
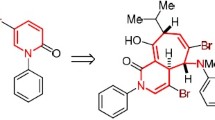
Bromination of the furanoquinoline alkaloid haplophyllidine by molecular bromine and N-bromosuccinimide was accompanied by intramolecular cyclization to form mixtures of new compounds containing additional penta-, hexa-, and spirocyclic rings incorporating the prenyl group of haplophyllidine. The structures and absolute configurations of the chiral centers of all four bromo-derivatives were elucidated using a combination of NMR spectroscopic methods and X-ray crystal structure analyses.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Quinoline is well known as the skeletal platform of several extremely interesting natural and synthetic biologically active compounds. Fluoroquinolones such as moxifloxacin [1] and ciprofloxacin [2] are currently widely used in medicine as antimicrobials; mefloquine (4-methanolquinoline derivative) [3] and chloroquine (4-aminoquinoline derivative) [4], as antimalarials. The last is undergoing clinical trials for treatment of COVID-19 [5, 6]. Pitavastatin, which contains a 4-fluorophenylquinoline moiety, is one of the newest statins and is used to reduce blood lipid levels and the risk of cardiovascular diseases [7].
The diverse high activity of quinoline compounds has prompted efforts to modify quinoline alkaloids. Halogenation, which can produce promising biologically active compounds and reactive intermediates, is one possible transformation pathway. Therefore, it seemed interesting to study the bromination products of haplophyllidine (1), the structure of which includes a prenyl group, by bromine and N-bromosuccinimide (NBS), which are widely used for bromination of alkenes in side chains of heteroaromatic compounds and for the synthesis of five-membered heterocyclic compounds [8]. Haplophyllidine, the structure and relative configuration of which were previously established [9], was isolated from seeds of Haplophyllum perforatum growing in Samarkand Region of Uzbekistan [10].
Herein, results from the bromination of 1 by molecular bromine in CHCl3 (method A) and by NBS in CHCl3 (method B) and DMF (method C) are reported. The addition of Br was facilitated by the two methyls on the prenyl double bond. The bromination turned out to be exceedingly interesting and promising for synthesizing a series of cyclic products. Bromination in all three reaction methods formed a mixture of products that could not always be successfully separated by column chromatography. Therefore, it seemed logical to use HPLC to estimate the product yields. As a result, it was found that four products (2–5) formed in the reactions according to methods A and B and three products (2, 4, and 5), method C (Table 1, Scheme 1). The dibromo-derivative of 1 was not observed to form in any of the reactions.

Bromination of haplophyllidine (1) by molecular bromine and N-bromosuccinimide (NBS).
Table 1 shows that bromination in CHCl3, regardless of the reagent, gave a mixture of compounds, the major constituents of which were 3 (41%, A) and bromo-derivatives 3 and 5 (~32% each, B) while 3 was not detected under the conditions of reaction C.
The structures of isolated 3–5 were proved by spectral methods and X-ray crystal structure analyses (XSAs). The chemical structure of 2, which was isolated as an oil, was elucidated by analyzing 1H and 13C NMR spectra and HSQC, HMBC, DQF-COSY, and NOESY experiments. Resonances of protons and C atoms in 1H and 13C NMR spectra were assigned by using HSQC and COSY experiments. The strong-field part of the 1H NMR spectrum of 2 showed two singlets for methyls at δ 1.33 (H-12) and 1.45 (H-13) and resonances for three methylenes at δ 1.99 (dddd, J = 13.4, 7.5, 6.7, 5.0 Hz, H-6a), 2.13 (dddd, J = 13.4, 8.7, 5.0, 3.4 Hz, H-6b), 2.65 (ddd, J = 16.6, 6.7, 5.0 Hz, H-5a), and 2.86 (ddd, J = 16.6, 8.7, 5.0 Hz, H-5b) and 2.80 (m, H-9). The middle of the proton spectrum displayed two doublets of doublets at δ 4.18 (dd, J = 7.4, 3.3 Hz, H-7) and 4.44 (dd, J = 7.3, 6.5 Hz, H-10), which were characteristic of protons on a C atom bound to oxygen, and 3H singlets at 3.14 and 4.27 ppm for C-8 and C-4 methoxyls, respectively. The aromatic part of the spectrum had only two 1H doublets with SSCC J = 2.6 Hz at δH 7.57 ppm (H-2) and 6.95 (H-3) that corresponded to protons of the furan ring [11].
An analysis of 13C NMR and HSQC spectra of 2 showed 18 resonances for C atoms as seven quaternary, including five on a heteroatom; four methine; three methylene; and four methyl C atoms. The weak-field region of the 13C NMR spectrum had resonances for three quaternary C atoms at δ 162.86 (C-1a), 158.01 (C-4), and 150.96 (C-8a) and for a protonated C atom at δ 142.90 (C-2), which was determined from an HSQC experiment of the furanopyridine motif. The aromatic range of C atoms revealed resonances of a second methine C atom at δ 104.82 (C-3) and two quaternary C atoms at δ 105.36 (C-3a) and 118.16 (C-4a).
Three resonances at stronger field that were characteristic of C atoms bound to oxygen were observed at δ 76.99 (C-8), 74.92 (C-11), and 71.57 (C-7). Next, the spectrum showed resonances of two methoxyls at δ 58.65 (C-14) and 51.32 (C-15), two methyls at 27.06 (C-12) and 25.63 (C-13), and the other aliphatic C atoms. The positions of the two methoxyls and two methyls were established from an HMBC experiment. The presence in the HMBC spectrum of cross-peaks H-14/C-4, H-15/C-8, and H-12(H-13)/C-10, C-11 indicated that the methoxyls were situated on C-4 and C-8, respectively; the two methyls, on C-11. The spectrum also had cross-peaks between H-7/C-8, C-11; H-9/C-8, C-8a, C-10, C-11; and H-10/C-8, C-9, C-11, C-12, C-13. Also, correlations were found in the DQF-COSY spectrum between H-2/H-3, H-5/H-6, H-6/H-7, and H-9/H-10. These data showed that an additional six-membered ring was present in 2 as compared to the starting alkaloid 1.
The relative configuration of 2 was established based on the NOESY spectrum. NOE correlations between protons H-7/H-10, H-13, H-15 were indicative of coplanarity and the β-orientation of these protons. Table 2 presents detailed 1H and 13C NMR spectral data and HMBC correlations for 2. Thus, the structure (7R,8R,10S)-10-bromo-4,8-dimethoxy-11,11-dimethyl-5,6,7,8,10,11-hexahydro-9H-furo[2,3-b]pyrano[2,3-h]quinoline was found for 2 based on the above data.
Resonances in 1H and 13C NMR spectra of 4 were assigned based on HSQC and HMBC experiments (Table 2). Products 2 and 4 were found to be epimers at C-10. A comparative analysis of the results revealed several differences in the PMR spectra of these compounds. Protons H-9 in cyclic product 4 resonated at δH 2.19 (dd, J = 12.0, 12.9 Hz) and 3.50 (dd, J = 12.0, 4.2 Hz) while the resonance of α-proton H-10 was shifted by 0.57 ppm to strong field. Also, the C-12 resonance of 4 was shifted (δ 18.23) as compared to that of 2 (δ 27.06). An XSA established the structure of 4 as (7R,8R,10R)-10-bromo-4,8-dimethoxy-11,11-dimethyl-5,6,7,8,10,11-hexahydro-9H-furo[2,3-b]pyrano[2,3-h]quinoline.
The structure of 3 was established as a bromo-derivative of an oxaspiroquinoline using 2D NMR spectroscopy and an XSA. The NMR spectroscopic data showed that 3 also contained a new ring. Thus, resonances of a methyl (H3CO-8 at δ 3.18 ppm) and an olefin [H-10 (δ 5.35 ppm) and C-10 (δ 54.64 ppm) and C-11 (δ 74.92 ppm)] disappeared from NMR spectra of starting 1. The disappearance of the C-10=C-11 double bond in 1 was compensated by the appearance in 3 of a new fivemembered ring based on the prenyl group of 1. The lack of a methyl resonance at 3.18 ppm indicated that the new ring closed through the oxygen on C-8. Also, the multiplicities of the 7-OH and H-7 resonances were observed to change and the H-9b resonance shifted to strong field (δ 3.26→2.77 ppm). Proton H-10 in 3 resonated at δ 5.04 (1H, dd, J = 11.4, 7.1 Hz). The resonances of the other protons of oxaspiroquinoline 3 and starting 1, including two singlets from magnetically nonequivalent geminal CH3 groups, appeared in analogous regions. The 13C NMR spectrum of 3 had resonances at δ 85.1 (spiro C), 84.0, and 53.9 ppm that were attributed to C-8, C-11, and C-10, which resonated in starting 1 at δ 79.2, 134.1, and 119.8 ppm, respectively. It is noteworthy that cycloaddition at an exocyclic double bond is one of the methods used to synthesize spirocyclic compounds [12] although the prenyl double bond and the methoxyl O atom of haplophyllidine were involved in the formation of spirocyclic 3.
The 1H NMR spectrum of 5 was missing the 7-OH resonance while H-9 resonated at δ 2.15 (dd, J = 10.6, 13.3 Hz) and 2.69 (dd, J = 5.3, 13.3 Hz). Also, the H-10 resonance underwent a strong-field shift by 0.83 ppm (δH 4.52, dd, J = 9.2, 4.9 Hz) as compared to 1. The 13C NMR spectrum of 5 had substantial changes in the chemical shifts of C-7, C-8, C-10, and C-11, which were observed at δ 86.5 (C-8), 85.5 (C-10), 80.4 (C-7), and 67.5 (C-11). The structure (7R,8R,10R)-10-(2′-bromopropan-2′-yl)-4,8-dimethoxy-5,6,7,8,9,10-hexahydrodifuro[2,3-b:2,3-h]quinoline was proposed for 5 based on the results and was confirmed by an XSA.
The formation of the variously structured intramolecular cyclization products 2–5 through the action of the brominating agents in CHCl3 provided the basis for a scheme of the reaction mechanism that explained well the experimental results using generally known concepts of the bromination mechanism of ethylene bonds (Scheme 2). The formation of two bromonium cations (A and B) is one reason for the different directions of the reactions. The presence of hydroxyl (7α-OH), methoxyl (8β-OCH3), and –N= groups in 1 that stabilized carbocations formed at C-10 and C-11 led to several cyclic reaction products.

Proposed bromination mechanism of haplophyllidine by molecular bromine and NBS in CHCl3.
The outcome of the final cyclization step depended on the degree of localization of negative charge on the oxide atoms and on the regioisomerization of the bromonium cations (A and B). The ionic charge of the bromonium ion and the ability of a methoxyl ion to attack it (bromination method C) decreased if the polarity of the solvent was increased (DMF as compared to CHCl3).
The structures of the bromination products were difficult to establish based on spectral methods, particularly NMR spectroscopy. Therefore, the structures of the obtained compounds were established using XSA analyses, which proved the structures of 3–5 and the absolute configurations of the chiral centers of the synthesized cyclic derivatives.
Figure 1 shows the molecular structures of 3–5, which agreed with the absolute configurations. The Flack parameters for these compounds were 0.009(13), 0.027(11), and 0.020(7). The presence of the heavy Br atom in the bromination products enabled the absolute configurations to be reliably determined as 7R,8R,10S (3), 7R,8R,10R (4), and 7R,8R,10R (5). The Br atom and C-11 methyl group were disordered in 5 and exchanged places in approximately equal ratios (one of the fixed positions of the molecule is shown in Fig. 1). Therefore, the temperature factors are greater for these atoms and the Csp3–Csp3 bond distances are anomalously elongated in this motif. Otherwise, the bond distances and angles fell within 3σ of the usual values [13].

Molecular structures of 3–5.
The core in the molecular structures of tetrahydrofuranoquinoline derivatives 3–5 was almost planar except for ring A. Ring A in 3 adopted a 6α-sofa conformation; the oxaspiro-bonded heterocycle, a half-chair. Ring A in 4 adopted a half-chair conformation with C-6 and C-7 deviating from the plane of the other four. The condensed six-membered oxa heterocycle had a chair conformation. Ring A in 5 had another half-chair shape that was related to the deviation of C-6 and C-7 to the opposite side; the five-membered heterocycle, an envelope conformation.
Anomalously short van-der-Waals intermolecular contacts were not observed in crystals of the molecules.
Thus, we showed that bromination of 1 by Br2 and NBS was associated with addition of Br and intramolecular cyclization involving the prenyl double bond and the O atom of the hydroxyl or methoxyl group to form additional penta-, hexa-, and spirocyclic structures.
Experimental
The yield and purity of compounds were determined using a Shimadzu LC-20 HPLC (Japan) and a C18 column (Shimadzu LC-20, Japan). IR spectra were recorded from KBr pellets on a System 2000 FT-IR spectrometer (PerkinElmer, USA). Mass spectra were measured in a CAMAG TLC-MS with an ACQUITY QDa detector. NMR spectra were recorded in CDCl3 on JNM-ECZ400R and JNM-ECZ600R spectrometers (JEOL, Japan) at operating frequencies 400 and 600 MHz for 1H. The internal standard in PMR spectra was TMS (0 ppm); in 13C NMR spectra, the solvent chemical shift (CDCl3, 77.16 ppm vs. TMS). NMR spectra were processed using MestReNova 14.2.0 software (Mestrelab Research S.L., Santiago de Compostela, Spain). The course of reactions and purity of products were monitored by TLC on Sigma-Aldrich Silufol L/W plates (10 × 20 cm) with a 254-nm fluorescent indicator (Germany) using benzine–EtOAc (2:1). Melting points of all synthesized compounds were measured on a Boetius apparatus.
Haplophyllidine (5,6,7,8-tetrahydro-4,8-dimethoxy-8-(3-methyl-2-butenyl)furo[2,3- b ]quinolin-7-ol (1) was isolated from seeds of H. perforatum by the literature method [10]. 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 1.68 (3H, s, H-12), 1.73 (3H, s, H-13), 1.96 (1H, br.s, 7-OH), 2.01 (1H, m, H-6a), 2.29 (1H, m, H-6b), 2.65 (1H, dd, J = 15.8, 8.4, H-9a), 2.77 (2H, m, H-5), 3.18 (3H, s, 8-OCH3), 3.26 (1H, dd, J = 15.8, 5.8, H-9b), 4.19 (1H, m, H-7), 4.27 (3H, s, 4-OCH3), 5.35 (1H, m, H-10), 6.96 (1H, d, J = 2.6, H-3), 7.56 (1H, d, J = 2.6, H-2). 13C NMR (100 MHz, CDCl3, δ, ppm): 162.49 (C-1a), 158.49 (C-4), 150.53 (C-8a), 142.64 (C-2), 134.13 (C-11), 119.82 (C-10), 117.23 (C-4a), 105.15 (C-3a), 104.84 (C-3), 79.19 (C-8), 70.03 (C-7), 58.57 (C-14), 50.84 (C-15), 30.32 (C-9), 26.24 (C-13), 24.16 (C-6), 18.78 (C-5), 18.21 (C-12).
Bromination of 1 by Molecular Br 2. A solution of 1 (0.5 g, 1.58 mmol) in CHCl3 (4 mL) was stirred, treated dropwise with a solution of Br2 (0.3 g, 1.875 mmol) in CHCl3 (2 mL) over 5 min, and stirred for 4 h at 20–25°C. The course of the reaction was monitored by TLC. When the reaction was finished, the solvent was distilled off. The oily residue containing four compounds (Rf 0.70, 0.67, 0.65, 0.50; benzine–EtOAc, 2:1) was purified over a column of silica gel using benzine–CHCl3 (100:1→100:10). Three pure products were obtained.
(7 R ,8 R ,10 S )-10-Bromo-4,8-dimethoxy-11,11-dimethyl-5,6,7,8,10,11-hexahydro-9 H -furo[2,3- b ]pyrano[2,3- h ]quinoline (2), oily compound, Rf 0.65, C18H22BrNO4. ESI-MS m/z: 364.1275 [M – OCH3]+ for Br79, 366.0756 [M – OCH3]+ for Br81 (calcd for C17H19BrNO3, 364.2584, 366.2563). Table 2 lists the 1H and 13C NMR spectral data.
(7 R ,8 R ,10 S )-10-Bromo-4-methoxy-11,11-dimethyl-6,7,10,11-tetrahydro-9 H ,5 H -spiro(furan-2,8-furo[2,3- b ]quinolin-7-ol) (3), colorless crystals, mp 216–218°C (hexane), Rf 0.67. IR (κBr, νmax, cm–1): 3598 (OH), 2981, 2954, 2931, 2862, 1609, 1581, 1541, 1459. 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 1.45 (3H, s, H-12), 1.59 (3H, s, H-13), 1.85 (1H, m, H-6b), 2.05 (1H, d, J = 6.0, 7-OH), 2.24 (1H, m, H-6a), 2.66 (1H, dd, J = 7.1, 2.5, H-9a), 2.77 (3H, m, H-5, 9b), 4.06 (1H, br.d, J = 8.8, H-7), 4.23 (3H, s, 4-OCH3), 5.04 (1H, dd, J = 11.4, 7.1, H-10), 6.90 (1H, d, J = 2.6, H-3), 7.51 (1H, d, J = 2.6, H-2). 13C NMR (100 MHz, CDCl3, δ, ppm): 163.12 (C-1a), 158.28 (C-4), 153.74 (C-8a), 142.55 (C-2), 114.97 (C-4a), 110.00 (C-3a), 104.76 (C-3), 85.08 (C-11), 84.03 (C-8), 72.58 (C-7), 58.57 (C-14), 53.93 (C-10), 42.13 (C-9), 26.81 (C-13), 26.36 (C-6), 25.72 (C-5), 19.26 (C-12).
(7 R ,8 R ,10 R )-10-Bromo-4,8-dimethoxy-11,11-dimethyl-5,6,7,8,10,11-hexahydro-9 H -furo[2,3- b ]pyrano[2,3- h ]quinoline (4), colorless crystals, mp 192–194°C (MeOH), Rf 0.70. IR (κBr, νmax, cm–1): 3126, 2984, 2942, 2903, 1607, 1578, 1459. C18H22BrNO4. ESI-MS m/z 363.9696 [M – OCH3]+ for Br79, 366.1808 [M – OCH3]+ for Br81 (calcd for C17H19BrNO3, 364.2584, 366.2563). Table 2 lists the 1H and 13C NMR spectral data.
Bromination of 1 by NBS in CHCl 3. A solution of 1 (0.3 g, 0.95 mmol) in CHCl3 (5 mL) was stirred, treated with a suspension of NBS (0.2 g, 1.12 mmol) in CHCl3 (2 mL) over 10 min, and stirred for 5 h at 20–25°C. The course of the reaction was monitored by TLC. When the reaction was finished, the solution was made basic with NH4OH to pH 9–10 and extracted with CHCl3. The solvent was distilled off. The crude product containing four compounds (Rf 0.70, 0.67, 0.65, 0.50) was purified over a column of silica gel using benzine–EtOAc (100:1→100:10). One pure product (5) was isolated.
(7 R ,8 R ,10 R )-10-(2′-Bromopropan-2′-yl)-4,8-dimethoxy-5,6,7,8,9,10-hexahydrodifuro[2,3-b:2,3-h]quinoline (5), colorless crystals, mp 135–136°C (hexane), Rf 0.50. C18H22BrNO4. ESI-MS m/z: 364.0749 [M – OCH3]+ for Br79, 366.0756 [M – OCH3]+ for Br81 (calcd for C17H19BrNO3, 364.2584, 366.2563). 1H NMR (600 MHz, CDCl3, δ, ppm, J/Hz): 1.66 (3H, s, H-12), 1.75 (3H, s, H-13), 1.90 (1H, dddd, J = 13.5, 10.4, 9.1, 4.3, H-6a), 2.10 (1H, m, H-6b), 2.15 (1H, dd, J = 13.3, 10.6, H-9a), 2.59 (1H, ddd, J = 16.5, 10.3, 3.9, H-5a), 2.69 (1H, dd, J = 13.3, 5.3, H-9b), 2.91 (1H, ddd, J = 16.5, 6.4, 4.3, H-5b), 3.08 (3H, s, 8-OCH3), 4.14 (1H, dd, J = 10.5, 5.3, H-7), 4.28 (3H, s, 4-OCH3), 4.52 (1H, dd, J = 9.2, 4.9, H-10), 6.96 (1H, d, J = 2.6, H-3), 7.58 (1H, d, J = 2.6, H-2). 13C NMR (150 MHz, CDCl3, δ, ppm): 163.49 (C-1a), 157.82 (C-4), 151.91 (C-8a), 143.00 (C-2), 118.66 (C-4a), 105.31 (C-3a), 104.86 (C-3), 86.46 (C-8), 85.49 (C-10), 80.43 (C-7), 67.50 (C-11), 58.73 (C-14), 51.66 (C-15), 43.25 (C-9), 30.89 (C-13), 30.46 (C-6), 29.47 (C-5), 18.98 (C-12).
Bromination of 1 by NBS in DMF. A cold (0°C) solution of 1 (0.5 g, 1.58 mmol) in DMF (5 mL) was stirred, treated with NBS (0.336 g, 1.89 mmol) over 5 min, and stirred for 4 h at 20–25°C. The course of the reaction was monitored by TLC. When the reaction was finished, the mixture was treated with ice water (5 mL). The resulting precipitate containing three compounds (Rf 0.70, 0.65, 0.50) was separated and purified over a column of silica gel using benzine–EtOAc (100:1→100:10). Only 4 was isolated pure. Crystals, mp 192–194°C, Rf 0.70 (benzine–EtOAc, 2:1).
Preparation of Solution of Reference Standard for HPLC. An accurate weight (1.25 mg) of reference (or test) standard was placed into a 10-mL volumetric flask, treated with EtOH (5 mL), mixed for 10 min in an ultrasound bath, diluted to the mark with the same solvent, stirred, and filtered through a 0.45-μm membrane filter.
Time, min | |||||||
|---|---|---|---|---|---|---|---|
Solvent | |||||||
0.01 | 20.00 | 40.00 | 50.00 | 51.00 | 55.00 | 55.00 | |
H2O | 70 | 70 | 0 | 0 | 70 | 70 | Stop |
MeCN | 30 | 30 | 100 | 100 | 30 | 30 | |
Chromatography conditions: chromatograph with manual injector, high-pressure pump, UV detector, Supelco C18 column (4.6 × 150 mm, 5 μm), room temperature, UV detection at 220 nm. The mobile phase was a gradient of H2O and MeCN at flow rate 1.0 mL/min. The injected sample volume was 20 μL; analysis time, 55 min. The column was equilibrated with H2O to achieve a stable baseline. For this, eluent was passed through the column for 30 min at flow rate 1.0 mL/min.
XSA. Single crystals were grown by slow evaporation of solvent at room temperature. They were transparent and prismatic. The unit cell constants of a crystal were determined and refined on an HPC XtaLAB Synergy diffractometer (Rigaku, Japan) using Cu Kα-radiation (T = 293 K, graphite monochromator). A three-dimensional dataset of reflections was obtained on the same diffractometer. Absorption corrections were applied by a semi-empirical method using the SADABS program [14]. Table 3 presents the main crystallographic parameters and characteristics of the XSA and refinement calculations for crystals of 3–5.
The structure was solved by direct methods using the SHELXS-97 program suite [15] and refined using calculations in the SHELXL-97 program [16]. All nonhydrogen atoms were refined by anisotropic full-matrix least-squares methods (over F2). Positions of H atoms were found geometrically and refined with fixed isotropic shift parameters Uiso = nUeq, where n = 1.5 for methyls and 1.2 for others and Ueq is the equivalent isotropic shift parameter of the corresponding C atoms. H atoms of hydroxyls were found in difference electron-density syntheses and refined isotropically.
References
A. S. Ginsburg, N. Hooper, N. Parrish, K. E. Dooley, S. E. Dorman, J. Booth, M. Diener-West, W. G. Merz, W. R. Bishai, and T. R. Sterling, Clin. Infect. Dis., 37 (11), 1448 (2003).
G. F. Zhang, X. Liu, S. Zhang, B. Pan, and M. L. Liu, Eur. J. Med. Chem., 146, 599 (2018).
A. M. Croft, J. R. Soc. Med., 100 (4), 170 (2007).
S. Vandekerckhove and M. D’hooghe, Bioorg. Med. Chem., 23 (16), 5098 (2015).
S. A. Shiryaev, P. Mesci, A. Pinto, I. Fernandes, N. Sheets, S. Shresta, Ch. Farhy, Ch.-T. Huang, A. Y. Strongin, A. R. Muotri, and A. V. Terskikh, Sci. Rep., 7 (1), Art. No. 15771 (2017); doi: https://doi.org/10.1038/s41598-017-15467-6.
M. Wang, R. Cao, L. Zhang, X. Yang, J. Liu, M. Xu, Z. Shi, Z. Hu, W. Zhong, and G. Xiao, Cell Res., 30 (3), 269 (2020).
S. M. Grundy and N. J. Stone, JAMA Cardiol., 2730287 (2019).
T. V. Lyubchuk and O. V. Gordienko, Khim. Geterotsikl. Soedin., 56, 1 (2020).
J. Buckingham, K. H. Baggaley, A. D. Roberts, and L. F. Szabo (eds.), Dictionary of Alkaloids, 2nd Ed. with CD-ROM, 2010.
L. T. Avazmukhamedov, T. T. Shakirov, and V. A. Tel′nov, Chem. Nat. Compd., 2, 114 (1966).
D. R. Kodirova, Kh. A. Rasulova, K. K. Turgunov, B. Tashkhodzhaev, Kh. M. Bobakulov, and N. D. Abdullaev, Chem. Nat. Compd., 47, 773 (2011).
S. Kotha, N. R. Panguluri, and R. Ali, Eur. J. Org. Chem., 5316 (2017).
F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen, and R. J. Taylor, Chem. Soc., Perkin Trans. 2, S1 (1987).
M. Sheldrick, Program for Empirical Absorption Correction of Area Detector Data, University of Goettingen, Goettingen, 1996.
G. M. Sheldrick, “A Short History of SHELX,” Acta Crystallogr., Sect. A: Found. Adv., 64, 112 (2008).
G. M. Sheldrick, “Crystal Structure Refinement with SHELXL,” Acta Crystallogr., Sect. C: Struct. Chem., 71, 3 (2015).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Prirodnykh Soedinenii, No. 6, November–December, 2022, pp. 927–933.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ubaidullaev, A.U., Vinogradova, V.I., Zhurakulov, S.N. et al. Intramolecular Cyclization During Bromination of the Quinoline Alkaloid Haplophyllidine. Chem Nat Compd 58, 1101–1107 (2022). https://doi.org/10.1007/s10600-022-03877-6
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-022-03877-6