyts J. Antiviral treatment of chronic hepatitis B virus (HBV) infections. Viruses. 2010;2:1279–1305.
Heijtink RA, Kruining J, de Wilde GA, Balzarini J, de Clercq E, Schalm SW. Inhibitory effects of acyclic nucleoside phosphonates on human hepatitis B virus and duck hepatitis B virus infections in tissue culture. Antimicrob Agents Chemother. 1994;38:2180–2182.
Marcellin P, Heathcote EJ, Buti M, et al. Tenofovir disoproxil fumarate versus adefovir dipivoxil for chronic hepatitis B. N Engl J Med. 2008;359:2442–2455.
Marcellin P, Gane E, Buti M, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet. 2013;381:468–475.
Kitrinos KM, Corsa A, Liu Y, et al. No detectable resistance to tenofovir disoproxil fumarate after 6 years of therapy in patients with chronic hepatitis B. Hepatology. 2013;59:434–442.
Heathcote EJ, Marcellin P, Buti M, et al. Three-year efficacy and safety of tenofovir disoproxil fumarate treatment for chronic hepatitis B. Gastroenterology. 2011;140:132–143.
Marcellin P, Chang TT, Lim SG, et al. Long-term efficacy and safety of adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. Hepatology. 2008;48:750–758.
Marcellin P, Chang TT, Lim SG, et al. Adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. N Engl J Med. 2003;348:808–816.
Lok AS, Lai CL, Leung N, et al. Long-term safety of lamivudine treatment in patients with chronic hepatitis B. Gastroenterology. 2003;125:1714–1722.
Tenney DJ, Rose RE, Baldick CJ, et al. Long-term monitoring shows hepatitis B virus resistance to entecavir in nucleoside-naive patients is rare through 5 years of therapy. Hepatology. 2009;49:1503–1514.
Zoutendijk R, Reijnders JG, Brown A, et al. Entecavir treatment for chronic hepatitis B: adaptation is not needed for the majority of naive patients with a partial virological response. Hepatology. 2011;54:443–451.
Park JW, Kim HS, Seo DD, et al. Long-term efficacy of entecavir in adefovir-refractory chronic hepatitis B patients with prior lamivudine resistance. J Viral Hepat.. 2011;18:e475–e481.
Berg T, Zoulim F, Moeller B, et al. Long-term efficacy and safety of emtricitabine plus tenofovir DF vs. tenofovir DF monotherapy in adefovir-experienced chronic hepatitis B patients. J Hepatol. 2014;60:715–722.
Fung S, Kwan P, Fabri M, et al. Randomized comparison of tenofovir disoproxil fumarate vs emtricitabine and tenofovir disoproxil fumarate in patients with lamivudine-resistant chronic hepatitis B. Gastroenterology. 2013;146:980–988.
Liaw YF, Sung JJ, Chow WC, et al. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med. 2004;351:1521–1531.
Gish RG, Chang TT, Lai CL, et al. Loss of HBsAg antigen during treatment with entecavir or lamivudine in nucleoside-naive HBeAg-positive patients with chronic hepatitis B. J Viral Hepat.. 2010;17:16–22.
Chang TT, Lai CL, Kew Yoon S, et al. Entecavir treatment for up to 5 years in patients with hepatitis B e antigen-positive chronic hepatitis B. Hepatology. 2010;51:422–430.
McMahon BJ. The natural history of chronic hepatitis B virus infection. Hepatology. 2009;49:S45–S55.
Acknowledgments
This study and the analyses presented were sponsored by Gilead Sciences, Inc. Anna Lau, PhD, and Evelyn Albu, PhD, of Percolation Communications LLC provided editorial assistance during manuscript development. Gilead Sciences, Inc., provided financial support for manuscript development.
Conflict of interest
The authors disclose the following financial relationships that may be viewed as potential conflict of interest.
Author | Employment | Stock ownership | Consultancy | Honoraria | Research grant | Paid expert testimony | Other |
|---|---|---|---|---|---|---|---|
MB | Bristol-Myers Squibb; Gilead | Bristol-Myers Squibb; Gilead | |||||
NT | AbbVie; Gilead; Janssen | AbbVie; Bayer; Bristol-Myers Squibb; Gilead; Janssen; Merck; Roche; Salix | AbbVIe; Beckman; Bristol-Myers Squibb; Gilead; Janssen; Roche; Vertex | ||||
JP | Abbott; AbbVie; Bristol-Myers Squibb; Boehringer; Gilead; GlaxoSmithKline; Kedrion; Janssen; Merck; Merck Sharp & Dohme; Novartis, Roche | Bristol-Myers Squibb; Novartis; Roche | Abbott; Bristol-Myers Squibb; Boehringer; Gilead; Kedrion; Janssen; Merck; Merck Sharp & Dohme; Novartis; Roche | ||||
RF | AbbVie; Bristol-Myers Squibb; Gilead; Janssen; Merck; Novartis; Roche | AbbVie; Bristol-Myers Squibb; Gilead; Janssen; Merck; Novartis; Roche | Roche | ||||
SG | AbbVie; Bristol-Myers Squibb; Gilead; Janssen; Merck Sharp & Dohme; Roche | AbbVie; Bristol-Myers Squibb; Gilead; Janssen; Merck Sharp & Dohme; Roche | |||||
ZK | Gilead; Idenix; Jansen; Novartis; Receptos; Roche | ||||||
RAS | Gilead | Gilead | |||||
JFF | Gilead | Gilead | |||||
EBM | Gilead | Gilead | |||||
PC | Gilead | Gilead | |||||
KMK | Gilead | Gilead | |||||
GMS | Gilead | Gilead | |||||
EG | Achillion; Gilead; Idenix; Janssen; Merck; Novartis; Novira; Roche | ||||||
PM | Abbott; Boehringer Ingelheim; Bristol-Myers Squibb; Gilead; Janssen/Tibotec; Merck; Novartis; Pfizer; Roche; Vertex | Bristol-Myers Squibb; Gilead; Janssen/Tibotec; Merck; Novartis; Roche |
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Buti, M., Tsai, N., Petersen, J. et al. Seven-Year Efficacy and Safety of Treatment with Tenofovir Disoproxil Fumarate for Chronic Hepatitis B Virus Infection. Dig Dis Sci 60, 1457–1464 (2015). https://doi.org/10.1007/s10620-014-3486-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-014-3486-7