
Abstract
Assessing patterns of functional connectivity among amphibian demes is crucial to unravel their population dynamics and prevent their isolation and eventual extinction. Integrative studies based on direct (capture-mark-recapture) and indirect (genetic) estimates of dispersal provide robust, biologically realistic inferences on population structure and connectivity, with applications for conservation efforts. We focused on two pond-breeding amphibians with contrasting life-history traits: the short-lived, semi-arboreal Hyla molleri and the long-lived, fossorial Pelobates cultripes. We PIT-tagged 2150 individuals of both species in two ponds (Laguna and Gravera, separated by 700 m) and monitored them from 2009 to 2021 to document the frequency and spatial extent of dispersal events. In addition, we genotyped individuals from these and two additional breeding populations at a maximum distance of 5 km with 15–16 microsatellites to characterize fine-scale patterns of genetic structure. We detected dispersal events connecting Laguna and Gravera in both species, albeit at low frequencies (4.8% and 7.7% of recaptured individuals of H. molleri and P. cultripes, respectively). However, both species were capable of covering long distances, with individual accumulated displacements up to 3.5 km (Hyla) and 1.8 km (Pelobates). Breeding populations > 2 km apart were genetically differentiated, indicating lower connectivity at this spatial scale. Estimates of pairwise migration rates differed between species and were asymmetrical, with different ponds representing “source” populations contributing more migrants to other populations in each species. We discuss the role of differences in life history traits and ecological preferences in shaping population dynamics in the two species and highlight management implications of our results.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Habitat loss and fragmentation are among the main causes of amphibian population declines in temperate regions (Wake and Koo 2018). These processes reduce the exchange of individuals and genes among demes (Johansson et al. 2007; Dixo et al. 2009), resulting in genetic drift and inbreeding, which increase the local extinction risk of small, isolated populations (Cushman 2006; Dixo et al. 2009; Reed and Frankham 2003). Due to their dependence on both terrestrial and aquatic habitats and their limited dispersal capacity (Bowne and Bowers 2004; Smith and Green 2005), amphibian population dynamics are strongly affected by habitat fragmentation and loss. However, the negative consequences of fragmentation can be counterbalanced by adopting measures promoting connectivity among demes (Johansson et al. 2007; Petranka et al. 2004). Habitat (= structural) connectivity facilitates individual dispersal, allowing gene flow among populations (functional connectivity) and sustaining levels of genetic diversity at the landscape scale. Thus, the long-term resilience of amphibian communities in fragmented landscapes relies on maintaining functional connectivity, and studies assessing fine-scale patterns of gene flow in amphibian populations provide key information to implement efficient conservation measures and prevent their genetic depauperation and eventual loss (Baguette et al. 2013; Luque et al. 2012; Peterman et al. 2015).
Characterization of functional connectivity requires information about the dispersal behaviour of the target species. Many amphibian species persist as spatially structured populations at the local scale (Capellà-Marzo et al. 2020; Fernández de Larrea et al. 2021; Smith and Green 2005), and rely on sporadic individual displacements to maintain gene flow among demes (local, randomly mating populations) (Marsh and Trenham 2001; Semlitsch 2008; Peterman et al. 2015). Effective migration can balance out local extinction processes through recolonization (Cayuela et al. 2020), making dispersal capacity an important factor in the dynamics of spatially structured populations (Gilpin 2012; Hanski and Gilpin 1991). In addition, since dispersal is a density-dependent process (Cayuela et al. 2020), accounting for population sizes is also important to characterize the role of different breeding nuclei for overall connectivity in fragmented habitats, because larger populations will produce a higher net number of migrants to neighbouring populations (Fahrig 1998, 2001).
The use of molecular markers to document regional patterns of genetic structure has produced valuable insights on the role of different landscape features in shaping patterns of connectivity (Dixo et al. 2009; Gutiérrez-Rodríguez et al. 2017a; Newman and Squire 2001; Wang 2009). However, assessing the frequency and spatial extent of individual dispersal events that result in actual gene flow requires the combination of direct observations of displacements, for instance based on individual-based capture-mark-recapture (CMR) data (Reyes-Moya et al. 2022), and indirect estimates of migration rates (based on molecular markers) at small spatial scales (Luque et al. 2012). This integrative approach provides robust, biologically realistic inferences about the spatial ecology of amphibians, informing on their population structure, dynamics and patterns of connectivity, with potential applications for the design of management actions (Capellà-Marzo et al. 2020; Fernández de Larrea et al. 2021; Gutiérrez-Rodríguez et al. 2017b; Olarte et al. 2020; Sánchez-Montes et al. 2017b, 2018).
Amphibian communities comprise assemblages of species with contrasting life-history traits, including differences in dispersal behaviour or ecological requirements that may be associated with differential responses to landscape features (Sánchez-Montes et al. 2018; Steele et al. 2009). Thus, comparative studies of fine-scale patterns of population structure in syntopic species are key to identify the main drivers of long-term community dynamics and provide valuable information for their conservation (Coster et al. 2015; Goldberg and Waits 2010; Gutiérrez-Rodríguez et al. 2017a; Igawa et al. 2013; Nowakowski et al. 2015; Richardson 2012; Steele et al. 2009; Whiteley et al. 2014). These comparative studies are a cornerstone of multispecies conservation planning (Nicholson and Possingham 2006; Schwenk and Donovan 2011).
Pond-breeding amphibian communities in the Mediterranean region include many endemic species that are threatened by habitat loss and fragmentation (Çiçek and Cumhuriyet 2017; Cuttelod et al. 2009). In the Iberian Peninsula, many species are in slow but continuous decline, and their long-term resilience relies on their ability to maintain functional connectivity in fragmented habitats. Unfortunately, little is known about the spatial ecology of most Iberian amphibians, which compromises attempts to preserve viable, well-connected networks of breeding populations. Here we present the results of an integrative, comparative assessment of fine-scale patterns of connectivity in two syntopic pond-breeding amphibians with contrasting life-history traits: the long-lived, fossorial spadefoot toad Pelobates cultripes (Cuvier, 1829) and the short-lived, semi-arboreal Iberian treefrog Hyla molleri Bedriaga, 1889. Although both species are syntopic, they differ in their use of terrestrial and aquatic habitats, dispersal capacity and demographic traits (Table 1). Our expectation is that higher dispersal capacity in H. molleri will translate into weaker genetic structure and increased functional connectivity. To test this hypothesis, we combined field-based observations of dispersal of marked individuals and microsatellite genotype data to unravel fine-scale patterns of population structure and functional connectivity in four breeding sites in Sierra de Guadarrama (Madrid, Spain). We discuss the implications of our results for the management of amphibian communities in fragmented habitats.
Materials and methods
Study area and target species
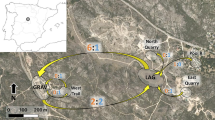
The study area is centred around two amphibian breeding ponds located in the southern slopes of Sierra de Guadarrama (Madrid, central Spain), separated by 700 m of terrestrial habitat, without artificial barriers potentially affecting amphibian dispersal (Fig. 1). The largest pond is Laguna de Valdemanco (hereafter Laguna, 40.8531°N, 3.6449°W, 1054 m a.s.l.), a temporary aquatic system with a maximum surface area of 12,800 m2 and 1 m of maximum depth. The surrounding vegetation is composed mainly of gum rockrose (Cistus ladanifer) and sparse willow trees (Salix sp.). This pond receives most of its water from rainfall (December-March) and dries up by summer (June–July). The second pond is Gravera (40.8526°N, 3.6538°W, 1006 m a.s.l.), a naturalized gravel pit occupying an area of 2100 m2 with a maximum depth of 3 m. The aquatic vegetation is sparser, including river water-crowfoot (Ranunculus fluitans), and the hydroperiod is longer, usually from November to August. The amphibian community inhabiting these ponds is composed of seven species (Pleurodeles waltl, Triturus marmoratus, Pelophylax perezi, Hyla molleri, Pelobates cultripes, Epidalea calamita and Alytes cisternasii), which have been the subject of an integrative monitoring program based on CMR since 2009 (see Capellà-Marzo et al. 2020; Fernández de Larrea et al. 2021; Gutiérrez-Rodríguez et al. 2017b; Sánchez-Montes et al. 2017b; Sánchez-Montes & Martínez-Solano 2011). Two additional amphibian breeding sites were sampled in the vicinities of this core area. Towards the East, Roblellano (40.8563°N, 3.6314°W, 1041 m a.s.l.) is a set of shallow, temporary aquatic systems close to the town of La Cabrera. The predominant flora includes oaks (Quercus pyrenaica and Q. ilex) and an undergrowth of C. ladanifer. At the West, Laguna de Navalengua, in the municipality of Bustarviejo (40.8528°N, 3.6823°W, 1080 m a.s.l.), is a temporary pond formed by the course of an intermittent stream (Arroyo de Navalengua). The maximum distance among the four sampled sites is 5 km, and the study area is crossed by two main roads (M-610 separating Roblellano from Laguna and Gravera, and M-633 separating Bustarviejo from the rest of sampling sites), and a temporary stream (Arroyo Albalá, Fig. 1).

Map of the study area in Sierra de Guadarrama (Madrid, Spain) showing the four sampled populations (B: Bustarviejo, G: Gravera, L: Laguna de Valdemanco and R: Roblellano, photographs of each pond below), roads M-610 and M-633, and temporary stream (Arroyo Albalá)
We focused on two syntopic anuran species: H. molleri and P. cultripes, which inhabit the four sampling locations. Although both species share the same breeding habitats, they have different life-history traits (Table 1), which may be potentially associated with different population dynamics. Some of these traits, such as longevity and age at sexual maturity, together with differences in dispersal behaviour, are expected to influence functional connectivity as inferred from local patterns of population structure (Sánchez-Montes et al. 2018).
Monitoring program and sampling methods
The monitoring program in Laguna and Gravera included year-round nocturnal surveys in the 2009–2021 period, especially focusing on the amphibian breeding season (January–June). The number of sampling sessions varied across years according to the breeding activity of the study species, which is strongly dependent on environmental conditions, ranging between 7 and 25 sessions per year for each species, with an average of 4 h per sampling session. Survey effort mainly concentrated in both breeding ponds, but we also covered trails connecting them across the terrestrial habitat matrix in order to detect dispersing individuals. In all surveys, we captured all detected adult individuals by hand, recorded their sex based on external morphological characters and measured their snout-to-vent length (SVL) and body mass. Individuals captured for the first time were marked with passive integrated transponders (PIT tags) providing unique alphanumeric codes. As a source of DNA for molecular analyses, we collected toe clips from the fourth toe of the right hind limb of adult individuals; these tissue samples were preserved in absolute ethanol until DNA extraction. We recorded the geographic coordinates of all observed individuals using a GARMIN GPS (see Fernández de Larrea et al. 2021). After processing, all individuals were released at the place of capture.
We also collected tissue samples from metamorphic individuals of H. molleri (2018) and tadpoles of P. cultripes (2019) in Laguna, which represented samples of the offspring cohort of the corresponding year. At the two breeding sites in the surroundings of the core area, we surveyed the ponds during the breeding seasons of 2019 (Roblellano) and 2020 (Bustarviejo), in search for adult individuals of H. molleri and P. cultripes. We collected a toe clip from each captured individual and stored the tissue samples in absolute ethanol until DNA extraction. All sampling procedures were approved by the Ethics Committee of Consejo Superior de Investigaciones Científicas, Spain (ref.: 710/2018) and Comunidad de Madrid (ref.: PROEX 040/19).
Genotyping by microsatellites
We genotyped 208 adult individuals of H. molleri and 225 of P. cultripes, captured in the four sampling locations during the 2018–2020 breeding seasons. This sample included 98% of all the adults of H. molleri captured in the core area in the 2018 and 2019 breeding seasons (114 in Laguna and 31 in Gravera) and 68.5% of the adult individuals of P. cultripes detected in the core area in the same two years (118 in Laguna and 21 in Gravera, Table 2; note that some individuals were detected in both years). Additionally, we genotyped cohort samples of 55 H. molleri metamorphic individuals and 103 P. cultripes tadpoles collected in Laguna in 2018 and 2019, respectively. Outside the core area, we genotyped 21 and 30 adult H. molleri individuals sampled in Roblellano and Bustarviejo, respectively, and 50 and 36 adult P. cultripes individuals from the same two localities (Table 2).
We used commercial kits (DNeasy 96 Blood & Tissue Kit, Qiagen) for DNA extraction, following the manufacturer’s protocol. We then genotyped individual DNA samples with polymorphic microsatellites optimized for each species: 15 for H. molleri (Sánchez-Montes et al. 2017a) and 16 for P. cultripes (Gutiérrez-Rodríguez & Martínez-Solano 2013), combined in 3 new multiplex reactions for each species (see Tables S1.1 and S1.2 in Supplementary Material for detailed information). PCR reactions included 7.5 µl Type-it mastermix for microsatellites (Qiagen), 1.2 µl primer mix (see Tables S1.1 and S1.2 in Supplementary Material), 5.3 µl RNase-free water and 1 µl of template DNA for a total volume of 15 µl per reaction tube. Thermal profiles were composed by initial denaturation (95 ºC for 5 min), 30 cycles of denaturation (95 ºC, 30 s), annealing (60 ºC, 90 s) and extension (72 ºC, 30 s), with a final extension step at 60 ºC during 30 min. Post-PCR DNA fragment sizes were analysed with an ABI PRISM 3720 sequencer using the GeneScan 500 LIZ size standard (Applied Biosystems). Finally, individual genotypes were assigned manually using GeneMapper v4.0 (Applied Biosystems).
Genetic diversity, N e and N b
We assessed the genetic diversity in the four sampling sites using the genotypes obtained in this work, together with the genotypes of 69 adult individuals of H. molleri and 95 of P. cultripes sampled in Laguna in 2013 and originally published in Sánchez-Montes et al. (2017b) and Gutiérrez-Rodríguez et al. (2017b). First, we tested for departures from Hardy–Weinberg proportions (HWP) at each locus and for linkage disequilibrium (LD) at pairs of loci for each population using Genepop v4.2 (Raymond and Rousset 1995; Rousset 2008), setting a dememorization number of 10,000, with 1000 batches and 10,000 iterations per batch. The significance of p-values in multiple tests was adjusted by applying the sequential Bonferroni correction (Rice 1989). We also used Genepop to test for significant evidence of heterozygote deficiency or excess in each population. We then used GenAlEx v6.503 (Peakall and Smouse 2012) to estimate four indices of genetic diversity: mean allelic richness per locus (AR), mean number of alleles exclusively found in one population (private alleles, PA) and observed and expected heterozygosity (HO and HE, respectively) in each population and temporal sample for both species.
We used the sibship frequency (SF) method implemented in Colony v2.0.6.5 (Jones and Wang 2010) to estimate the effective size (Ne) of all populations and the effective number of breeders (Nb) during the 2018 (for H. molleri) and 2019 (for P. cultripes) breeding seasons in Laguna. The SF method reconstructs pedigrees to estimate Ne, which in local and exhaustively surveyed areas such as our study system, can readily be compared with independent evidence of breeding activity (Sánchez-Montes et al. 2017b). To estimate Ne, we used the genotypes of all adults of each population as ‘offspring sample’ with no parental candidates. In the case of Laguna and Gravera, adults from 2018 and 2019 were analysed together for each species. To estimate Nb, we used the genotypes of metamorphic individuals of H. molleri (sampled in 2018) and larvae of P. cultripes (2019) as offspring, and all genotyped males and females captured during 2018 and 2019 as candidate parents, assuming prior probabilities of parentship of 0.2, 0.5 and 0.8, respectively. We ran two replicates with different random seed numbers for each Ne and Nb analysis, in order to test the consistency of results. We used the Full-Likelihood method, with ‘long’ run length and ‘high’ precision settings, a priori genotyping error rates of 0.05 for all markers (based on Sánchez-Montes et al. 2017a) and allowing for the possibility of polygamy for both sexes. Analyses were performed with and without a ‘weak’ sibship size prior = 1 for both paternal and maternal families, in order to aid but not force family reconstruction (see Sánchez-Montes et al. 2017b).
Genetic structure, dispersal and migration rates
We calculated Jost’s D and Hedrick’s GST indices as measures of pairwise genetic differentiation between populations (Hedrick 2005; Jost 2008), using the R-package ‘diveRsity’ (Keenan et al. 2013). These indices provide a good description of allelic differentiation/fixation among populations, reflecting demographic properties like the effective number of migrants (Alcala et al. 2014; Jost et al. 2018). To test if genetic differentiation between populations follows a pattern of isolation by distance, we conducted Mantel tests (Rousset 1997) using mantel.rtest function in the R-package ‘adegenet’ (Jombart 2008), with 1000 permutations. Genetic (Nei’s distance) and geographic distances were calculated using dist and dist.genpop functions, respectively, from R-package ‘adegenet’. Further, to characterize genetic structure in each species, we performed Bayesian genetic clustering analyses using Structure v2.3.4 (Pritchard et al. 2000). We conducted two types of analyses, using or not the information of the localities where samples were collected (option: locprior). In both cases we ran 10 replicates for each value of K (number of clusters) from 1 to 6, with 1,000,000 iterations as burn-in and 500,000 of post-burn-in, assuming an admixture model and correlated allelic frequencies. Results were inspected with Structure Harvester to assess the value of K that best fits the genetic data (Earl and vonHoldt 2012), based on the original method (increasing values of likelihood (L), Pritchard et al. 2000) and on the ΔK method of Evanno et al. (2005). Individual barplots were obtained with Clumpak (Kopelman et al. 2015).
In order to compile direct evidence of functional connectivity, we summarized the frequency and direction of recorded dispersal events of all marked individuals from Laguna and Gravera based on their capture histories in the period 2009–2021. Additionally, we calculated accumulated distances travelled by dispersing individuals as a minimum estimate of their dispersal potential, for both species. To infer whether observed displacements resulted in gene flow, we estimated migration rates per generation among the four populations based on individual adult genotypes from the 2018 and 2019 breeding seasons in Laguna and Gravera, 2019 in Roblellano and 2020 in Bustarviejo, using the software BayesAss v3.0.4 (Wilson and Rannala 2003). This program assumes migration rates to be small and constant over short periods of time (Faubet et al. 2007). Ten replicate analyses with different random seeds were run for 30,000,000 iterations, discarding the first 6,000,000 iterations as burn-in, and mixing parameters for allelic frequencies (a), inbreeding coefficients (f) and migration rates (m) set to 0.5, 0.2 and 0.25, respectively. These values provided acceptance rates between 20 and 60% in MCMC chains, which are optimal according to the software manual. We assessed convergence of migration rate estimates across replicate runs by checking the logfiles with software Tracer v1.7.1 (Rambaut et al. 2018). Finally, the probability of ancestry for each individual according to the BayesAss run with a best likelihood was classified into three categories: non-migrant (resident), first-generation migrant, and second-generation migrant (see Capellà-Marzo et al. 2020; Fernández de Larrea et al. 2021).
Results
Genetic diversity, N e and N b
None of the genetic markers employed showed significant and consistent deviations from HWP across all populations in any of the two species. However, three of the P. cultripes markers showed significant deviations from HWP after applying Bonferroni correction (Pc4.11: 1 population, Pc4.3: 2 populations, and Pc4.7: 3 populations), all of them caused by heterozygote deficiency (Table 2). In H. molleri, only markers Hmol3.3 (Laguna 2019 and Gravera 2018) and Hmol3.28 (Laguna 2018 and 2019, Roblellano and Bustarviejo) showed significant departures from HWP in more than one population after Bonferroni correction, caused by heterozygote excess in all cases. We found no significant and consistent evidence of linkage disequilibrium in any pair of loci, suggesting that allelic associations are not caused by physical linkage, and thus, all loci were used for subsequent analyses.
The four populations of both species showed moderate levels of genetic diversity, with all H. molleri populations presenting slight heterozygote excess (HO > HE), and most P. cultripes populations showing the opposite pattern (HO < HE, Table 2). Genetic diversity was higher in Hyla compared to Pelobates in all sites (Table 2). The population of Laguna scored the highest values of allelic richness and private alleles in both species, as compared with the other three localities. We found a decrease in genetic diversity (mainly AR) through time in Laguna and Gravera for both species (Table 2).
The highest estimates of Ne for H. molleri were found in Laguna (48–67, with minor differences across years) followed by Gravera (32–41), with Roblellano and Bustarviejo having Ne estimates ≤ 20 (Fig. 2, Table S2). In P. cultripes, the highest Ne estimate was also obtained in Laguna (31–61), with all other populations having values < 30. In all analyses, estimates using or not the sibship size prior returned similar results, albeit slightly lower values were recovered in the former case (Table S2). Finally, estimates of Nb in Laguna were similar across analyses assuming different parentship probabilities and using or not the sibship size prior, and showed higher values for H. molleri (55–72) than for P. cultripes (33–35) (Fig. 2, Table S2).

Estimates of Ne for H. molleri (left) and P. cultripes (right) in the four populations analysed (top) and Nb estimates in Laguna (bottom). Point estimates are shown with 95% confidence intervals (bars), representing the use (triangles) or not (dots) of a sibship size prior in the analyses and under different parentship probabilities for Nb. Detailed values can be found in Table S2 in Supplementary Material
Genetic structure, dispersal and migration rates
The populations of H. molleri showed higher genetic differentiation (D: 0.020–0.056; GST: 0.032–0.095) than those of P. cultripes (D: 0.005–0.018; GST: 0.018–0.066). Overall, similar patterns of genetic differentiation were found in both species, with the lowest estimates corresponding to the closest populations (Gravera and Laguna), whereas the most genetically differentiated populations were also the most geographically separated (Roblellano and Bustarviejo, Table 3). Mantel tests did not support a pattern of isolation by distance, as the relationships between genetic and geographic distances were not significant (R = 0.758, p-value = 0.130 for H. molleri; R = 0.622, p-value = 0.073 for P. cultripes).
Genetic structure analyses for H. molleri yielded increasing likelihood values with increasing K, regardless of using or not the information from sampling locations. The number of genetic clusters best explaining the data corresponded to K = 5 when using the locprior option, while results were not conclusive (albeit K = 5 was the best according to the likelihood method) when sampling location information was not taken into account (Fig. 3, Figs. S1 and S2). For P. cultripes, the ΔK method yielded K = 2 as the optimal partition when using locprior, but the likelihood method supported four clearly differentiated clusters (K = 4, as also suggested in analyses without locprior), with little to no genetic admixture in the cases of Roblellano and Bustarviejo (Fig. 3, Figs. S1 and S2). Overall, populations of each site were clearly differentiated in both species, although less clearly so in Laguna and Gravera, which showed more or less extensive admixture (Fig. 3).

Maps showing locations of study populations (B: Bustarviejo, G: Gravera, L: Laguna de Valdemanco and R: Roblellano), with pie charts coloured according to inferred genetic ancestry proportions at the values of K best explaining the genetic data in each species (K = 5 for Hyla molleri and K = 4 for Pelobates cultripes) in Bayesian genetic clustering analyses. Below, assignment probabilities for each individual (columns) of H. molleri (left) and P. cultripes (right) to each genetic cluster as inferred with software Structure (locprior: ON)
Over the study period (2009–2021) we PIT-tagged 944 individuals of H. molleri (recapture rate: 37.2%) and 1206 individuals of P. cultripes (recapture rate: 55.9%). Considering all individuals from Laguna and Gravera with one or more recaptures throughout the study period, we recorded a total of 22 displacements by 17 individuals of H. molleri (4.8% of all recaptured individuals, N = 351) and 64 displacements of 48 individuals of P. cultripes (7.1%, N = 674). In H. molleri, most displacements were carried out by males that were captured in the two breeding ponds in the same or in different years, with the exception of one single event of 1 km travelled between Gravera and an additional, non-systematically surveyed locality between Gravera and Bustarviejo that represents the single largest recorded displacement in the study area (Fig. 4). Most recorded displacements of P. cultripes corresponded to short distances between a breeding site and a terrestrial shelter, performed by both males and females; only a few individuals (most of them males) covered over 700 m to disperse between breeding sites, preferentially from Laguna towards Gravera (Fig. 4). Despite their small size, some adult treefrogs covered accumulated distances over 3.5 km, whereas spadefoot toads also performed long cumulative displacements up to 1.8 km (Fig. 4).

Maps showing the direction and frequency of dispersal events between the two main breeding ponds (G: Gravera and L: Laguna) recorded during the 2009–2021 CMR monitoring program for both species (top: H. molleri, bottom: P. cultripes), separated by sex (males:females). Additional displacements to non-breeding terrestrial habitats are represented with orange dots. Right: barplots of individual cumulative distances recorded for each species
Results from BayesAss analyses were consistent across the 10 runs, as all replicate analyses converged on likelihood scores around -7155 for H. molleri and -8053 for P. cultripes. Based on the run with the best likelihood, we recovered low migration rates per generation among H. molleri populations (< 10%), with the exception of geographically closest localities, Laguna and Gravera (Fig. 5, Table S3). In these localities, migration rates were asymmetric, with higher migration from the smaller to the larger population (based on Ne estimates): 24.6% from Gravera to Laguna and 14.2% in the opposite direction. We recovered higher migration rates in P. cultripes, especially from Laguna towards Gravera and Roblellano (21% in both cases), followed by migration rates from Bustarviejo to Gravera (9%; Fig. 5, Table S3).

Circos plots representing inferred residence and migration rates per generation between the four sampled populations of H. molleri (left) and P. cultripes (right) based on microsatellite genotypes (raw BayesAss results are displayed in Table S3 in Supplementary Material). Dashed arrows indicate migration directionally from the source population to the recipient one, with widths proportional to migration rate estimates
Almost all individuals of P. cultripes from Laguna were classified as residents (99%), whereas most individuals of H. molleri in this population were classified as second-generation migrants from Gravera (53.51%) (Fig. S3). Most individuals (71.4%) of P. cultripes from Gravera were classified as first-generation migrants from Laguna, and the 24% remaining were classified as first-generation migrants from Bustarviejo. In the case of H. molleri, 38.71% of the individuals in Gravera were classified as second-generation migrants from Laguna, with a lower number of individuals (3.24%) inferred to be second generation migrants from Roblellano and Bustarviejo. In Bustarviejo, most individuals of both species were classified as residents (81% in P. cultripes; 67% in H. molleri), with a small contribution of second-generation migrants from other populations. Finally, only 2% of P. cultripes individuals from Roblellano were classified as resident, while most individuals were classified as first (10%) or second (44%) generation migrants from Laguna. In contrast, most individuals of H. molleri from Roblellano were classified as residents (52.4%), with minor contributions of first- and second-generation migrants from Gravera (19%) and Laguna (3.3%), respectively.
Discussion
Robust characterization of population structure and functional connectivity is critical to understand the demographic dynamics of amphibian populations in fragmented habitats. Reduced landscape connectivity in organisms with low dispersal capacity results in more structured and isolated populations, with less possibility of genetic exchange. However, high migration rates can mitigate the negative effects of demographic fluctuations on genetic diversity in spatially structured populations (Jangjoo et al. 2016; McEachern et al. 2011). It is thus important to characterize fine-scale patterns of gene flow in amphibians and identify the spatial scale at which breeding populations are demographically connected. Our integrative study provides evidence of recent genetic exchange between populations < 1 km apart (Gravera and Laguna, Figs. 3 and 5) in both species, in agreement with estimates of functional connectivity (individual displacements) obtained from field-based observations (CMR data) (Fig. 4). In contrast, more distant populations (> 2 km) of H. molleri and P. cultripes were significantly differentiated, and Bayesian clustering analyses and migration rate estimates indicated reduced connectivity at this spatial scale. Despite these similarities, each species showed different patterns of connectivity among breeding sites that can be interpreted in terms of their life-history traits (see below), highlighting the added value of comparative studies of syntopic species.
While providing solid evidence, direct approaches often underestimate actual frequencies and distances of spatial displacements because detectability of individuals is seldom perfect and long distance movements are more prone to pass unnoticed. Furthermore, without evidence of breeding success in the destination population, the proportion of dispersal events with consequences on gene flow is unknown. Conversely, estimates based on genetic markers are indirect and potentially miss relevant demographic information, and are difficult to interpret in terms of actual numbers of dispersing individuals. Combining both types of evidence provides a comprehensive overview of the dispersal process, including its demographic context. In our study system, geographic distance alone does not fully explain observed patterns of genetic structure, and so other factors including local effective population sizes, dispersal capacity, and ecological preferences likely contribute to shape functional connectivity in each species. Comprehensive interpretation of functional connectivity in H. molleri and P. cultripes was thus only feasible by integrating multiple-source evidence of dispersal, based both on direct individual displacement records and indirect gene flow assessments, with genetic diversity, effective size and migration rate estimates.
Assessing patterns of genetic diversity in spatially structured amphibian populations can provide early insights on potential population declines and helps delineate breeding units connected by migration. In our study area, Laguna harboured the highest values of allelic richness and heterozygosity for both species, with a slight genetic impoverishment through the years (Table 2). The regional importance of Laguna for both species is also supported by higher Ne estimates, which can be interpreted as a proxy of the capacity of a population to maintain genetic diversity in the long term (Frankham 1996). Nevertheless, Ne estimates in all breeding populations of the two species studied are far below minimum threshold values that have been proposed to avoid inbreeding depression (Ne > 50–100) and to maintain the evolutionary potential of populations (Ne > 1000, Frankham et al. 2014). This makes these populations vulnerable to genetic drift and local extinction (Phillipsen et al. 2011; Lourenço et al. 2018), especially if habitat fragmentation is sufficient to promote their isolation. However, successful migration can mitigate the negative effects of demographic fluctuations on genetic diversity in spatially structured populations (Jangjoo et al. 2016; McEachern et al. 2011). This seems to be the case in our study system, where genetic connectivity between Laguna and Gravera suggests that both populations have inter-dependent metapopulation dynamics mediated by frequent dispersal of individuals.
The low Ne estimates of both species in Bustarviejo and Roblellano, coupled with results from Bayesian clustering analyses and pairwise genetic differentiation estimates, that suggest demographic isolation, highlight the vulnerability of these populations to genetic drift. Changes in land use and vegetation cover (Gutiérrez-Rodríguez et al. 2017a) or landscape factors such as roads M-610 and M-633 may have created barriers reducing gene flow among breeding units (Richardson 2012). In contrast, we found evidence of recent genetic exchange between Gravera and Laguna for both species (Fig. 3), which are separated 700 m in a straight line, based on direct records of individual displacements obtained from field-based observations (Fig. 4) and genetic data. Estimated migration rates are in agreement with these results, showing a higher proportion of migrants of both species between these ponds, but with different populations being identified as the major source of migrants (Fig. 5). Notably, BayesAss results are not fully in agreement with CMR data regarding the main directionality of dispersal in each species (Figs. 4 and 5). Nevertheless, this is expected because CMR data represent spatial displacements that are recorded during a specific study period and that can result (or not) in gene flow, whereas BayesAss directly quantifies gene flow as migration rates during an entire generation. Since migration is a density-dependent process, we expected a higher flux of migrants from the larger to the smaller population, but we only found this to be the case in P. cultripes (as also found in syntopic Pleurodeles waltl, Fernández de Larrea et al. 2021). In H. molleri, Gravera supplied the most migrants to Laguna (Fig. 5 and Supplementary Material). This is similar to results in Pelophylax perezi (Capellà-Marzo et al. 2020) and is probably related to higher recruitment rates associated with the longer hydroperiod in Gravera. This highlights the important role of Gravera, a temporary pond of artificial origin which is not legally protected (unlike the regionally listed Laguna de Valdemanco), as a demographic reservoir in local amphibian community dynamics. Disparity in directional patterns of gene flow between species supports the need to preserve both ponds as part of a well-connected pond network, and to design management actions taking into consideration the different role of each pond in the population dynamics of each species.
Contrasting patterns of migration rates between species can be associated with differences in habitat preferences and/or life-history traits. Due to their semi-arboreal habits, individuals of H. molleri probably rely more on the presence of abundant vegetation cover in areas around breeding sites, whereas P. cultripes, which have fossorial habits, prefer open areas (Gutiérrez-Rodríguez et al. 2017a). These differences are probably of minor importance at the spatial scale of our study, but may play a role in combination with other factors like topography at larger distances (Sánchez-Montes et al. 2018). On the other hand, dispersal capacity is perhaps the biological trait with most leverage on population structure. The connectivity of amphibian populations mainly depends on juvenile post-metamorphic dispersal (Rothermel 2004), which may contribute more to the regional persistence of populations than adult dispersal (Sinsch 1992, 1997). However, our CMR dataset involves only adult individuals, because juveniles are difficult to mark and track with PIT-tags due to their small size, high mortality and secretive life style during their maturation stage. More information about juvenile dispersal is necessary to obtain more accurate estimates of migration rates and their impact on local patterns of population differentiation at fine spatial scales. Furthermore, CMR data suggest male-biased dispersal in both species, although additional evidence from both longer time series and detailed genomic analyses of male and female genotypes is required to test this hypothesis.
Besides dispersal capacity, other life-history traits can promote genetic differentiation in amphibians (Nowakowski et al. 2015; Richardson 2012; Whiteley et al. 2014). For instance, breeding site fidelity is associated with lower migration rates and increased genetic differentiation between populations. Based on the literature (Table 1), we expected to find stronger genetic structure in P. cultripes compared to H. molleri. While we obtained similar recapture rates for both species, our field records suggest P. cultripes have stronger breeding site fidelity than H. molleri, because they performed less displacements among breeding sites (most recorded individual movements of P. cultripes were between a breeding site and an autumn/winter terrestrial shelter) and they covered shorter accumulated distances. In contrast, some individuals of H. molleri performed up to two journeys between Gravera and Laguna in a single breeding season. However, BayesAss estimates of migration rates and individual ancestries and, to a lesser extent, clustering analyses seem in contradiction with these expectations. This may be partly explained by differences in other life-history traits, including life span and age to sexual maturity. Hyla molleri is a short-lived species (up to 4–5 years, Sánchez-Montes and Martínez-Solano 2023), and consequently has a shorter reproductive lifespan. Thus, breeding site fidelity in H. molleri may be disadvantageous in fragmented landscapes with distant and disconnected breeding habitats that are subject to strong interannual variation in hydroperiod. In contrast, long-lived species like P. cultripes, which can have reproductive lifespans > 10 years (Sánchez-Montes and Martínez-Solano 2023) may be less affected by stochasticity in regional rainfall patterns and adopt a more sedentary lifestyle, with strong breeding site fidelity and occasional long-distance dispersal maintaining gene flow. The longer generation time of P. cultripes as compared to H. molleri may thus explain higher gene flow in the former, despite less recorded dispersal events during the study timeframe.
Our comparative approach highlights how the different ecological requirements and life-history traits of the species can play a key role in local population structure and dynamics. The disparity in migration rates between H. molleri and P. cultripes shows the different role of each breeding site on local population dynamics of each species, and provides relevant information for the design and implementation of conservation measures promoting population connectivity, for instance focusing on groups of functionally connected ponds instead of single breeding sites. Integrative approaches based on direct and indirect estimates of dispersal like this study provide a broader, demographically contextualized overview of the dispersal process, with robust estimates of gene flow among populations, and identify breeding habitats that represent key nodes for the functional connectivity of amphibian communities.
Data availability
All data generated and analysed during the current study are available from the authors upon reasonable request.
References
Alcala N, Goudet J, Vuilleumier S (2014) On the transition of genetic differentiation from isolation to panmixia: what we can learn from GST and D. Theor Popul Biol 93:75–84. https://doi.org/10.1016/j.tpb.2014.02.003
Baguette M, Blanchet S, Legrand D, Stevens VM, Turlure C (2013) Individual dispersal, landscape connectivity and ecological networks. Biol Rev 88:310–326. https://doi.org/10.1111/brv.12000
Bowne DR, Bowers MA (2004) Interpatch movements in spatially structured populations: a literature review. Landsc Ecol 19:1–20. https://doi.org/10.1023/B:LAND.0000018357.45262.b9
Capellà-Marzo B, Sánchez-Montes G, Martínez-Solano I (2020) Contrasting demographic trends and asymmetric migration rates in a spatially structured amphibian population. Integr Zool 15:482–497. https://doi.org/10.1111/1749-4877.12449
Cayuela H, Valenzuela-Sánchez A, Teulier L, Martínez-Solano Í, Léna JP, Merilä J, Muths E, Shine R, Quay L, Denoël M, Clobert J, Schmidt BR (2020) Determinants and consequences of dispersal in vertebrates with complex life cycles: a review of pond-breeding amphibians. Q Rev Biol 95:1–36
Çiçek K, Cumhuriyet O (2017) Amphibians and reptiles of the Mediterranean basin. In: Fuerst-Bjelis B (ed) Mediterranean identities: environment, society, culture. InTech, Rijeka, pp 203–237
Coster SS, Babbitt KJ, Cooper A, Kovach AI (2015) Limited influence of local and landscape factors on finescale gene flow in two pond-breeding amphibians. Mol Ecol 24:742–758. https://doi.org/10.1111/mec.13062
Cushman SA (2006) Effects of habitat loss and fragmentation on amphibians: a review and prospectus. Biol Conserv 128:231–240. https://doi.org/10.1016/j.biocon.2005.09.031
Cuttelod A, García N, Malak DA, Temple HJ, Katariya V (2009) The Mediterranean: a biodiversity hotspot under threat. In: Jean-Christophe V, Hilton-Taylor C, Stuart SN (eds) Wildlife in a changing World–an analysis of the 2008 IUCN Red List of Threatened Species, vol 89. IUCN, Switzerland, p 9
Dixo M, Metzger JP, Morgante JS, Zamudio KR (2009) Habitat fragmentation reduces genetic diversity and connectivity among toad populations in the Brazilian Atlantic Coastal Forest. Biol Conserv 142:1560–1569. https://doi.org/10.1016/j.biocon.2008.11.016
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Fahrig L (1998) When does fragmentation of breeding habitat affect population survival? Ecol Modell 105:273–292. https://doi.org/10.1016/S0304-3800(97)00163-4
Fahrig L (2001) How much habitat is enough? Biol Conserv 100:65–74. https://doi.org/10.1016/S0006-3207(00)00208-1
Faubet P, Waples RS, Gaggiotti OE (2007) Evaluating the performance of a multilocus Bayesian method for the estimation of migration rates. Mol Ecol 16:1149–1166. https://doi.org/10.1111/j.1365-294X.2007.03218.x
Fernández de Larrea I, Sánchez-Montes G, Gutiérrez-Rodríguez J, Martínez-Solano Í (2021) Reconciling direct and indirect estimates of functional connectivity in a Mediterranean pond-breeding amphibian. Conserv Genet 22:455–463. https://doi.org/10.1007/s10592-021-01345-2
Frankham R (1996) Relationship of genetic variation to population size in wildlife. Conserv Biol 10:1500–1508. https://doi.org/10.1046/j.1523-1739.1996.10061500.x
Frankham R, Bradshaw CJ, Brook BW (2014) Genetics in conservation management: revised recommendations for the 50/500 rules, red list criteria and population viability analyses. Biol Conserv 170:56–63. https://doi.org/10.1016/j.biocon.2013.12.036
Gilpin M (ed) (2012) Metapopulation dynamics: empirical and theoretical investigations. Academic press, London
Goldberg CS, Waits LP (2010) Comparative landscape genetics of two pond-breeding amphibian species in a highly modified agricultural landscape. Mol Ecol 19:3650–3663. https://doi.org/10.1111/j.1365-294X.2010.04673.x
Gutiérrez-Rodríguez J, Martínez-Solano I (2013) Isolation and characterization of sixteen polymorphic microsatellite loci in the Western Spadefoot, Pelobates cultripes (Anura: Pelobatidae) via 454 pyrosequencing. Conserv Genet Resour 5:981–984. https://doi.org/10.1007/s12686-013-9948-y
Gutiérrez-Rodríguez J, Gonçalves J, Civantos E, Martínez-Solano I (2017a) Comparative landscape genetics of pond-breeding amphibians in Mediterranean temporal wetlands: the positive role of structural heterogeneity in promoting gene flow. Mol Ecol 26:5407–5420. https://doi.org/10.1111/mec.14272
Gutiérrez-Rodríguez J, Sánchez-Montes G, Martínez-Solano I (2017b) Effective to census population size ratios in two near threatened Mediterranean amphibians: Pleurodeles waltl and Pelobates cultripes. Conserv Genet 18:1201–1211. https://doi.org/10.1007/s10592-017-0971-5
Hanski I, Gilpin M (1991) Metapopulation dynamics: brief history and conceptual domain. Biol J Linn Soc 42:3–16. https://doi.org/10.1111/j.1095-8312.1991.tb00548.x
Hedrick PW (2005) A standardized genetic differentiation measure. Evolution (N Y) 59:1633–1638. https://doi.org/10.1111/j.0014-3820.2005.tb01814.x
Igawa T, Oumi S, Katsuren S, Sumida M (2013) Population structure and landscape genetics of two endangered frog species of genus Odorrana: different scenarios on two islands. Heredity (Edinb) 110:46–56. https://doi.org/10.1038/hdy.2012.59
Jangjoo M, Matter SF, Roland J, Keyghobadi N (2016) Connectivity rescues genetic diversity after a demographic bottleneck in a butterfly population network. Proc Natl Acad Sci 113:10914–10919. https://doi.org/10.1073/pnas.1600865113
Johansson M, Primmer CR, Merilä J (2007) Does habitat fragmentation reduce fitness and adaptability? A case study of the common frog (Rana temporaria). Mol Ecol 16:2693–2700. https://doi.org/10.1111/j.1365-294X.2007.03357.x
Jombart T (2008) Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Jones OR, Wang J (2010) Molecular marker-based pedigrees for animal conservation biologists. Anim Conserv 13:26–34. https://doi.org/10.1111/j.1469-1795.2009.00324.x
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026. https://doi.org/10.1111/j.1365-294X.2008.03887.x
Jost L, Archer F, Flanagan S, Gaggiotti O, Hoban S, Latch E (2018) Differentiation measures for conservation genetics. Evol Appl 11:1139–1148. https://doi.org/10.1111/eva.12590
Keenan K, Mcginnity P, Cross TF et al (2013) DiveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788. https://doi.org/10.1111/2041-210X.12067
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191. https://doi.org/10.1111/1755-0998.12387
Lourenço A, Antunes B, Wang IJ, Velo-Antón G (2018) Fine-scale genetic structure in a salamander with two reproductive modes: does reproductive mode affect dispersal? Evol Ecol 32:699–732. https://doi.org/10.1007/s10682-018-9957-0
Luque S, Saura S, Fortin MJ (2012) Landscape connectivity analysis for conservation: insights from combining new methods with ecological and genetic data. Landsc Ecol 27:153–157. https://doi.org/10.1007/s10980-011-9700-5
Marsh DM, Trenham PC (2001) Metapopulation dynamics and amphibian conservation. Conserv Biol 15:40–49. https://doi.org/10.1046/j.1523-1739.2001.00129.x
McEachern MB, Van Vuren DH, Floyd CH, May B, Eadie JM (2011) Bottlenecks and rescue effects in a fluctuating population of golden-mantled ground squirrels (Spermophilus lateralis). Conserv Genet 12:285–296. https://doi.org/10.1007/s10592-010-0139-z
Newman RA, Squire T (2001) Microsatellite variation and fine-scale population structure in the wood frog (Rana sylvatica). Mol Ecol 10:1087–1100. https://doi.org/10.1046/j.1365-294X.2001.01255.x
Nicholson E, Possingham HP (2006) Objectives for multiple-species conservation planning. Conserv Biol 20:871–881. https://doi.org/10.1111/j.1523-1739.2006.00369.x
Nowakowski AJ, Dewoody JA, Fagan ME et al (2015) Mechanistic insights into landscape genetic structure of two tropical amphibians using field-derived resistance surfaces. Mol Ecol 24:580–595. https://doi.org/10.1111/mec.13052
Olarte O, Sánchez-Montes G, Martínez-Solano I (2020) Integrative demographic study of the Iberian painted frog (Discoglossus galganoi): inter-annual variation in the effective to census population size ratio, with insights on mating system and breeding success. Integr Zool 15:498–510. https://doi.org/10.1111/1749-4877.12452
Peakall R, Smouse PE (2012) GenALEx 6.5: genetic analysis in excel. population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Peterman WE, Anderson TL, Ousterhout BH et al (2015) Differential dispersal shapes population structure and patterns of genetic differentiation in two sympatric pond breeding salamanders. Conserv Genet 16:59–69. https://doi.org/10.1007/s10592-014-0640-x
Petranka JAW, Smith CHK, Scott AF (2004) Identifying the minimal demographic unit for monitoring pond-breeding amphibians. Ecol Appl 14:1065–1078
Phillipsen IC, Funk WC, Hoffman EA et al (2011) Comparative analyses of effective population size within and among species: ranid frogs as a case study. Evolution (N Y) 65:2927–2945. https://doi.org/10.1111/j.1558-5646.2011.01356.x
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1007/s10681-008-9788-0
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol 67:901–904
Raymond M, Rousset F (1995) GENEPOP (Version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Reed DH, Frankham R (2003) Correlation between fitness and genetic diversity. Conserv Biol 17:230–237. https://doi.org/10.1046/j.1523-1739.2003.01236.x
Reyes-Moya I, Sánchez-Montes G, Martínez-Solano I (2022) Integrating dispersal, breeding and abundance data with graph theory for the characterization and management of functional connectivity in amphibian pondscapes. Landsc Ecol 37:3159–3177. https://doi.org/10.1007/s10980-022-01520-x
Rice WR (1989) Analyzing tables of statistical tests. Evolution (N. Y) 43:223–225
Richardson JL (2012) Divergent landscape effects on population connectivity in two co-occurring amphibian species. Mol Ecol 21:4437–4451. https://doi.org/10.1111/j.1365-294X.2012.05708.x
Rothermel BB (2004) Migratory success of juveniles: a potential constraint on connectivity for pond-breeding amphibians. Ecol Appl 14:1535–1546. https://doi.org/10.1890/03-5206
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228. https://doi.org/10.1002/ajmg.c.30221
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Sánchez-Montes G, Martínez-Solano I (2011) Population size, habitat use and movement patterns during the breeding season in a population of Perez’s frog (Pelophylax perezi) in central Spain. Basic Appl Herpetol 25:81–96. https://doi.org/10.11160/bah.11013
Sánchez-Montes G, Ariño AH, Vizmanos JL, Wang J, Martínez-Solano I (2017a) Effects of sample size and full sibs on genetic diversity characterization: a case study of three syntopic Iberian pond-breeding amphibians. J Hered 108:535–543. https://doi.org/10.1093/jhered/esx038
Sánchez-Montes G, Wang J, Ariño AH, Martínez-Solano I (2017b) Reliable effective number of breeders/adult census size ratios in seasonal-breeding species: opportunity for integrative demographic inferences based on capture–mark–recapture data and multilocus genotypes. Ecol Evol 7:10301–10314. https://doi.org/10.1002/ece3.3387
Sánchez-Montes G, Wang J, Ariño AH, Martínez-Solano Í (2018) Mountains as barriers to gene flow in amphibians: quantifying the differential effect of a major mountain ridge on the genetic structure of four sympatric species with different life history traits. J Biogeogr 45:318–331. https://doi.org/10.1111/jbi.13132
Sánchez-Montes G, Martínez-Solano I (2023) Seguimiento de poblaciones de anfibios mediante la integración de historiales de captura y herramientas moleculares. Munibe Monographs. Nat Ser 5:83–93
Schwenk WS, Donovan TM (2011) A multispecies framework for landscape conservation planning. Conserv Biol 25:1010–1021. https://doi.org/10.1111/j.1523-1739.2011.01723.x
Semlitsch RD (2008) Differentiating migration and dispersal processes for pond-breeding amphibians. J Wildl Manage 72:260–267. https://doi.org/10.2193/2007-082
Sinsch U (1992) Structure and dynamic of a natterjack toad metapopulation (Bufo calamita). Oecologia 90:489–499. https://doi.org/10.1007/BF01875442
Sinsch U (1997) Postmetamorphic dispersal and recruitment of first breeders in a Bufo calamita metapopulation. Oecologia 112:42–47. https://doi.org/10.1007/s004420050281
Smith MA, Green DM (2005) Dispersal and the metapopulation paradigm in amphibian ecology and conservation: are all amphibian populations metapopulations? Ecography (cop) 28:110–128. https://doi.org/10.1111/j.0906-7590.2005.04042.x
Steele CA, Baumsteiger J, Storfer A (2009) Influence of life-history variation on the genetic structure of two sympatric salamander taxa. Mol Ecol 18:1629–1639. https://doi.org/10.1111/j.1365-294X.2009.04135.x
Wake DB, Koo MS (2018) Amphibians. Curr Biol 28:R1237–R1241. https://doi.org/10.1016/j.cub.2018.09.028
Wang IJ (2009) Fine-scale population structure in a desert amphibian: Landscape genetics of the black toad (Bufo exsul). Mol Ecol 18:3847–3856. https://doi.org/10.1111/j.1365-294X.2009.04338.x
Whiteley AR, McGarigal K, Schwartz MK (2014) Pronounced differences in genetic structure despite overall ecological similarity for two Ambystoma salamanders in the same landscape. Conserv Genet 15:573–591. https://doi.org/10.1007/s10592-014-0562-7
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191. https://doi.org/10.1093/genetics/163.3.1177
Acknowledgements
We thank the editor and two anonymous reviewers for their constructive comments, which improved the manuscript. We also thank C. Caballero, B. Capellà, M. Durán, A. Carramolino, I. Reyes, D. Jiménez, D. Fernández and M. Pinilla for help during the field work. Special thanks to Claudia and Nicolás for their effort sampling H. molleri. This study was funded by FEDER/Ministerio de Ciencia, Innovación y Universidades-Agencia Estatal de Investigación, Spain, grants PID2020-116289GB-I00 and CGL2017-83131-P.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This study was funded by FEDER/Ministerio de Ciencia, Innovación y Universidades–Agencia Estatal de Investigación, Spain, Grants CGL2017-83131-P and PID2020-116289 GB-I00. HMG has received support from Fondo Social Europeo, Programa Operativo de Empleo Juvenil (European Union, Comunidad de Madrid, PEJ-2019-AI/AMB-13486).
Author information
Authors and Affiliations
Contributions
Project design: IMS, GSM; Sample Collection: IMS, GSM, PGM, GU; Laboratory work: PGM, GU, HMG, Data analyses: PGM, GU, HMG; Paper writing: HMG, with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors have no conflicts of interest to declare.
Research involving human and animals rights
All procedures were approved by the Ethics Committee of Consejo Superior de Investigaciones Científicas, Spain (ref.: 710/2018) and Comunidad de Madrid (ref.: PROEX 040/19).
Informed consent
All authors consent to submitting this article to Conservation Genetics.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Martínez-Gil, H., Sánchez-Montes, G., Montes-Gavilán, P. et al. Fine-scale functional connectivity of two syntopic pond-breeding amphibians with contrasting life-history traits: an integrative assessment of direct and indirect estimates of dispersal. Conserv Genet 24, 361–374 (2023). https://doi.org/10.1007/s10592-023-01506-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-023-01506-5