
Abstract
Knowledge of genetic diversity and structure is essential for developing conservation strategies for endangered species. Blue whales were hunted to near extinction in the mid-twentieth century. Not-withstanding almost 380,000 animals killed globally, much remains unknown about their population structure and migration patterns. Herein, we use whole genome sequencing to elucidate the poorly understood population genetics of North Atlantic (NA) blue whales. We generated a de novo genome assembly for a NA blue whale to analyze 19 other whole genomic sequences and 31 complete mitochondrial genomes. Present-day and historical samples (earliest from 1876) from the Atlantic and Antarctic Oceans were included to understand the impact of whaling on the genetic diversity of this species. We found low but statistically significant population structuring and high genetic diversity. Demographic modeling using fastsimcoal2 rejected an absence of gene flow between eastern and western NA blue whales and suggested an asymmetric west to east gene flow. Introgression estimated using D-statistics between blue and fin whales (Balaenoptera physalus), was observed in all present-day samples. This gene flow was found to be unidirectional from fin whales to blue whales and accounts for ~ 3.5% of the NA blue whale’s genome. Our sequencing and population structure analyses provide a genomic baseline to inform ongoing conservation strategies for this iconic species.
Similar content being viewed by others
Introduction
The blue whale (Balaenoptera musculus) is the largest animal that has ever lived, with individuals reaching 30 m in length and weighing up to 150 tonnes. They are found in oceans across the globe but were historically most abundant in the Southern Ocean (Sears and Perrin 2018). These whales were too fast and powerful for early whalers to catch using traditional methods and it was not until technological advancements in the 1860s that commercial exploitation of the large “rorqual” whales became possible. Whaling for blue whales began in the Northeast Atlantic (NEA) and subsequently spread rapidly to all other oceans on an industrial scale (Thomas et al. 2016).
The blue whales’ large size made them a lucrative target as they provided a more profitable yield per unit of hunting effort than other whales. By the second half of the 20th century, commercial whaling had brought this species to the brink of extinction. A worldwide pause on the hunting of blue whales was put into effect by the International Whaling Commission (IWC) in 1966. Although a complete moratorium on all commercial whaling was implemented in 1985, some illegal hunting persisted. It has been estimated that between 1900 and the late 1970s, over 379,000 blue whales were harvested globally (Rocha et al. 2014).
Blue whales are currently classified as Endangered by the International Union for Conservation of Nature (Cooke 2019) and in North America they are listed under the Canadian Species at Risk Act (SARA) and the US Endangered Species Act (ESA). While no longer hunted, they continue to be threatened by ship strikes, fishing gear entanglement, marine noise, pollutants, and climate change (COSEWIC 2012). Though blue whale numbers are thought to be slowly increasing globally, this growth has been described as spotty and equivocal (Branch et al. 2004; Thomas et al. 2016; Cooke 2019). Today there are an estimated 5,000–15,000 blue whales, only 3–11% of the 1926 global species estimate (Cooke 2019). Despite the modest growth for the species, several aggravating factors continue to challenge recovery of some blue whale populations. For NA blue whale stocks these challenges include reduced calving and recruitment rates, persistent ecological disruptions (Beauchamp et al. 2009; Koubrak et al. 2022), as well as lagging governance on recovery efforts (Koubrak et al. 2022). In fact, there is no evidence of recovery from the impact of whaling on this species in the NA (Ramp et al. 2006). Understanding population structure is of critical value for the conservation of blue whales, considering the regional trends.
Knowledge of the distribution, genetic structure, and population ecology of blue whales is essential for the protection of this vulnerable species. Blue whales are currently divided into at least four distinct subspecies (Balaenoptera musculus brevicauda, B. m. intermedius, B. m. indica and B. m. musculus). Population structure, genetic diversity and connectivity in the Pacific and Southern oceans have been studied extensively using genetic markers (Attard et al. 2010, 2016, 2018; Barlow et al. 2018; Costa-Urrutia et al. 2013; Leduc et al. 2017; Sremba et al. 2012; Torres-Florez et al. 2014). No similar genetic data has been generated thus far for blue whales in the NA (Balaenoptera m. musculus). However, numerous studies have looked at contemporary blue whale movements, distribution and population size within the NA using sightings, photoidentification, vocalizations, satellite tracking and isotope analyses (Davis et al. 2020; Delarue et al. 2022; Lesage et al. 2017; Pike et al. 2019; Silva et al. 2013, 2019; Storrie et al. 2018; Trueman et al. 2019). While data from tagged animals helps provide information on contemporaneous movements (Lesage et al. 2017), a large gap remains in our understanding of blue whale migrations, population ecology and interconnectivity, particularly across the NA.
One outstanding conservation question is whether blue whales in the eastern and western portions of the NA comprise a single population, as homogeneous versus discrete populations may merit different recovery strategies. Early whalers thought that there were two distinct stocks of blue whales in the NA (Lesage et al. 2017). Long-term photo-identification data coupled with satellite telemetry data suggests a low degree of admixture between eastern and western NA blue whales (Ramp and Sears 2013; Sears and Calambokidis 2002; Sears and Perrin 2018; Silva et al. 2013). However, blue whale songs recorded from the Northeast Atlantic (NEA) and Northwest Atlantic (NWA) are similar, yet distinct from blue whales in other oceans, suggesting that population structure, if it exists, is likely minor and recently evolved (Clark 1994). In the NWA, blue whales presently only number about 250 adults (COSEWIC 2012), whereas in the NEA there are ~ 3,000 individuals (Pike et al. 2019). Currently, the National Oceanic and Atmospheric Administration’s (NOAA) revised Recovery Plan for the Blue Whale (2020) (https://repository.library.noaa.gov/view/noaa/27399) makes clear that the question of whether eastern and western whales in the NA constitute one population or two is unresolved. However, in that Recovery Plan, all blue whales in the NA are considered a single management unit based on the International Whaling Commission’s (IWC) blue whale stock definition and the similarities in blue whale song found across the NA, pending further evidence.
An additional conservation issue for blue whales is introgression and hybridization (Allendorf et al. 2001; Rhymer and Simberloff 1996). Purported blue whale/fin whale hybrids have been reported by whalers from the coast of Lapland and Alaska for more than a century (Cocks 1887; Doroshenko 1970). In the last few decades, hybrids caught near Iceland and Spain have been verified using molecular evidence (Árnason et al. 1991; Bérubé and Aguilar 1998; Spilliaert et al. 1991). It is not clear whether these hybridization events represent regular and sustained gene flow between the two species (Westbury et al. 2019; but see Árnason et al. 2018). However, if gene flow is occurring from the far more abundant fin whales (population size > 80,000, Pampoulie et al. 2020) to blue whales, this presents a threat to NA blue whale population(s) due to loss of genetic integrity. This is of particular concern if opportunities for contact between competing populations and species occur (Rhymer and Simberloff 1996).
Herein, we constructed the first de novo assembly of a NWA blue whale genome and used it as a reference for assembling the nuclear and mitochondrial genomes of a collection of present-day and historical blue whales from across the NA, as well as an Antarctic blue whale and NA fin whales. Our objectives are to use these data to advance the current understanding of blue whale population structure across the NA and provide information on population size changes and migrations through time. Additionally, these genomes can provide information relevant to conservation, issues such as quantifying the potential genetic threat posed to blue whales through introgression with fin whales, seen in both contemporary and historical samples, and provided genetic evidence to determine the number of distinct populations in the NA.
Methods
Samples
We sampled 26 blue whales from across the NA, one from the Southern Atlantic and one from the Antarctic. Of the NA samples, four were from strandings along the east coast of Canada between 2014 and 2019 and seven were sampled using biopsy darts near Svalbard, Norway between 2014 and 2017. The remaining samples were from historical museum skeletons in Canada, the USA, Iceland and Norway, with collection dates ranging from 1876 to 1975 (Table 1). Seven present-day fin whales from Norwegian waters were also sampled using biopsy darts. In addition, archived genomic sequences from a NA blue whale (SRR5665644) and two sei whales (SRR5665645 and SRR5665646) were downloaded from GenBank.
De novo genome assembly
The DNA used for the de novo blue whale genome assembly came from muscle taken from a female blue whale (NW-M6, ROMM125066; Table 1) salvaged from Newfoundland in 2014 by the Royal Ontario Museum (ROM), under permit (SARA permit ref: NLSAR-003-14). The Illumina and PacBio reads from the NA blue whale were assembled using the hybrid assembler MASURCA v 3.2.8 (Zimin, et al. 2017). Assembly completeness was assessed using BUSCO (Simão et al. 2015) and genome size was also estimated independently from Illumina short reads using PREQC (Simpson 2014). Sex chromosome linked contigs were detected by aligning to the cow X chromosome (CM008197.2). The genome was screened for repeat regions using REPEATMASKER v4.0.7 and REPEATMODELER (see Supporting Information for more assembly details).
Transcriptome assembly
RNA for the transcriptome assembly was extracted from skin/blubber tissue, collected from a whale sampled in the Svalbard Archipelago (79°N), Norway (Fig. 1A). Transcripts were assembled from paired-end RNAseq data using TRINITY (Grabherr et al. 2011) and TOPHAT (Trapnell et al. 2009) assemblers and redundancies in the predicted transcripts were removed using CD-HIT (Fu et al. 2012). The masked NA blue whale genome was annotated with the predicted transcripts using the MAKER2 pipeline (Holt and Yandell 2011) (for details see Supporting Information). The annotation quality was assessed using BUSCO and INTERPROSCAN v5.23-62.0 (Mulder and Apweiler 2007).
Mitochondrial genome assembly
Mitochondrial genomes for present-day and historical whales were assembled from the trimmed and merged Illumina paired-end reads by mapping them to a reference mitochondrial genome (NC_00160136) using BOWTIE2 2.3.3.1 (Langmead and Salzberg 2012). This analysis had a larger sample size including several individuals from which mitochondrial DNA was successfully recovered but had low concentration for whole genome analyses (Table 1).
Whole genome sequencing
The DNA extraction from present-day samples (after 2010) was done from frozen tissue and short reads were sequenced as detailed in Supporting Information. For the historical samples (1876–1975) the DNA was extracted from the bones at the ROM’s ancient DNA facility following the methodology detailed in the Supporting Information. The paired-end reads were trimmed and merged using SeqPrep v 1.1 (https://github.com/jstjohn/SeqPrep) with default settings and quality score cut-off for mismatches in overlap > 20 (-q 20). The first and last two bases of the merged reads were trimmed, removing potentially damaged sites in ancient DNA (Dabney et al. 2013).
The trimmed sequences from the present-day and historic samples were mapped to the masked de novo assembled NA blue whale genome autosome contigs (Table 1) using BWA 0.7.17 (Li and Durbin 2009) and genome-wide variants were detected for analyses. The SNPs for all the whole genome analyses for present day samples were filtered for quality score and mapping quality > 30, depth 10X to 130X, and MAF > 0.1 (see Supporting Information for further details).
Population structure analysis
Genomic population structure analyses were conducted on blue whales from present-day NA, historic NWA and Antarctica (Table 1). The six historic NWA blue whales (NWa-R4, NWa3, NWa4, NWa5, NWa6, and NWa-CM1) dated from the early days of whaling of this species through to post-whaling. Population structure was investigated using principal component analysis (PCA), Hudson’s Fst (Bhatia et al. 2013), and Jost’s D (Jost 2008). The blue whales were also checked for kinship using PLINK (--genome) (Purcell et al. 2007). The phylogenetic relationships between individuals were explored using RAxML-NG (Kozlov et al. 2019) and SVDQuartets (Chifman and Kubatko 2014). Additionally, phylogenetic relationships among blue whale samples were examined using maternally inherited mitochondrial genomes from a larger set of blue whales from NA, Antarctic and the Pacific.
Principal component analysis
PCA was performed using LASER v 2.04 (Wang et al. 2015) which uses projection Procrustes analysis for samples with low depth of coverage. The samples with low depth (< 10X) of coverage were placed in the context of a reference PCA space constructed using genotypes of reference individuals with higher coverage depth. The first PCA analysis included present-day and historical blue whales from NA and Antarctica (Table 1) along with seven NA fin whales, and two sei whales.
A second PCA analysis visualized the genetic relationship among just the blue whales, which included present-day and historical blue whale samples (NWa-R4, NWa3 and NWa4). The SNPs for the PCA were also filtered for being genotyped in > 50% samples and r2 > 0.8 within a 1 kb window to filter for linkage disequilibrium. The first and second PCA analyses included 4,136,458 and 2,517,406 SNPs, respectively. The reference PCA space for the first PCA analysis was constructed using present-day NA blue whales (> 15X) and two present-day fin whales (~ 15X) and two sei whales (~ 10X). The reference space for the second PCA analysis was computed from the present-day NA blue whales (> 15X).
Genomic phylogenetic analysis
Phylogenetic relationships were estimated for the blue whales from present-day, historical (NWa3, NWa4 and NWa-R4) and Antarctica using RAxML-NG and SVDQuartets with the NA fin whale as the root for an alignment of 176,382 SNPs. SNPs were filtered as in PCA analysis. For RAxML-NG, the substitution model GTR + G was used with the Lewis ascertainment bias correction and bootstrapped 500 times. The best maximum likelihood tree was visualized using FigTree v.1.4.4 (http://tree.bio.ed.ac.uk/software/figtree). SVDQuartets was run in PAUP* v.4a168 (Swofford 2002) sampling all quartets and inferred with 1,000 nonparametric bootstraps. These analyses were repeated for a reduced subset of the blue whales, excluding the lower coverage assemblies below 6X, which included the NA historical samples, to assess their impact on the accuracy and resolution of the reconstructed trees.
Whole genome fst and population statistics estimates
Hudson’s Fst was estimated for the present-day (> 15X coverage) NEA and NWA blue whale samples. Hudson’s Fst estimate is not biased by differing sample size between populations (Bhatia et al. 2013). Fst was estimated for 906,598 genome-wide biallelic SNPs also filtered for being present in > 50% samples and 1 kb apart. Fst was estimated in R using a custom script and the 95% confidence interval was estimated using 1,000 bootstrap replicates. The Jost’s D, also a measure for genetic differentiation among populations, was estimated using the basic.stats function of hierfstat in R.
Mitochondrial genomes: genetic diversity and matrilineal population structure
The 27 mitochondrial genomes sequenced here, along with the four that are publicly available (CM018075, MF409242, X72204 and assembled from SRR5665644) include 28 unique mitochondrial haplotypes. The unique haplotypes were aligned with a sei whale (NC_006929) using MAFFT v7 (Katoh and Standley 2013), the sister group to blue whales (Árnason et al. 2018). Gene regions (including codon positions) were identified and PartitionFinder v.2.1.1 (Guindon et al. 2010; Lanfear et al. 2017) used to determine the best partitioning scheme and substitution models with the Bayesian Information Criterion. MrBayes v3.2.7 (Ronquist et al. 2012) was used to reconstruct the phylogenetic relationships between the blue whales with the sei whale as the outgroup. From the resultant phylogenetic tree, a blue whale sequence from each of the three clades descended from the two deepest nodes in the tree were selected and combined with the mitochondrial genome sequences of the sei, fin (NC_001321), humpback (Megaptera novaeangliae, NC_006927) and minke (Balaenoptera acutorostrata, NC_005271) whales. This second mitochondrial dataset was also partitioned using PartitionFinder2 but excluded the control region due to alignment ambiguity. A time-scaled phylogeny was estimated using the species divergence dates from Árnason et al. (2018) and the program BEAST v2.6.6 (Bouckaert et al. 2019) utilizing a relaxed lognormal clock with a Calibrated Yule model tree prior. The ages determined for the two earliest blue whale divergences then served as calibration points for another BEAST analysis using a lognormal relaxed clock and a coalescent constant population tree prior for the 28 blue whale mitochondrial haplotypes producing a time-scaled blue whale phylogeny. All Bayesian analyses were run for 100 million generations multiple times and Tracer v1.7.1 (Rambaut et al. 2018) was used to check convergence and that equivalent samples sizes (ESS) were greater than 200.
A haplotype median-joining network was created using the program POPART (Leigh and Bryant 2015) with the default settings for the 31 mitochondrial genomes. Descriptive statistics including the number of segregating sites, haplotypes, haplotype diversity, nucleotide diversity and Tajima’s D were calculated for the mitochondrial genomes using DnaSP v. 6.12 (Rozas et al. 2017).
Population history and gene flow analysis with demographic model fitting
Three demographic models were tested using whole-genome data and FASTSIMCOAL v 2.7 (Excoffier et al. 2021) with each model estimating the time in generations of the split between the NEA and NWA blue whale populations. Separate effective population size parameters were estimated for both populations as well as for the common ancestor. The first model (“no gene flow”) had no gene flow between the populations and had a total of four parameters. The second model (“symmetric gene flow”) estimated a fifth parameter, the per capita rate of gene flow. The third model (“asymmetric gene flow”) had six parameters and allowed asymmetric rates of gene flow. Autosomal biallelic SNPs from the noncoding region (112,902) of the genome were utilized to generate the site frequency spectrum (SFS) for FASTSIMCOAL2. The SNPs were also filtered for any missing data, 10 kb apart and 10 kb from coding regions. ARLECORE from the ARLEQUIN software suite (Excoffier and Lischer 2010) was used to generate a folded minor allele SFS together with 200 bootstrapped SFS (for the non-parametric bootstrap analysis below). The number of invariant sites was estimated from our SNP dataset and manually inserted into SFS. We used a mutation rate of 1.39 × 10− 8 substitutions per nucleotide per generation (Árnason et al. 2018) to calibrate the models. For each model we ran 100 independent runs of FASTSIMCOAL2, each with different initial starting parameters. Starting parameters were drawn from log-uniform distributions ranging from 100 to 1,500,000 generations for the three effective population size parameters and 1 × 10− 8 to 0.01 for per capita migration rate parameters. The time of divergence was drawn from a uniform distribution ranging from 100 to 1,500,000 generations. All parameters were set to be unconstrained allowing FASTSIMCOAL2 to explore parameter values outside the initial parameter ranges. Each run proceeded through 40 ECM cycles of hill climbing, with 200,000 coalescent simulations used to estimate the likelihood corresponding to each set of parameter value combinations tested. We re-estimated the likelihood of the best supported set of parameter values for each run using 10 million coalescent simulations. Simulation estimates were then used to choose the best set of parameter values and to determine the maximum likelihood value for each model. Support for the three models was assessed using Akaike weights. For the best supported model, 200 non-parametric bootstraps were used to estimate the 95% confidence intervals for model parameters. Each bootstrap had 100 independent runs, each starting from the initial maximum-likelihood parameter values. Fewer ECM cycles (20 rather than 40) were thus required as initial parameters were already in the right region of parameter space. Due to the computationally intensive nature of bootstrapping, fewer simulations (100,000 per set of parameter combinations) were used. Values of effective population size reported by fastsimcoal2 are for haploid individuals. We divided these values by two to transform them to effective population size for diploid individuals.
Heterozygosity
Genome-wide heterozygosity for present-day NA blue whale samples (> 15X) and the Antarctic historical sample was estimated in ANGSD (Korneliussen et al. 2014) based on the site frequency spectrum (SFS) using the infinite sites model. The SNPs for the present-day sample were filtered as in PCA and the SNPs from the historical sample was also filtered for deaminated cytosine residues (-noTrans). The inbreeding coefficients were estimated using PLINK (--het) in present-day NA blue whale samples (> 15X). The population-level mean diversity (total population heterozygosity) in the NA blue whale population (HT) was estimated using the basic.stats function of hierfstat in R.
Introgression
Gene flow between present-day and historical blue to fin whale, humpback and sei whales was investigated using D-statistics (Green et al. 2010) and the direction of gene flow and percent of introgression was estimated by Dfoil (Pease and Hahn 2014) with minke whale as the outgroup (SRS439234). D-statistics was estimated using the four-taxon phylogeny of (((Antarctic Blue, NA Blue), Fin), Minke). The ABBA/BABA tests, where “A” is the ancestral allele and “B” is the derived allele, were performed in ANGSD (Korneliussen et al. 2014) with option -doAbbababa with mapping quality > 30 and quality score > 20 test. This test avoids bias due to differences in depth of coverage by sampling sites at each position of the genome (Korneliussen et al. 2014). The historical samples were also filtered for deaminated cytosine (-rmTrans). The jackknife procedure was used for standard error estimations. Similarly, to study blue-humpback (SRS4201634) whale and blue-sei (SRR5665645) whale introgression, analyses were conducted for (((Antarctic Blue, NA Blue), Humpback), Minke) and (((Antarctic Blue, NA Blue), Sei), Minke).
Dfoil was estimated using the phylogenetic relationship of (((Sei, present-day NA Blue), (Fin, Humpback)), Minke). Sites for these analyses were also filtered for genotype in > 0.50 samples. The genetic identity of the fin whale used in the introgression analysis was verified by testing against another known fin and clustering in the first PCA analysis (Fig. 1B).
Results
Genome assembly
The NA blue whale genome of 2.49 Gbp comprising 11,400 contigs was assembled (N50 of 1.46 Mb and L50 of 449). The genome contained 94.8% complete and 2.6% fragmented mammalian single copy genes. The total size of the genome estimated from Illumina reads was ~ 2.7Gb indicating 92.6% had been assembled with 255 contigs mapped to the sex chromosome and one contig aligned to the mitochondrial DNA (Árnason and Gullberg 1993). Repetitive elements comprised 46.2% of the genome. Predicted transcriptome included 30,867 genes that represented 82.7% of complete mammalian single copy genes and 65.7% of which had known Pfam domains.
Population structure
Principal component analysis
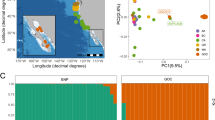
The first PCA grouped blue, fin and sei whales into species specific clusters (Fig. 1C), except for the NWa-CM1 which was intermediate between the blue and fin whale clusters, suggesting a blue-fin hybrid. The D-statistics tests also revealed that two other historical blues, NWa5 and NWa6, had significant fin whale introgression. All three whales were removed from further population analyses using nuclear sequences.
The kinship analysis did not identify any closely related blue whales and the closest blue whale were NW9/M6 (pi-hat = 0.11). Most NA samples clustered together in the second PCA, (Fig. 1D) on PC1 except for NW9/M6 and NE-Ar/NE3. On PC2, NE4/NE73 was an outlier to the main cluster of NA samples. The PC1 and PC2 accounted for 11.42% and 10.80% of total variability, respectively.
Phylogenetics analysis
The genetic relationships estimated using RAxML and SVDQuartest while not fully resolved, did indicate that all the NA blue whales were allied and distinct from the Antarctic blue whale (Fig. 1B and Fig. S2 for higher coverage whales only). The resultant tree placed the NEA whales nested within NWA whales. The bootstrap support values were strong for the basal nodes in the tree for several NWA branches in both analyses but were weaker for the shallower nodes including all the NEA whales and two NWA whales. For the analyses using a subset of the whales with the lower coverage historical NA whales removed, both resultant tree topologies again placed the NEA whales nested within the NWA whales but not agree on which NWA whale sequence was the more divergent (Fig. S2).
Fst and population differentiation statistics
Genetic divergence estimated using Hudson’s Fst suggested moderate (0.21) genome-wide differentiation between NEA and NWA blue whales, with a 95% confidence interval of 0.21 to 0.22. Population allelic differentiation (Jost’s D) between NEA and NWA blue whales was 0.0077.

Population structure of present-day and historical blue whales from North Atlantic based on whole-genome sequencing data. A. Map of blue whale sampling locations. (B) Phylogenetic trees estimated using RAxML-NG and SVDQuartets with only bootstrap support values above 50% presented, (RAxML above and SVDQuartets below). (C) Principal component analysis (PCA) of seven Northwest Atlantic (NWA) blue whales, eight Northeast Atlantic (NEA) blue whales, one Antarctic blue whale, seven fin whales from the North Atlantic and two sei whales using LASER. The circled sample represents the blue-fin hybrid historical sample (NWA-CM1). The variability accounted by PC1(43.89%) and PC2(7.63%) (D) PCA of seven NWA blue whales and eight NEA blue whales. The variability accounted by PC1(11.42%) and PC2(10.80%) (see Supplement Figure S1 for PCA with NA blue whales and the Antarctic whale)
Mitochondrial genetic diversity and matrilineal population structure
We examined the complete mitochondrial genome sequences from 31 blue whales (Table 2). Genetic relationships among the blue whales reconstructed using these sequences revealed a tree distinct from that estimated using the nuclear sequences in that the Antarctic whale nested in amongst the NA blue whales (Fig. 2A). The tree has potentially five haplogroups, with the majority of the NEA and NWA whales sampled being in haplogroup A. Sister to haplogroup A were two poorly resolved clades consisting of an Antarctic, a Pacific, and a Southern Atlantic whale and one NWA whale (haplogroups C & B). Haplogroups D and E represented much more divergent mitochondrial lineages sharing successive common ancestors with haplogroups A, B and C dated at 167,000 and 201,000 years ago, respectively. The mitochondrial genome median joining network revealed consistent results to the mitochondrial phylogenetic tree with widely separated clusters among the blue whales sampled (Fig. 2B). Due to variation in mitochondrial genome coverage between modern and historical samples, sequence coverage was mapped onto the tree and network to verify clustering was not due to low coverage (see Figure S3).

(a) Time-calibrated phylogeny of 25 unique North Atlantic, one South Atlantic, one Antarctic and one Pacific blue whale mitochondrial genome haplotypes. Scale bar represents 95% highest posterior density (HPD) for divergence estimates. Numbers at nodes denote posterior probabilities (only support for the basic nodes is reported). Lineages are coloured according to the region where the whale was sampled. (b) Mitochondrial haplotype median-joining network for blue whales with ticks along the branch lengths denoting nucleotide differences between haplotypes. The two longest branches have 30 and 32 substitutions respectively
Population history and gene flow analysis with demographic model fitting
Among the three gene flow models (‘no gene flow’, ‘symmetric gene flow’ and ‘asymmetric gene flow’) compared between blue whales from NEA and NWA, the model with no gene flow received almost no support (Table 3) and was rejected in favour of models with gene flow. A model with symmetric gene flow received an Akaike Weight of only 0.10 while a model with asymmetric gene flow received the greatest support with an Akaike Weight of 0.90. This best fit model suggested that the populations diverged 499 generations ago and that subsequent gene flow has been primarily unidirectional with substantial gene flow from the northwest to the northeast (Fig. 3) but very little gene flow in the reverse direction. The per capita rates of gene flow reported in Fig. 3 translate to 21.8 (18.6 to 25.5, 95% CI) individuals migrating from the west to the east each generation and 0.039 (0.00046 to 1.49; 95% CI) from the east to the west. Effective population sizes between the northwest and northeast were comparable at around 5,000 individuals while ancestral values were about four times these values.

The best fit demographic model for the blue whale samples from the Northwest Atlantic (NW) and Northeast Atlantic (NE). Effective population sizes (Ne), divergence in generations ago (Tdiv), and rates of gene flow per 1000 individuals per generation are shown. 95% confidence of model parameters are shown in brackets based on 200 bootstrap replicates. Time flows from the top of the diagram to the bottom
Heterozygosity and population history of NA blue whales
Genome-wide heterozygosity for the present-day NA and historical Antarctic blue whales was ~ 0.0025 (0.00250–0.00254) and 0.0053, respectively. The inbreeding coefficients for the present-day whales were low (< 0.02) The total population heterozygosity (HT) estimated for the NA population was 0.3952.
Introgression
All present-day NA blue whale samples showed significant fin whale introgression (Z-score > 3) (Table 4). Four of six historical blue whale samples also had evidence of fin whale introgression. The sample NWa-CM1 (from 1974) had a D-statistic value of 0.94 indicating a recent hybridization between fin and blue whale. Likewise, NWa5 and NWa6 (from early-1900s) also had high D-statistics values indicating they were hybrids. None of these hybrids carried fin whale mitochondria, indicating their mothers were blue. None of the blue whales sampled showed significant introgression with humpback whales. This is also the case with blue and sei whales, except for NE-Ar (D-statistic = 0.013; Z-score = 5.71). Dfoil statistics revealed unidirectional gene flow, from fin to blue whale (Table S1) and that fin whale sequences constitutes ~ 3.5% of the genomes of these NA blue whales.
Discussion
Our results provide the first insights into the population structure, and demographic history of blue whales from the NA, and document levels of introgression with fin whales.
North Atlantic blue whale population structure
There has been uncertainty whether NA blue whales consist of one or more populations. Photoidentification studies from both sides of the NA over the last several decades support more than one population, with only a single occurrence of an overlap between eastern and western whales (Sears and Calambokitis 2002; Ramp and Sears 2013; Sears and Perrin 2018; see also Silva et al. 2013). However, similarities in whale song across the NA, relative to blue whale calls in other ocean basins, suggests a single population or at most two with minimal differentiation (Clark 1994). Our findings based on the nuclear sequences examined here showed a moderate and statistically significant Fst between eastern and western blue whales but a low Jost’s D value indicating low allele differentiation between the two. Fst is influenced by within-subpopulation heterogeneity whereas the Jost’s D relies on numbers of alleles within and among subpopulations (Alcala and Rosenberg 2019). The phylogenetic reconstructions using nuclear SNPs for both modern (high coverage) NA whales alone and for modern and historical samples combined do not support the premise of two reciprocally monophyletic clusters separated by geography, rather the NEA blue whales sampled here were nested within the NWA whales. This was supported by PCA analyses which showed overlap between the sampled whales and modelling using FASTSIMCOAL2, which rejected a scenario of no gene flow between whales from both sides of the NA.
The model favoured in FASTSIMCOAL2 has asymmetric gene flow with many more whales migrating from west to east each generation than in the opposite direction. The underlying causation of this asymmetric gene flow is intriguing in an environment with no known barriers and suggests the possibility of an underlying driver. One possibility is oceanic circulation. The North Atlantic Current is a strong ocean current flowing west to east. Whales conceivably use ocean currents to conserve energy during long migrations (Lesage et al. 2017), though they are unlikely limited by them. However oceanic circulation also contributes to clinal plankton biogeography in the NA, including a west-east zonal gradient, with the NWA less biodiverse, notably for calanoid species (Kléparski et al. 2021), important in the feeding ecology of NA krill (Schmidt 2010). Blue whales of the northeast Pacific are known to track spring/summer plankton blooms over time, as they forage northward along the coast (Abrahms et al. 2019). It is possible that blue whales in the NA similarly track resource blooms spatially over time, contributing to patterns of gene flow over generations. Recent warming in the Barrents Sea has led to an increase in krill biomass, secondary to changes in the Atlantic advection (Eriksen et al. 2017) which could have implications for population recovery. Additional research is required to explore this further.
The phylogeny reconstructed from the maternally inherited mitochondrial genomes agreed with the tree estimated from the nuclear sequences in that there was no clear distinction between eastern and western whales. Where it differed, is that the Antarctic blue whale sample was not basal to the NA whales but nested in amongst them. This may either represent unsorted ancestral polymorphism present in the population or possibly that females may have a dispersal behaviour that periodically involves long-range matrilineal dispersals, including individuals moving into the NA from other blue whale populations.
Heterozygosity
We observed relatively high genome-wide heterozygosity in present-day NA blue whales as was reported by Árnason et al. (2018) and in the one historical Antarctic whale sample. Population-level heterozygosity was also high, which was consistent with reports of high heterozygosity observed in Chilean and other Antarctic whales (Torres-Florez et al. 2014). While blue whales were hunted to the brink of extinction, they currently exhibit high genetic variability. However, these animals are long lived and with the bottleneck event having happened so recently, a reduction in heterozygosity would likely not be observable for several generations. High variability along with low inbreeding has been associated with healthier reproductive outcomes and greater adaptability (but see Teixeira and Huber 2021) which could assist in the species’ recovery, as long as low population numbers do not persist for an extended number of generations.
Blue whale / fin whale hybridization
Blue and fin whales are the two largest animals on earth and hybrids between these species have been observed on multiple occasions (Árnason et al. 1991; Pampoulie et al. 2020). This is remarkable as they are not sibling species and diverged about ~ 8.35 million years ago (Árnason et al. 2018). We demonstrated gene flow between NA blue and fin whales in all our present-day and four (of six) historical samples, including a first-generation hybrid. The recent gene flow that we recorded between fin whales and NA blue whales was not detected in the previous study by Westbury et al. (2019). Our findings are partially in agreement with Árnason et al. (2018), but we only detected gene flow between blue and fin whales not between humpbacks and blues. Also, we found the contribution to the blue whale genome of fin whale DNA was larger than reported in Árnason et al. (2018) at 3.5% and the gene flow was unidirectional, from fin whale to blue whale. Important differences in our analyses are a larger sample size, using a de novo assembled blue whale genome as the reference and comparing between different blue whale populations, namely the Antarctic and NA, rather than between different whale species as was done in these other studies. Only two of our whale samples from the early 20th century lacked any sign of introgression. Our results indicate recent introgression observed in NA blue whales took place after the separation between NA and Antarctic subspecies. Hybridization between species can occur naturally or result from breeding disruptions promoted by anthropogenic activities and effects. While the abundance of fin whales was negatively impacted by whaling (Wolf et al. 2022), their numbers still greatly exceed that of blue whales globally. While male fin whales are smaller than their blue whale counterparts, they have comparable cruising and sprinting speeds (Sears and Perrin 2018; Aguilar & García-Vernet 2018), which could make male fin whales competitive during courtship chases where blue and fin whales are sympatric.
Hybridization can in some cases lead to the extinction of distinct species through introgressive swamping of the genome (Rhymer and Simberloff 1996). Our relatively small sample sizes from early whaling and post-whaling blue whales preclude drawing conclusions on whether hybridization rates are changing. However, the high frequency of occurrence indicates a need to examine greater numbers of both recent and historical samples to ascertain if there is a threat of genetic swamping of NA blue whales by fin whales.
Conclusions
Here we present the first collection of present day and historical blue whale genomes, both nuclear and mitochondrial, from samples across the NA. Results of our study indicate that the genetic structuring of blue whales in the NA, although statistically significant, is more nuanced than simple models of one or two populations. Fst analyses shows statistically significant genetic structuring between eastern and western blue whales, however asymmetric gene flow from west to east is occurring across the NA and phylogenetic reconstruction place the eastern whales nested within the western whales. Future conservation actions and management policies should be informed by this complexity. The NEA appears to be an area for potential blue whale population recovery given the dramatic increases in krill stocks in the Barents Sea. Hybridization of blue whales with fin whales appears frequent in the NA and future rates of hybridization should be monitored. While genetic variability in NA blue whales is high, this is likely due to the recency of the bottleneck and the species long generation time, since it takes time to see the impact of population reduction on the genomes. If blue whale populations do not increase from their current low levels, reduction in heterozygosity probably will occur over time. Further studies with larger sample sizes for present-day and historical blue whales from around the world should be conducted to better define populations and subspecies and examine gene flow to help plan global conservation efforts for this endangered species.
Data Availability
Genome sequences and assembly have been submitted to Gen-Bank (BioProject ID PRJNA704862 and, assembled mitochondrial sequences ON257884-ON257910) and a git repository has been created (http://github.com/SushmaJossey/BlueWhale) which includes the genome and transcriptome assembly workflow code, as well as, the repeat masking for the North Atlantic blue whale.
Change history
17 February 2024
A Correction to this paper has been published: https://doi.org/10.1007/s10592-024-01607-9
References
Abrahms B, Hazen EL, Aikens EO, Savoca MS, Goldbogen JA, Bograd SJ, Jacox MG, Irvine LM, Palacios DM, Mate BR (2019) Memory and resource tracking drive blue whale migrations. Proc Nat Acad Sci 116(12):5582–5587
Aguilar A, Garcia-Vernet R (2018) Fin whale. In: Würsig B, Thewissen JGM (eds) Encyclopedia of marine mammals. Academic Press, pp 368–371
Alcala N, Rosenberg NA (2019) Jost’s D, and FST are similarly constrained by allele frequencies: a mathematical, simulation, and empirical study. Mol Ecol 28:1624–1636
Allendorf FW, Leary RF, Spruell P, Wenburg JK (2001) The problems with hybrids: setting conservation guidelines. Trends Ecol Evol 16:613–622
ambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst Biol 67:901–904
Árnason Ú, Gullberg A (1993) Comparison between the complete mtDNA sequences of the blue and the fin whale, two species that can hybridize in nature. J Mol Evol 37:312–322
Árnason U, Spilliaert R, Palsdottir Á, Árnason A (1991) Molecular identification of hybrids between the two largest whale species, the blue whale (Balaenoptera musculus) and the fin whale (B. Physalus). Hereditas 115:183–189
Árnason Ú, Lammers F, Kumar V, Nilsson MA, Janke A (2018) Whole-genome sequencing of the blue whale and other rorquals finds signatures for introgressive gene flow. Sci Adv 4:eaap9873
Attard CRM, Beheregaray LB, Jenner C, Gill P, Jenner M, Morrice M, Bannister J, LeDuc R, Möller L (2010) Genetic diversity and structure of blue whales (Balaenoptera Musclus) in Australian feeding aggregations. Conserv Genet 11:2437–2441
Attard CR, Beheregaray LB, Möller LM (2016) Towards population-level conservation in the critically endangered Antarctic blue whale: the number and distribution of their populations. Sci Rep 6:22291
Attard CR, Beheregaray LB, Sandoval-Castillo J, Jenner KCS, Gill PC, Jenner M-NM, Morrice MG, Möller LM (2018) From conservation genetics to conservation genomics: a genome-wide assessment of blue whales (Balaenoptera musculus) in Australian feeding aggregations. R Soc Open Sci 5:170925
Barlow DR, Torres LG, Hodge KB, Steel D, Baker CS, Chandler TE, …, Klinck H (2018) Documentation of a New Zealand blue whale population based on multiple lines of evidence. Endanger Species Res 36:27–40
Beauchamp J, Bouchard H, de Margerie P, Otis N, Savaria J-Y (2009) Recovery strategy for the Blue Whale (Balaenoptera musculus), Northwest Atlantic population, in Canada [FINAL]. Species at Risk Act Recovery Strategy Series. Fisheries and Oceans Canada, Ottawa, ON. 62 pp
Bérubé M, Aguilar A (1998) A new hybrid between a blue whale, Balaenoptera musculus, and a fin whale, B. Physalus: frequency and implications of hybridization. Mar Mamm Sci 14:82–98
Bhatia G, Patterson N, Sankararaman S, Price AL (2013) Estimating and interpreting FST: the impact of rare variants. Genome Res 23:1514–1521
Bouckaert R, Vaughan TG, Barido-Sottani J, Duchêne S, Fourment M, Gavryushkina A, …, Drummond AJ (2019) BEAST 2.5: an advanced software platform for bayesian evolutionary analysis. PLoS Comput Biol 15:e1006650
Branch TA, Matsuoka K, Miyashita T (2004) Evidence for increases in Antarctic blue whales based on bayesian modelling. Mar Mamm Sci 20:726–754
Chifman J, Kubatko L (2014) Quartet inference from SNP data under the coalescent model. Bioinformatics 30:3317–3324
Clark CW (1994) Blue deep voices: insights from the Navy’s Whales ‘93 program. Whalewatcher 28:6–11
Cocks AH (1887) The fin whale fishery of 1886 on the Lapland coast. Zoologist 11:207–222
Cooke JG (2019) Balaenoptera musculus (errata version published in 2019)’.The IUCN Red List of Threatened Species 2018: e.T2477A156923585, https://doi.org/10.2305/IUCN.UK.2018-2.RLTS.T2477A156923585.en
COSEWIC (2012) COSEWIC status appraisal summary on the Blue Whale Balaenoptera musculus, Atlantic population, in Canada. Committee on the Status of Endangered Wildlife in Canada. Ottawa.xii pp. www.registrelep-sararegistry.gc.ca/default
Costa-Urrutia P, Sanvito S, Victoria-Cota N, Enríquez-Paredes L, Gendron D (2013) Fine-scale population structure of blue whale wintering aggregations in the Gulf of California. PLoS ONE 8:e58315
Dabney J, Meyer M, Pääbo S (2013) Ancient DNA damage. Cold Spring Harb. Perspect Biol 5:a012567
Davis GE, Baumgartner MF, Corkeron PJ, Bell J, Berchok C, Bonnell JM, …, Van Parijs SM (2020) Exploring movement patterns and changing distributions of baleen whales in the western North Atlantic using a decade of passive acoustic data. Glob Chang Biol 26:4812–4840
Delarue JJY, Moors-Murphy H, Kowarski KA, Davis GE, Urazghildiiev IR, Martin SB (2022) Acoustic occurrence of baleen whales, particularly blue, fin, and humpback whales, off eastern Canada, 2015–2017. Endanger Species Res 47:265–289
Doroshenko VN (1970) A whale with features of the fin and the blue whale. Izvestia TINRO 70:225–257
Eriksen E, Skjoldal HR, Gjøsæter H, Primicerio R (2017) Spatial and temporal changes in the Barents Sea pelagic compartment during the recent warming. Prog Oceanogr 151:206–226
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Excoffier L, Marchi N, Marques DA, Matthey-Doret R, Gouy A, Sousa VC (2021) fastsimcoal2: demographic inference under complex evolutionary scenarios. Bioinformatics 37:4882–4885
Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, …, Regev A (2011) Trinity: reconstructing a full-length transcriptome without a genome from RNA–Seq data. Nat Biotechnol 29:644–652
Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M …, Pääbo S (2010) A draft sequence of the neanderthal genome. Science 328:710–722
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Holt C, Yandell M (2011) MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12:491
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Kléparski L, Beaugrand G, Edwards M (2021) Plankton biogeography in the North Atlantic Ocean and its adjacent seas: species assemblages and environmental signatures. Ecol Evol 11(10):5135–5149
Korneliussen TS, Albrechtsen A, Nielsen R (2014) ANGSD: analysis of next generation sequencing data. BMC Bioinformatics 15:356
Koubrak O, VanderSwaag DL, Worm B (2022) Endangered Blue Whale Survival in the North Atlantic: lagging scientific and governance responses, charting future courses. The Internl JMarine & Coastal Law 37:89–136
Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A (2019) RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35:4453–4455
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B (2017) PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol 34:772–773
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357
Leduc RG, Archer FI, Lang AR, Martien KK, Hancock-Hanser B, Torres-Florez JP, …, Taylor BL (2017) Genetic variation in blue whales in the eastern pacific: implication for taxonomy and use of common wintering grounds. Mol Ecol 26:740–751
Leigh JW, Bryant D (2015) POPART: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116
Lesage V, Gavrilchuk K, Andrews RD, Sears R (2017) Foraging areas, migratory movements and winter destinations of blue whales from the western North Atlantic. Endanger Species Res 34:27–43
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760
Mulder N, Apweiler R (2007) InterPro and InterProScan: tools for protein sequence classification and comparison. Methods Mol Biol 396:59–70
Pampoulie C, Gíslason D, Ólafsdóttir G, Chosson V, Halldórsson SD, Marianiet S, …, Víkingsson GA (2020) Evidence of unidirectional hybridization and second-generation adult hybrid between the two largest animals on Earth, the fin and blue whales. Evol Appl 14:314–321
Pease JB, Hahn MW (2014) Detection and polarization of introgression in a five-taxon phylogeny. Syst Biol 64:651–662
Pike DG, Gunnlaugsson T, Mikkelsen B, Halldórsson SD, Víkingsson GA (2019) Estimates of the abundance of cetaceans in the Central North Atlantic based on the NASS Icelandic and Faroese Shipboard surveys conducted in 2015. NAMMCO Sci Publications 11. https://doi.org/10.7557/3.4941
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, …, Sham PC (2007) PLINK: a toolset for whole-genome association and population-based linkage analysis. Am J Human Genet 81:559–575
Ramp C, Sears R (2013) Distribution, densities, and annual occurrence of individual blue whales (Balaenoptera musculus) in the Gulf of St. Lawrence, Canada from 1980–2008. DFO Can Sci Advis Sec Res Doc. 2012/157. vii + 37 p
Ramp C, Bérubé M, Hagen W, Sears R (2006) Survival of adult blue whales Balaenoptera musculus in the Gulf of St. Lawrence, Canada. Mar Ecol Prog Ser 319:287–295
Rhymer JM, Simberloff D (1996) Extinction by hybridization and introgression. Ann Rev Ecol& System 27:83–109
Rocha RC, Clapham PJ, Ivashchenko YV (2014) Emptying the oceans: a summary of industrial whaling catches in the 20th century. Mar Fish Rev 76:37–48
Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S, …, Huelsenbeck JP (2012) MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542
Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sanchez-Gracia A (2017) DnaSP v6: DNA sequence polymorphism analysis of large datasets. Mol Biol Evol 34:3299–3302
Schmidt K (2010) Chapter five - food and feeding in Northern Krill (Meganyctiphanes norvegica Sars). In: Tarling GA (ed) Advances in Marine Biology, vol 57. Academic Press, pp 127–171
Sears R, Calambokidis J (2002) Status of the blue whale, Balaenoptera musculus. Canada. Report to the Committee on the Status of Endangered Wildlife in Canada (COSEWIC). Canadian Wildlife Service, Ottawa
Sears R, Perrin WF (2018) Blue Whale: Balaenoptera musculus. In: Würsig B, Thewissen JGM (eds) Encyclopedia of marine mammals. Academic Press, pp 110–114
Silva MA, Prieto R, Jonsen I, Baumgartner MF, Santos RS (2013) North Atlantic blue and fin whales suspend their spring migration to forage in middle latitudes: building up energy reserves for the journey? PLoS ONE 8:e76507
Silva MA, Borrell A, Prieto R, Gauffier P, Bérubé M, Palsbøl PJ, Colaço A (2019) Stable isotopes reveal winter feeding in different habitats in blue, fin and sei whales migrating through the Azores. R Soc Open Sci 6:181800
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212
Simpson JT (2014) Exploring genome characteristics and sequence quality without a reference. Bioinformatics 30:1228–1235
Spilliaert R, Vikingsson G, Arnason U, Palsdottir A, Sigurjonsson J, Arnason A (1991) Species hybridization between a female blue whale (Balaenoptera musculus) and a male fin whale (B. Physalus): molecular and morphological documentation. J Hered 82:269–274
Sremba AL, Hancock-Hanser B, Branch TA, LeDuc RL, Baker CS (2012) Circumpolar diversity and geographic differentiation of mtDNA in the critically endangered Antarctic blue whale (Balaenoptera musculus intermedia). PLoS ONE 7:e32579
Storrie L, Lydersen C, Andersen M, Wynn RB, Kovacs KM (2018) Determining the species assemblage and habitat use of cetaceans in the Svalbard Archipelago, based on recorded observations from 2002–2014. Polar Res 37:1463065
Swofford D (2002) PAUP*: phylogenetic analysis using parsimony (* and other methods). Version 4. Sinauer Associates Sunderland, MA
Teixeira JC, Huber CD (2021) The inflated significance of Neutral genetic diversity in conservation genetics. Proc Natl Acad Sci, 118(10), e2015096118
Thomas PO, Reeves RR, Brownell RL (2016) Status of the world’s baleen whales. Mar Mamm Sci 32:682–734
Torres-Florez JP, Hucke-Gaete R, Rosenbaum H, Figueroa CC (2014) High genetic diversity in a small population: the case of Chilean blue whales. Ecol Evol 4:1398–1412
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111
Trueman CN, Jackson AL, Chadwick KS, Coombs EJ, Feyrer LJ, Magozzi S, …, Cooper N (2019) Combining simulation modeling and stable isotope analyses to reconstruct the last known movements of one of Nature’s giants. PeerJ 7:e7912
Wang C, Zhan X, Liang L, Abecasis GR, Lin X (2015) Improved ancestry estimation for both genotyping and sequencing data using projection Procrustes analysis and genotype imputation. Am J Hum Genet 96:926–937
Westbury MV, Petersen B, Lorenzen ED (2019) Genomic analyses reveal an absence of contemporary introgressive admixture between fin whales and blue whales, despite known hybrids. PLoS ONE, e0222004
Wolf M, De Jong M, Halldórsson SD, Árnason Ú, Janke A (2022) Genomic impact of whaling in North Atlantic fin whales. Mol Biol Evol 39(5):msac094
Zimin AV, Puiu D, Luo MC, Zhu T, Koren S, Marçais G, Yorke JA, Dvořák J, Salzberg SL (2017) Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome research. 1;27(5):787–792
Acknowledgements
Thanks to Lois A. Harwood and Jack Lawson (Department of Fisheries and Oceans) for facilitating our recovery of the two Newfoundland blue whales and many thanks to the people of Trout River, Rocky Harbour and Woody Point who assisted in this effort. The authors also thank Tonya Wimmer (Marine Animal Rescue Society) for tissues and Kamal Khidas (CMN), Michael McGowen, Darrin Lunde and John Ososky (NMNH) for allowing us to sample their museum collections, and Veronica Chevasco in assisting in ancient DNA recovery. Additionally, we thank Hanne Garmel (WM), Haakon Livland (SIM), and Hanneke Meijer (UMBNH) for help in providing NEA historical samples, David William Peter Dolan (Department of Informatics, University of Bergen) for bioinformatic/infrastructure support, Edgardo M. Ortiz (Technical University of Munich) for data conversion and Jeffrey Macdonald (The Hospital for Sick Children) for data submission.
Funding
The genome sequencing and bioinformatic analysis was funded by the Alan and Patricia Koval Foundation, The Centre for Applied Genomics at The Hospital for Sick Children, CanSeq150 by Canada’s Genomics Enterprise (a Canada Foundation of Innovation Major Science Initiative award), and a computing grant ‘Resource for Research Groups’ from Compute Canada (2018–2020) and the Norwegian Research Council ICE-whales programme (no. 244488/E10). The sequencing activities at the Genomics Core Facility of the University of Bergen was supported by the Fram Center Hazardous Substances Flagship Program (grant no. 602018), the Research Council of Norway (grant no. 245979/F50) and the Trond Mohn Foundation (grant no. BFS2016-genom). Bioinformatic analysis at the University of Bergen was provided through ELIXIR Norway, Research Council of Norway grant 270068. The funding for salary of S.J. was provided by a Rebanks post-doctoral fellowship and the Alan and Patricia Koval Foundation.
Author information
Authors and Affiliations
Contributions
This study was conceived and supervised by M.D.E. Sample collection was undertaken by M.D.E., B.K.L., J.M., A.G., R.L., K.M.K., C.L. and H.R. Genome and transcriptome assembly, annotation, introgression, heterozygosity and PCA and introgression analyses was done by S.J., phylogenetic analysis, and mitochondrial analysis by O.H. and Fastsimcoal2 and Fst analysis by J.T.W. Guidance on genome assembly and analysis was provided by L.L and S.W.S. The manuscript was drafted by S.J., O.H., J.M. and J.T.W. All the authors provided invaluable editorial inputs on multiple versions of the manuscript, including the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Consent for publication
All authors have consented to this paper’s publication.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised due to the BioProject ID in the Data Availability section was incorrectly given as “PRJNA70482” and should have read “PRJNA704862”.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jossey, S., Haddrath, O., Loureiro, L. et al. Population structure and history of North Atlantic Blue whales (Balaenoptera musculus musculus) inferred from whole genome sequence analysis. Conserv Genet 25, 357–371 (2024). https://doi.org/10.1007/s10592-023-01584-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-023-01584-5