
Abstract
Rumex lunaria is an endemic shrub of the Canary Islands, which is colonizing the Timanfaya National Park (TNP) in Lanzarote. Whether the arrival of R. lunaria to Lanzarote has been natural or by human intervention is still a matter of debate. To address this question, 100 specimens of R. lunaria were collected from the seven main Canary Islands, and genetic analysis of four chloroplast DNA loci were performed, covering a total length of 4809 nucleotide positions. Multiple alignments revealed 49 nucleotide substitutions, which define 30 different haplotypes. Island-specific haplotypes were found in Tenerife, La Gomera, La Palma and Gran Canaria, with the greatest diversity found in the first island. Interestingly, the unique haplotype detected in El Hierro is shared with almost all plants from Lanzarote (95%), including all individuals sampled in the TNP. The most frequent haplotype present in Gran Canaria was detected in only one sample from Lanzarote (5%). These results were corroborated by a robust phylogenetic analysis, which supports the hypothesis of a common origin of R. lunaria populations from El Hierro and the vast majority of those from Lanzarote. In addition, this study rules out the genetic singularity of the R. lunaria specimens that are colonizing the TNP.
Similar content being viewed by others

Introduction
Canary Islands represent an excellent model for phylogeographic analysis because, as in other oceanic archipelagos, the geographical barriers of island-specific populations are clearly defined (Crawford and Stuessy 1997). The Canary Islands have a west–east age gradient, with El Hierro (HI) as the youngest island, followed by La Palma (LP), La Gomera (GO), Tenerife (TF), Gran Canaria (GC), and Fuerteventura (FU) - Lanzarote (LZ) (Anguita and Hernán 2000; Carracedo and Troll 2013) (Fig. 1a). The Canary archipelago belongs to the “Mediterranean basin”, one of the most remarkable biodiversity hotspots worldwide (Myers et al. 2000), and contains more than 570 endemic plant species (about 40% of all plant species described in the islands). Some of these endemic species are distributed throughout the whole archipelago, while others are restricted to certain islands (Francisco-Ortega et al. 2000). The spread of species between islands may be influenced by several factors, such as dispersal vectors, number of introduced individuals, adaptation to a new environment and biotic interactions with native species (Bruno et al. 2005; Jones and Gomulkiewicz 2012). Human activity is transferring island-endemic species from one island to another, causing environmental issues, which are even more serious in fragile island ecosystems. Therefore, to assess whether an island-specific endemism has been naturally dispersed or transported by human action to other islands, represents a challenge for conservation biology in this region. In this scenario, analysis of genetic variability of invasive plant species represents a valuable tool, since results can be used to estimate the human influence over the species distribution, allowing to achieve the best decision for the species management (Kardos 2021).

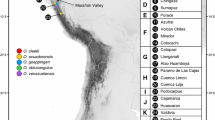
Sampling of Rumex lunaria populations in Canary Islandsand PCR amplification of cpDNA loci. a Geographical location of theCanary archipelago and estimated geological ages for each island, which are highlighted with a color code. >b Number (n) and location of R. lunaria individuals analyzed in this study, including five individuals sampled inside the TNP (LZ). Ma: Millions of years; HI: El Hierro; LP: La Palma; TF: Tenerife; GO: La Gomera; >GC: Gran Canaria; FU: Fuerteventura; LZ: Lanzarote; TNP: Timanfaya National Park.
Rumex lunaria L., is an endemic shrubby plant from Canary Islands. It has been described as a polygamous species with a gynodioecious reproductive system, considered an intermediate evolutionary step between monoicy and dioicy (Löve 1943; Navajas-Pérez et al. 2005). The genus Rumex belongs to Polygonaceae family, in which the species R. lunaria is included in the subgenus Acetosa (section Hastati). This species has been cited among the rare edible plant species of the Canary Islands (Darias et al. 1993), and was found to produce phytochemical compounds of medical interest (Rodríguez de Vera et al. 2004, 2007; Tonny et al. 2017; Mishra et al. 2018; Froldi et al. 2020). R. lunaria has been traditionally used as forage, mainly as a consequence of its good adaptation to arid environments (Ventura et al. 2004; Alvarez et al. 2008; Arévalo et al. 2012).
Currently, R. lunaria is widespread distributed in western islands (HI, LP, GO, and TF), as well as in GC. Oral sources mention the transport of R. lunaria seeds from El Hierro to Lanzarote for crop, in the early 1900s. People emphasize that the common name of “calcosa”, by which this species was known at that time in Lanzarote, is used by people from El Hierro only, while in the rest of the islands it was called, and is still called, “vinagrera” (López 2006).The first known reference to the presence of Rumex lunaria on LZ comes from the Norwegian botanist Per Sunding (Sunding 1970). Shortly after, it was considered a putative invasive species in LZ, because new plant individuals were detected in 1987 near the Timanfaya National Park (TNP) (Wildpret et al. 1995; Betancort 1999). Due to its ability to colonize volcanic territories, especially pyroclastic areas, R. lunaria is nowadays transforming the singular and fragile landscape of the TNP. However, objective evidences of human intervention are needed before considering the expansion of R. lunaria in LZ as an invasive process, before setting up control and management actions. Faced with this situation, we carried out a genetic study aimed to trace the origin of R. lunaria populations in LZ, and to determine if these populations show any genetic peculiarity that is worthy of protection.
Methods
Sample collection and DNA purification
A total of 100 R. lunaria individuals were sampled from the seven main Canary Islands. The area of each island, and the relative abundance of R. lunaria populations (Gobierno de Canarias 2022) were considered to collect a representative number of individuals. The low number of individuals sampled in FU reflects the extremely low abundance of R. Lunaria specimens. In fact, the four samples from FU were collected in urban gardens or in farmland next to houses. One individual from TF belonging to the closely related species R. vesicarius was included as outgroup for the indicated genetic analysis. GPS coordinates and additional information on each sample can be found in Supplementary Information (Table S1).
DNA was extracted from 30 mg of silica gel-dried leaf tissue and homogenized in 2 ml Lysing Matrix-A tubes (M.P. Bio-medicals, USA) by vigorous shaking (5 m/s; 30 s), in a FastPrep-24 system (M.P. Biomedicals, USA). An initial homogenization cycle without adding lysis buffer was included, and genomic DNA was purified from the lysate using the E.Z.N.A DS Plant DNA Kit (Omega Bio-Tek, USA), following manufacturer’s recommendations. DNA was recovered in 50 µl of elution buffer supplied with the kit, and DNA concentration and purity were determined with a DeNovix DS-11 spectrophotometer (Denovix, USA). Each DNA sample was diluted to a final concentration of 10 ng/µl using 10 mM Tris-HCl, pH 8.0, and stored at − 20 °C. For PCR amplification dilutions were prepared at 0.4 ng/µl in the same buffer.
PCR amplification and DNA sequencing
Four chloroplast DNA (cpDNA) loci were selected for this study. Oligonucleotides, used as primers for PCR amplification and/or for amplicon sequencing, were taken from the existing literature or designed specifically for this work (Table 1). The first amplicon includes the spacer region between genes encoding leucine-tRNA for UAA codon (trnL-UAA) and threonine-tRNA for UGU codon (trnT-UGU). The second amplicon includes the intron of lysine-tRNA gene for UUU codon (trnK-UUU), as well as the coding region of the maturase protein (matK). The third amplicon encompasses the DNA fragment placed between genes encoding aspartic-tRNA for GUC codon (trnD-GUC) and threonine-tRNA for GGU codon (trnT-GGU), including three intergenic sequences and two tRNA genes (trnE-UUC and trnY-GUA). The last fragment includes the cpDNA region between cysteine-tRNA gene for GCA codon (trnC-GCA), and the gene encoding B subunit of RNA polymerase (rpoB).
PCR reactions contained 2 ng of template genomic DNA, 1X Phire Hot Start II Reaction Buffer (Thermo Fisher Scientific, USA), 0.2 mM each dNTP (VWR, USA), 0.2 µM of each amplification primer, 0.2 µl Phire Hot Start II DNA polymerase (Thermo Fisher Scientific, USA), and H2O to obtain a final volume of 20 µl. A ProFlex PCR System (Applied Biosystems, USA) was used for incubation of PCR reactions with the following thermal cycling conditions: initial denaturation step at 98 °C for 30 s; 35 amplification cycles with denaturation at 98 °C for 10 s, annealing at the temperature indicated in Table 1 for 10 s; extension at 72 °C during the time indicated in Table 1; and a final extension step at the same temperature and time that previous amplification cycles.
Before DNA sequencing, PCR products were checked by gel electrophoresis in 2% agarose gels prepared in 1X TBE buffer. PCR products were enzymatically purified with the ExoCleanUp reagent (VWR, USA), and sequenced by the Sanger method with the primers indicated in Table 1, making use of an external service (Macrogen Inc). Sequencing electropherograms were manually inspected with the MEGA X software (Kumar et al. 2018), in order to discard low quality reads and to trim primer sequences when present.
DNA sequence alignment and data analysis
Sequences obtained for each cpDNA locus were separately aligned with the ClustalW algorithm (Thompson et al. 1994; Larkin et al. 2007) included in the MEGA X software (Kumar et al. 2018). Alignments were manually inspected to confirm gap positions and, when necessary, were trimmed from both ends to generate a rectangular-matrix dataset. A concatenated alignment was then obtained using DnaSP software 6.0 (Rozas et al. 2017) by putting together sequences from the four cpDNA loci.
The number of polymorphic (segregating) sites (S), number of haplotypes (H), haplotype diversity (Hd), and nucleotide diversity (π) were calculated with DnaSP 6.0 (Rozas et al. 2017). Relationships among the haplotypes were depicted with median-joining (MJ) networks (Bandelt et al. 1999) using PopART v1.7 (Leigh and Bryant 2015), excluding indel mutations and setting Epsilon to zero. Pairwise FST values among islands, as well as AMOVA analysis, were carried out with Arlequin v3.5.2.2 software (Excoffier et al. 2007).
Phylogenetic inferences were carried out at the CIPRES portal (Miller et al. 2010), using the concatenated alignment. F81 was selected as the best substitution model based on the Bayesian Information Criterion (BIC) and Akaike information criteria (AIC), calculated separately for each loci, using both ModelFinder (Kalyaanamoorthy et al. 2017) implemented on IQ-TREE 2 tool (Minh et al. 2020). Maximum Likelihood (ML) phylogeny was generated using RaxML v8.2.12 (Stamatakis 2014), including 1000 bootstrap replicates, and unlinked the four cpDNA loci. Additionally, Bayesian phylogenetic tree was reconstructed using MrBayes v3.2.7 (Ronquist et al. 2012), using two independent runs of four calculation chains, ran simultaneously for 107 generations each, and sampling every 100 generations. The first 2.5 × 104 strings were discarded as burn-in, and the four cpDNA loci were unkinked during calculations. The consensus tree was then visualized and edited with FigTree software v1.4.4 (Rambaut 2018). Orthologous sequences of R. vesicarius were used as outgroups.
Results
Phylogeographic patterns of cpDNA variation in R. lunaria populations
One hundred individuals of R. lunaria, which represent the main populations of this species in the Canary Islands (Gobierno de Canarias 2022), were sampled in the present study. Among them, five individuals from the TNP (LZ) were included (Fig. 1b). Optimized PCR conditions allowed us to obtain the expected amplicon for each cpDNA loci marker (Table 1), without contamination or unspecific amplification products. High-quality sequences for all samples were obtained and deposited at the NCBI GenBank (Table S1).
The alignment of concatenated DNA sequences from the four cpDNA loci, and the 100 R. lunaria individuals, resulted in 4,809 aligned sites. Among them, 50 positions were affected by gaps of different length, which were excluded from subsequent analysis. In the final dataset (4759 sites) a total of 49 positions were variable, being 33 of them parsimony-informative and 16 singleton variants (Table S2). FU and LP showed the highest nucleotide diversity while HI and LZ the lowest, being the overall nucleotide diversity 1.54 × 10−3 ± 7.0 × 10−5 (Table 2). Considering the 49 nucleotide substitutions, a total of 30 cpDNA haplotypes were detected (Table S2), which led to a high haplotype diversity when populations from all seven islands were considered together (0.859 ± 0.029) (Table 2). TF and LP showed the highest haplotype diversity while HI and LZ, again, the lowest. Is noteworthy that both nucleotide and haplotype diversity were zero in HI because only one cpDNA haplotype was detected in this island, whereas the high values for theses genetic diversity parameters encountered in FU are due to the fact that 2 highly divergent haplotypes were detected with only four sampled individuals.
Interestingly, between 4 and 11 island-specific haplotypes were found in LP, GO, TF and GC islands, whereas the unique haplotype present in HI (H1) was shared with LZ and FU. Additionally, in these last two islands was detected the haplotype most frequently found in GC (H25; Fig. 2a; Table S2). Intriguingly, while the presence of a single haplotype is consistent with the relatively young geological origin of HI island, the presence of only 2 haplotypes is discordant with the longer evolutionary time in the cases of LZ and FU (Fig. 1a).

Phylogeographicanalysis of R. lunaria populationsin the Canary Islands. a Median Joining haplotype network for R. lunaria cpDNA. The most probable connection is shown for the 30 different haplotypes detected ( Table S2 ). Circle sizes are proportional to the number of individuals with each haplotype (in brackets). Haplogroups (HG1-5) are highlighted in grey, and the percentage of individuals from each island within each haplogroup is shown in brackets. Colors represent the geological age of each island as indicated in Fig. 1a. The mutational events necessary to generate a haplotype from its predecessor are shown with crossed lines, and the most probable ancestral haplotypes are shown with black circles. b Pairwise FST values (below diagonal) and statistical significance values (above diagonal), obtained for each possible island combination. c Models proposed for AMOVA analysis. Total FRT values are shown at the bottom of each model, and their statistical significance between brackets. Percentage of variation within regions (blue) and among regions (grey) are shown at the left. HI: El Hierro; LP: La Palma; TF: Tenerife; GO: La Gomera; GC: Gran Canaria; FU: Fuerteventura; LZ: Lanzarote; **p < 0.01; *p < 0,05; n.s.: not significant
Evolutionary relationships among haplotypes were inferred by construction of a median-joining haplotype network (Fig. 2a), which again evidences the high haplotype diversity in LP, GO, TF, and GC, and the absence of island-exclusive haplotypes in HI, LZ and FU. The most basal divergence event seems to have occurred between GC and the most western islands (TF, GO, LP and HI), which segregates with at least seven mutational events. The next inter-island divergence process appears to have generated the cpDNA lineages of TF, GO and LP/HI. More recently, the cpDNA lineages of LP and HI separated from a common ancestor. The cpDNA haplotypes of R. Lunaria has experienced an intra-island diversification in the populations of GC, TF, GO and LP. Moreover, in TF and LP 2 clearly separated cpDNA lineages are currently found. This genetic analysis suggests that haplotype H1, present in HI, LZ and FU (100, 95 and 50% of samples, respectively) had a unique origin, as well as haplotype H25 detected in GC, LZ and FU (48, 5 and 50% of samples, respectively). Finally, 5 clear haplogroups were detected (Fig. 2a), being haplogroups HG2, HG3 and HG4 island-exclusive (LP, GO and TF islands, respectively), while certain members of haplogroups HG1 and HG5 are present in several islands, as mentioned before (haplotypes H1 and H25). Therefore, these results reveal a clear genetic structure of R. lunaria populations.
The pairwise FST estimations support the interpretation of the haplotype network given above (Fig. 2b). A clear differentiation was detected between GC and the western islands (FST = 0.76–0.9), but also between GC and LZ (FST = 0.87), while populations from HI and LZ showed no differentiation (FST = 0). To test this hypothesis, AMOVA analysis were conducted under two possible models, excluding FU in both cases due to the low number of individuals sample in this island. The first model groups LZ and GC islands as one region, while the second considered LZ in the same region than western islands (Fig. 2c). In the first model, 75.8% of genetic variation was found within regions (i.e. between islands inside each region), while in the second model the differences inside each region decreases to 38.5%, being the differences between regions the most important source of genetic variation. Thus, AMOVA results confirms the hypothesis that R. lunaria population from LZ is more genetically related to those from the geographically distant western islands than to the population of the neighboring GC.
Phylogenetic inferences
Topologies of the phylogenetic trees obtained by ML and Bayesian approaches were identical (Fig. 3), revealing the existence of six clades statistically well-supported (Bayesian posterior probabilities between 0.97 and 1). The strong differentiation between R. lunaria population from GC (clade-6) and the populations from western islands was corroborated, since HI, LP, GO and TF appear to share a common ancestor. Clade-1 accommodates all individuals from HI, while clades-2 and − 5 gather all the R. lunaria specimens from LP and GO, respectively. Individuals from TF were distributed in the clades-3 and − 4, largely in line with their geographical location (south or north, respectively). These clear phylogeographic signals were notably disrupted by samples from FU and LZ, since individuals from these neighbor islands were assigned to 2 phylogenetically distant clades. In the case of LZ, 20 out of 21 samples (including the 5 from TNP) were grouped in clade-1 together with those from HI, while the sample LZ-3, which was collected in an urban area near the locality of Femés, was assigned to clade-6 along with samples from GC. Similarly, R. lunaria individuals from FU were half distributed between clade-1 and − 6.

Phylogenetic relationships detected among the 100 R. Lunaria individuals included in the study. The consensus tree obtained by Bayesian inference is displayed. Values over the branches are ML bootstrap percentages and Bayesian posterior probabilities, which indicate the statistical robustness of the tree nodes. Numbers inside colored circles identify the clades referred in the text. The percentage of individuals from each island belonging to a particular clade is shown in brackets. The geological age of each island is indicated by the same color code as in Fig. 1a. Individuals from Timanfaya National Park are highlighted in red. R. vesicarius sequences were included as outgroup. Branch lengths reflect genetic distances as indicated by the scale. HI: El Hierro; LP: La Palma; TF: Tenerife; GO: La Gomera; GC: Gran Canaria; FU: Fuerteventura; LZ: Lanzarote
Discussion
R. lunaria is causing important concerns in LZ island due to the recent colonization of the TNP volcanic landscapes (Wildpret et al. 1995; Betancort 1999). Local oral sources indicate that, in the early 1900’s, R. lunaria was deliberately introduced in Máguez, a rural region in the northeast of LZ, to be used as fodder for goat farming (López 2006; Gil et al. 2009). These sources pointed to HI as the origin of seeds or specimens, citing as evidence the fact that the same common name (calcosa) is used in HI and LZ, while this plant species is known under a different popular name (vinagrera) in the other islands. In addition, we were unable to find any descriptive work that mentions the presence of R. lunaria in LZ prior to that date, which is in consonance with this hypothesis. However, alternative explanations should be considered, as the presence in the island of old seeds with high resilience that have germinated recently (Pérez-García et al. 2008), or long-distance transportation of the winged seeds by a natural vector (Nogales et al. 2012). In the present study, a genetic analysis of R. lunaria populations in the Canary Islands has been performed, searching for evidence that either support a natural or human-modified distribution in the Canary Islands, and in order to advise local government on making the right choices for management of this species.
In our study, the highest number of different cpDNA haplotypes was observed in TF, followed by GC, LP and GO. In contrast, in the most western island (HI) we found a single haplotype, which is consistent with being the youngest island of the Canary Archipelago. Paradoxically, despite they are the geologically oldest islands, only 2 different haplotypes were detected in LZ and FU. All haplotypes present in TF, GC, LP and GO, with only one exception in GC, are island specific. However, this peculiarity was not observed in LZ and FU. The biogeographical signal we observed for R. lunaria in most of the Canary Islands fits with the predominant pattern described for biological species in this archipelago, which is the result of a stepwise colonization from the older islands in the east, to the younger islands in the west (Juan et al., 2000). This principle has been designed as the “island progression rule”, in which older land masses donate migrants to younger islands (Whittaker et al. 2017). Indeed, this distribution has been established for many taxa, not only in the Canary Islands, but in different archipelagos (Carvalho et al. 2015). Therefore, species dispersion in volcanic archipelagos usually occurs as a directional process, from older to younger islands, and this distribution has been associated with lower genetic diversity in younger island populations than in older ones. Examples of this distribution pattern can be found in the R. lunaria related species R. bucephalocepharus (Talavera et al., 2013), but also in distant species, such as Olea europaea (García-Verdugo et al. 2009).
Both haplotype network and phylogenetic analysis support a common origin for the vast majority (95%) of the R. lunaria individuals sampled in LZ (including all specimens from the TNP), 50% of the individuals from FU and all individuals from HI. Only one sample from LZ, as well as the other 50% from FU, share evolutionary ancestry with the most common haplotype from GC. These observations indicate that evolutionary time has not been enough for the diversification of original haplotypes in LZ and FU. This inconsistency between geological time and haplotype diversity, suggests that R. lunaria must have colonized LZ and FU at a much more recent time in the evolutionary scale, thus in line with the oral history in the case of LZ.
Anemochory is the unique natural mechanism of seed dispersal described for R. lunaria (Barquin and Wildpret 1975). The lack of haplotypes shared among TF, GC, LP and GO, suggests that natural dispersal of R. lunaria between islands is an extremely rare event, even between islands as close as TF and GO which are separated by about 30 km. Further evidence of its limited dispersal capacity is the genetic differentiation of R. lunaria populations observed in TF. Moreover, it is highly improbable that a natural migration has occurred between the two most distant islands of the archipelago with no signs of having been in intermediate islands, contrary to the typical case that has been previously observed for other Rumex species (Talavera et al. 2013).
Therefore, the migration of R. lunaria between HI, LZ and FU has probably happened through human intervention. In this regard, the fact that the most frequent cpDNA haplotype found in LZ is evolutionarily more related to those found in the western islands than to haplotypes from geographically closer islands, is in opposition to the theory that natural and long-distance dispersal increases genetic dissimilarity (Wright 1943). Considering together the distances observed in the haplotype network, phylogenetic relationships and AMOVA results, the most plausible scenario is the migration of R. lunaria from HI to LZ, and from HI or LZ to FU. Overall, if genetic analyses performed in this study were considered, there is no evidence that supports the existence of a genetic singularity that is worthy of protection in the R. lunaria population from LZ.
Although initially it was not an objective of the study, our data reveal a fingerprint of human intervention in the distribution of R. lunaria in FU. The few plants from this island that could be incorporated to our analyses were collected in highly anthropized areas. Specimens of R. lunaria are much less abundant in FU than in LZ. Since FU has not experienced historical or sub-recent volcanic processes, and R. lunaria is a pioneer colonizer of this type of areas (Wildpret et al. 1995), a rapid expansion of R. lunaria in LZ (worryingly in its national park) might have been favored in comparison to FU. This would be an example of how different ecological factors can drive local adaptation in biological invasions (Rosenthal et al. 2021).
It is well known that human activities exert an important effect over biogeography of island species (Helmus et al. 2014; Graham et al. 2017; Hofman and Rick 2017). In the Canary Islands, translocation of endemic plants with ornamental purposes is a generalized practice. One clear example is the dragon tree (Dracaena spp.), which has been cultivated in public or private gardens, and also along main roads between different urban centers, resulting in a non-random distribution of specific haplotypes (Durán et al. 2020). Recently, it has been reported that traffic along roads near the Timanfaya National Park, is modifying distribution of native and non-native species, including R. lunaria (Bernardos et al. 2023).
Data availability
All data generated and analyzed in this study are included in the published version of the article and its supplementary information files. Sequences have been deposited at NCBI GenBank (Acc. Numbers can be found at Table S2).
References
Alvarez S, Mendez P, Díaz C et al (2008) Forage from the Canary isles (Spain) adapted to arid lands. J Anim Vet Adv 7:359–363
Anguita F, Hernán F (2000) The Canary islands origin: a unifying model. J Volcanol Geotherm Res 103:1–26. https://doi.org/10.1016/S0377-0273(00)00195-5
Arévalo JR, Mora JL, Chinea E (2012) Forage quality of native pastures on Lanzarote island (Canary islands). J Food Agric Environ 10:696–701. https://doi.org/10.1234/4.2012.2760
Bandelt H-J, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036
Barquín Díez E, de la Wildpret W (1975) The dispersal of Canary plants. First data. Vieraea 5:38–60
Bernardos M, Cornejo NS, Torres Hassan AD, Cabrera R, Arévalo JR (2023) Road impact on plant colonization in the arid timanfaya national park. Plants 12:3538. https://doi.org/10.3390/plants12203568
Betancort JAR (1999) Flora y vegetación de la isla de Lanzarote (reserva de la biosfera). PhD Thesis, Universidad de La Laguna
Bruno J, Fridley J, Bromberg K, Bertness M (2005) Insights into biotic interactions from studies of species invasions. In: Sax DF, Stachowicz JJ, Gaines SD (eds) Species invasions: insights into ecology, evolution, and biogeography. Sinauer Associates, Oxford, pp 13–40
Carracedo JC, Troll VR (2013) Teide volcano: Geology and eruptions of a highly differentiated oceanic stratovolcano. Springer, Berlin
Carvalho JC, Cardoso P, Rigal F, Triantis KA, Borges PAV (2015) Modeling directional spatio-temporal processes in island biogeography. Ecol Evol 5:4671–4682. https://doi.org/10.1002/ece3.1632
Crawford DJ, Stuessy TF (1997) Plant speciation on Oceanic islands. In: Iwatsuki K, Raven PH (eds) Evolution and diversification of land plants. Springer, Tokyo, pp 249–267
Darias V, Bravo L, Sánchez-Mateo CC et al (1993) Aperitive and rare edible species of flora of Canary islands. Acta Hortic 333:275–282. https://doi.org/10.17660/ActaHortic.1993.333.33
Demesure B, Sodzi N, Petit RJ (1995) A set of universal primers for amplification of polymorphic non-coding regions of mitochondrial and chloroplast DNA in plants. Mol Ecol 4:129–134. https://doi.org/10.1111/j.1365-294x.1995.tb00201.x
Durán I, Marrero A, Msanda F et al (2020) Iconic, threatened, but largely unknown: biogeography of the macaronesian dragon trees (Dracaena spp.) as inferred from plastid DNA markers. Taxon 69:217–233. https://doi.org/10.1002/tax.12215
Excoffier L, Laval G, Schneider S (2007) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform 1:47–50
Francisco-Ortega J, Santos-Guerra A, Kim S-C, Crawford DJ (2000) Plant genetic diversity in the Canary islands: a conservation perspective. Am J Bot 87:909–919. https://doi.org/10.2307/2656988
Froldi G, González DL, Rosteghin F et al (2020) α-Glucosidase and glycation inhibitory activities of Rumexlunaria leaf extract: a promising plant against hyperglycaemia-related damage. Nat Prod Res 34:3418–3422. https://doi.org/10.1080/14786419.2019.1569655
García-Verdugo C, Fay MF, Granado-Yela C et al (2009) Genetic diversity and differentiation processes in the ploidy series of Oleaeuropaea L.: a multiscale approach from subspecies to insular populations. Mol Ecol 18:454–467. https://doi.org/10.1111/j.1365-294X.2008.04027.x
Gil J, Peña M, Niz R (2009) Usos culturales de las yerbas en Los campos de Lanzarote. Bases orales para la reconstrucción del conocimiento etnobotánico tradicional. Aderlan, Lanzarote
Gobierno de Canarias (2022) Banco de Datos de Biodiversidad de Canarias (BDBC). https://www.biodiversidadcanarias.es/biota/especie/F00295. Accessed Jan, 2023.
Graham NR, Gruner DS, Lim JY, Gillespie RG (2017) Island ecology and evolution: challenges in the anthropocene. Environm Conserv 44:323–335. https://doi.org/10.1017/S0376892917000315
Helmus MR, Mahler DL, Losos JB (2014) Island biogeography of the anthropocene. Nature 513:543–546. https://doi.org/10.1038/nature13739
Hofman CA, Rick TC (2017) Ancient biological invasions and island ecosystems: tracking translocations of wild plants and animals. J Archaeol Res 26:65–115. https://doi.org/10.1007/s10814-017-9105-3
Jones EI, Gomulkiewicz R (2012) Biotic interactions, rapid evolution, and the establishment of introduced species. Am Nat 179:E28–E36. https://doi.org/10.1086/663678
Juan C, Emerson BC, Oromí P, Hewitt GM (2000) Colonization and diversification: towards a phylogeographic synthesis for the Canary islands. Trends Ecol Evol 15:104–109. https://doi.org/10.1016/s0169-5347(99)01776-0
Kalyaanamoorthy S, Minh BQ, Wong TK et al (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589. https://doi.org/10.1038/nmeth.4285
Kardos M (2021) Conservation genetics. Curr Biol 31:R1185–R1190. https://doi.org/10.1016/j.cub.2021.08.047
Kumar S, Stecher G, Li M et al (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Larkin MA, Blackshields G, Brown NP et al (2007) Clustal W and clustal X version 2.0. Bioinformatics 23:2947–2948. https://doi.org/10.1093/bioinformatics/btm404
Leigh JW, Bryant D (2015) POPART: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116. https://doi.org/10.1111/2041-210X.12410
López JP (2006) Los nombres comunes de plantas, animales y hongos de El Hierro. Academia Canaria de la Lengua
Löve Á (1943) Rumex Lunaria L., a gynodioecious tetraploid species. Nature 151:559–560. https://doi.org/10.1038/151559b0
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES science gateway for inference of large phylogenetic trees. In: 2010 gateway computing environments workshop (GCE). IEEE, pp 1–8. https://doi.org/10.1109/GCE.2010.5676129
Minh BQ, Schmidt HA, Chernomor O et al (2020) IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 37:1530–1534. https://doi.org/10.1093/molbev/msaa015
Mishra AP, Sharifi-Rad M, Shariati MA et al (2018) Bioactive compounds and health benefits of edible Rumex species-A review. Cell Mol Biol 64:27–34. https://doi.org/10.14715/cmb/2018.64.8.5
Myers N, Mittermeier RA, Mittermeier CG et al (2000) Biodiversity hotspots for conservation priorities. Nature 403:853–858. https://doi.org/10.1038/35002501
Navajas-Pérez R, de la Herrán R, López González G et al (2005) The evolution of reproductive systems and sex-determining mechanisms within Rumex (Polygonaceae) inferred from nuclear and chloroplastidial sequence data. Mol Biol Evol 22:1929–1939. https://doi.org/10.1093/molbev/msi186
Nogales M, Heleno R, Traveset A, Vargas P (2012) Evidence overlooked mechanisms of long-distance seed dispersal to and between Oceanic islands. New Phytol 194:313–317. https://doi.org/10.1111/j.1469-8137.2011.04051.x
Pérez-García F, González-Benito ME, Gómez-Campo C (2008) Germination of fourteen endemic species from the Iberian Peninsula, Canary and balearic islands after 32–34 years of storage at low temperature and very low water content. Seed Sci Technol 36:407–422. https://doi.org/10.15258/sst.2008.36.2.14
Rambaut A (2018) FigTree: Tree Figure Drawing Tool. Available at: http://tree.bio.ed.ac.uk
Rodríguez de Vera BC, Jiménez Díaz JF, Navarro García E et al (2004) Componentes fitoquímicos de las especies botánicas de Rumex, plantas de uso medicinal. Canarias Médica Y Quirúrgica 2:48–58
Rodríguez de Vera BC, Navarro García E, Jiménez Díaz JF, Navarro García R (2007) Efectos farmacológicos y cicatrizantes de extracto de Rumexlunaria L. Canar Méd Quir 5:33–35
Ronquist F, Teslenko M, Van Der Mark P et al (2012) MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542. https://doi.org/10.1093/sysbio/sys029
Rosenthal WC, McIntyre PB, Lisi PJ et al (2021) Invasion and rapid adaptation of guppies (Poecilia reticulata) across the Hawaiian archipelago. Evol Appl 14:1747–1761. https://doi.org/10.1111/eva.13236
Rozas J, Ferrer-Mata A, Sánchez-Del Barrio JC et al (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302. https://doi.org/10.1093/molbev/msx248
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Sunding P (1970) First records and new combinations in the vascular flora of Lanzarote and Fuerteventura. Nytt Magasin for Botanikk 17:77–80
Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol 17:1105–1109. https://doi.org/10.1007/BF00037152
Talavera M, Navarro-Sampedro L, Ortiz PL, Arista M (2013) Phylogeography and seed dispersal in islands: the case of Rumex bucephalophorus subsp. canariensis (Polygonaceae). Ann Bot 111:249–260. https://doi.org/10.1093/aob/mcs284
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of Progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. https://doi.org/10.1093/nar/22.22.4673
Tonny TS, Sultana S, Siddika F (2017) Study on medicinal uses of Persicaria and Rumex species of polygonaceae family. J Pharmacogn Phytochem 6:587–600. https://doi.org/10.22271/phyto
Ventura MR, Castañón JIR, Pieltain MC, Flores MP (2004) Nutritive value of forage shrubs: Bituminaria bituminosa, Rumex Lunaria, Acacia salicina, Cassia sturtii and Adenocorpus foliosus. Small Rumin Res 52:13–18. https://doi.org/10.1016/S0921-4488(03)00225-6
Whittaker RJ, Fernández-Palacios JM, Matthews TJ, Borregaard MK, Triantis KA (2017) Island biogeography: taking the long view of nature’s laboratories. Science. https://doi.org/10.1126/science.aam8326
Wildpret W, Beltrán Tejera E, González-Mancebo JM, Centellas Bodas A (1995) Pelargonium capitatum Y Rumex Lunaria, Dos Plantas invasoras en El Parque Nacional De Timanfaya (Lanzarote, Islas Canarias). Consideraciones ecológicas y fitosociológicas. Anuario Del Instituto De Estudios Canarios 39:9–16
Wright S (1943) Isolation by distance. Genetics 28:114–138. https://doi.org/10.1093/genetics/28.2.114
Acknowledgements
Special thanks to Consejería de Transición Ecológica, Lucha contra el Cambio Climático y Planificación Territorial (Gobierno de Canarias) for funding this study. Additionally, many thanks to Pascual Gil Muñoz, director and chief conservator of the Timanfaya National Park, for providing access to R. lunaria samples at the Timanfaya National Park. The authors are grateful to Gabinete de Estudios Ambientales (GEA Tenerife) for the help with the field activities and first-hand knowledge. Finally, RC and MBH thank to Andreea Cosoveanu, researcher at the Universidad de La Laguna, for her valuable contributions to improve the manuscript, and Guacimara Espinel, for the help with the field activities and first-hand knowledge.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This work has been supported by a research contract between Parque Nacional de Timanfaya and Fundación General de la Universidad de La Laguna (FGULL), with the Grant Reference Number A21100106.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Sample collection was carried out by RC and MBH. Laboratory procedures and data collection were performed by MGC and JPP. Data analysis was carried out by MGC, MHF and JPP. The first draft of the manuscript was written by MGC. All authors read and edited the initial version of the manuscript and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
González Carracedo, M.A., Hernández Ferrer, M., Cabrera, R. et al. Phylogeographic analysis points toward invasion of the Timanfaya National Park (Lanzarote; Canary Islands) by a translocated native plant (Rumex lunaria). Conserv Genet 25, 621–630 (2024). https://doi.org/10.1007/s10592-023-01592-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-023-01592-5