
Abstract
Restoring levels of genetic diversity in small and declining populations is increasingly being considered in biodiversity conservation. Evidence-based genetic management requires assessment of risks and benefits of crossing populations. Because risks are challenging to assess experimentally, e.g. through multi-generational crosses, decision-support approaches utilize proxy risk factors such as time since separation of lineages. However, the paucity of empirical datasets on fitness consequences of longer separation times tends to favour crossing lineages with conservatively short separations, restricting wildlife managers’ options. Here, we assessed the genetic outcomes of interbreeding in the wild between lineages of a threatened Australian freshwater fish (Macquarie perch) separated by an estimated 119,000–385,000 years of evolution in distinct environments. Fish belonging to the Murray-Darling Basin (MDB) lineage escaped from Cataract Dam—into which they were translocated in ~ 1915—into the Cataract River, where they interbred with the local Hawkesbury-Nepean Basin (HNB) lineage. Analyses of reduced-representation genomic data revealed no evidence of genetic incompatibilities during interbreeding of the two lineages in the Cataract River: assignment to genotypic clusters indicated a spectrum of hybrid types including second generation hybrids and backcrosses to both parental lineages. Thus, no adverse effects were detected from genetic mixing of populations separated by > 100,000 years. We are not advocating purposely crossing the two lineages for management purposes under present cost–benefit considerations, because there are currently sufficient intra-lineage source populations to beneficially mix. Instead, this study presents a useful calibration point: two morphologically different lineages evolved in different habitats for 119,000–385,000 years can successfully interbreed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Restoring levels of genetic diversity in populations of conservation concern is increasingly considered in threatened species management because low genetic diversity can lead to loss of fitness due to inbreeding depression and reduced adaptive potential (Harrisson et al. 2014; Weeks et al. 2015; Ralls et al. 2020). Genetic management, such as assisted gene flow between isolated populations, can help alleviate some of these problems by increasing the genetic diversity of populations of concern (Frankham et al. 2017; Pavlova et al. 2017a). However, evidence-based application of genetic management requires assessment of the genetic risks and benefits of crossing populations (Frankham et al. 2017; Hoffmann et al. 2021; Liddell et al. 2021).
Appropriate mixing of populations for increasing genetic diversity and/or reproductive fitness of threatened species has positive conservation outcomes (Chan et al. 2019; Hoffmann et al. 2021; Lutz et al. 2021). A meta-analysis found that mixing populations was beneficial in terms of fitness effects and evolutionary potential for more than 90% of 156 inbred populations in which the likelihood of outbreeding depression was assessed to be low, and none of the cases assessed showed clear harmfulness (Frankham 2015). Furthermore, benefits of genetic mixing typically persist beyond the F2 generation (Frankham 2016). However, there are few empirical datasets informing what length of separation might maximize genetic diversity while not affecting fitness levels deleteriously (e.g. Wells et al. 2019; Hoffmann et al. 2021).
Harmful effects of mixing genetic lineages, including loss of locally adapted traits and outbreeding depression—a reduction in fitness due to crossing of genetically divergent populations—are generally thought to be more likely and/or of greater effect size for populations that have been separated for many generations, particularly if they are adapted to different environments (Frankham et al. 2011). Although strategies to avoid negative consequences of mixing gene pools have been proposed (e.g., evaluating fitness consequences over multiple generations of experimental crosses) the risks of such crossings are challenging to assess directly (Weeks et al. 2011; Frankham et al. 2019). Instead, proxy risk factors such as the length of evolutionary separation are used to assess the potential compatibility of populations that might be mixed. However, generally studies have not estimated outcrossing success of populations with known times of divergence. Accordingly, decision-support approaches have tended to propose conservatively short thresholds for times of divergence to avoid outbreeding depression, such as 500 years, even though much longer times of separation might not be problematic (Frankham et al. 2011; Weeks et al. 2017; Ottenburghs 2021). Such short thresholds can severely limit wildlife managers’ options of source populations for genetic management, or could preclude genetic management altogether as would have been the case if applied to the highly successful genetic rescue of the mountain pygmy possum (Burramys parvus; Weeks et al. 2017). Accordingly, there is a need for research assessing the outcomes of crossing lineages with longer, well-estimated times of separation to inform when it might be appropriate to relax conservative thresholds.
The threatened Macquarie perch (Macquaria australasica Cuvier 1830) was widespread and common in south-eastern Australia until the early twentieth century, but is now restricted to a few isolated headwaters in the inland Murray Darling Basin (MDB), and the coastal Hawkesbury-Nepean Basin (HNB) and Georges Basin (Cadwallader 1981; Lintermans 2007; Faulks et al. 2010; Trueman 2011). Population declines and extinctions are attributed to a range of threats, including habitat loss and fragmentation, increased sedimentation, the introduction of alien fish, changes to flow-regimes and overexploitation (Cadwallader 1978; Ingram et al. 1990, 2000; Faulks et al. 2011).
The Macquarie perch lineages in the MDB and HNB experience different environmental conditions (Pavlova et al. 2017a). The inland MDB tends to exhibit greater variability of environmental metrics, including large temperature fluctuations and semi-arid to arid conditions, while the coastal HNB experiences more stable temperatures and water-supplies (Byrne et al. 2011). The two lineages differ genetically and in morphology, including colour, weight and size at sexual maturity, contributing to calls for recognition of separate species status (Dufty 1986; Faulks et al. 2010; Pavlova et al. 2017a, b). Phylogenetic estimates place the split between the MDB and HNB Macquarie perch lineages at 119,000–385,000 years ago, in the mid- to late-Pleistocene (Pavlova et al. 2017b). In 1915, < 500 Macquarie perch sourced from the MDB were translocated to Cataract Dam in the HNB (Legislative Assembly of New South Wales 1916). There is genetic evidence that some of these fish escaped the dam into the Cataract River where they have hybridised with local individuals of HNB lineage (Faulks et al. 2010; Pavlova et al. 2017a). Escapee Macquarie perch have also been recorded downstream from other dams, such as Cotter Dam, and the species seems to be able to survive overtopping of dams or passage through dam release structures (Lintermans 2012). While it is unknown exactly when translocated individuals first escaped, overtopping of the dam in periods of high water levels and deliberate water releases have occurred frequently since 1915, so it is likely that the lineages have been sympatric in the Cataract River for much of the period since 1915. It is likely that Macquarie perch of the HNB lineage were present in the upper Cataract River before the construction of the dam as suitable habitat was available. However, HNB Macquarie perch must have gone extinct soon after the construction of the dam or been outcompeted by translocated MDB fish as all sampled Cataract Dam Macquarie perch clearly group with MDB lineages (Faulks et al. 2010; Pavlova et al. 2017a).
The Cataract River population of Macquarie perch presents a rare opportunity to investigate the extent of genomic incompatibilities between lineages that diverged under different environmental conditions since the mid- to late-Pleistocene then were reconnected for up to ~ 100 years (~ 7 to 14 generations; Pavlova et al. 2017a; Lintermans et al. 2019). Mixing of populations separated for tens of thousands of years and/or adapted to different environments can have positive fitness effects (e.g. mountain pygmy possum populations seperated for more than 20,000 years and Trinidadian guppy (Poecilia reticulata) populations; Weeks et al. 2017; Fitzpatrick et al. 2020), but such crossings are considered risky under precautionary thresholds (Frankham et al. 2011; Kronenberger et al. 2017). The Cataract River scenario comprises a rare inadvertent ‘field experiment’ of population genomic consequences of such risky gene mixing for up to ~ 14 Macquarie perch generations between populations that diverged > 100,000 years ago (Pavlova et al. 2017a, b).
Here, we aim to test whether the MDB and HNB lineages hybridise without apparent reduced fitness caused by genomic incompatibilities, by addressing the following questions: (1) Are the MDB and HNB lineages hybridising in both directions, i.e. are MDB mitochondrial genomes (mitogenomes) found in fish that also have HNB nuclear ancestry, and the other way around? (2) Are there any signs of restrictions to gene flow between the lineages based on the numerical distributions of hybrid classes? Detection of sets of individuals bearing each mitolineage and showing a spectrum of degrees of inter-lineage nuclear admixture would indicate low likelihood of strong barriers to gene flow, and that morphologically different lineages evolved in different habitats for 119,000–385,000 years can successfully interbreed over multiple generations.
Materials and methods
Study system and genetic sampling
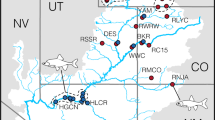
The Cataract River, a tributary of the Nepean River (southern HNB), was dammed in 1907 by the 56 m high Cataract Dam to supply water to Greater Sydney (Fig. 1). In 1915, < 500 Macquarie perch from Murrumbidgee River drainage (MDB) were translocated to the 8.5 km2 reservoir (Legislative Assembly of New South Wales 1916). Macquarie perch were sampled by electrofishing at three sites in the lower Cataract River (Jordans Pass, Broughtons Pass Weir, and below the Cataract Dam wall) in September 2013 and the Cataract Dam Reservoir in June 2017 and February 2018 (Fig. 1). Fin-clips for DNA sampling were collected from each individual, and Cataract River fish were released at their collection site.

Map of study sites on the Cataract River and Cataract Dam, New South Wales, Australia. Study area is highlighted in red on inset map. MDB Murray-Darling Basin; HNB Hawkesbury-Nepean Basin. Site 1: Below Cataract Dam; Site 2: Jordans Pass; Site 3: Broughtons Pass Weir
Sequencing, annotation, extraction and genotyping of mitogenome and DNA
A DNeasy Blood and Tissue Kit (Qiagen) was used to extract DNA from fin-clip samples of 51 fish sampled from lower Cataract River and 58 from Cataract Dam Reservoir. DNA was sent to Diversity Arrays Technology Pty. Ltd., Australia, Australia, where DArTseq—a reduced genome representation sequencing approach—and calling of single nucleotide polymorphism (SNP) were conducted (DartSeqTM; see Lutz et al. 2021 for details). Mitochondrial assignments of lower Cataract River fish to inland (MDB) or coastal (HNB) lineages were obtained from Pavlova et al. (2017b) to allow testing of whether fish of the two lineages interbreed in both directions (see details below on how hybrids are identified).
Locus-selection and data-filtering
Genotype data for 109 (58 Cataract Dam and 51 Cataract River) individuals at 5585 single biallelic SNPs were obtained. Single biallelic SNP loci (called from 69 bp sequencing reads) with reproducibility > 0.99 were retained, while loci missing genotyping scores in > 25% of individuals and two Cataract Dam individuals missing > 25% of genotypes were removed. This filtering resulted in 2777 loci for 56 Cataract Dam and 51 Cataract River fish being retained. Filters were applied using the package dartR (Gruber et al. 2018) in R (R Core Team 2019).
Next, we generated a dataset that included only those loci that should best discriminate the two populations. To achieve this, allele frequencies in the two populations were calculated using the tab function in the package adegenet (Jombart 2008). Loci bearing alleles that were undetected in Cataract Dam samples but common in the Cataract River samples (frequency of > 0.42) were retained. This was done because no known ‘pure’ Cataract River fish could be sourced, and indeed may not exist. This procedure resulted in 417 loci, of which the 370 most diagnostic of lineage (i.e. with the highest allele frequencies specific to the Cataract River) were retained to fit within the computational constraints of NEWHYBRIDS (Anderson and Thompson 2002): see section “Population structure and hybridisation”.
Population structure and hybridisation
First, to assess the admixture status of fish sampled from the Cataract River, genetic similarities between individuals were visualised for the genotype dataset using a principal coordinate analysis implemented in gl.pcoa function of dartR package (PCoA, Georges 2018) for the full 2777 loci and the potentially more-discriminating 370 loci. Admixed individuals were expected to occupy intermediate positions between Cataract River and Cataract Dam on principal coordinate axes. Admixed fish were expected to be detected only in the Cataract River population, because fish could escape from Cataract Dam into Cataract River, but not the other way around, because the 56 m dam wall represents a barrier to natural movement of fish from Cataract River to Cataract Dam. Based on habitat suitability, it is likely that HNB lineage Macquarie perch inhabited the upper Cataract River before the construction of the dam.
To further investigate levels of admixture between Cataract River and Cataract Dam individuals, we assessed the population genetic structure using STRUCTURE analysis (Pritchard et al. 2000), a model-based clustering method, to quantify individual memberships to the two ‘parental’ genotypic lineages. A set of 51 Cataract River and 56 Cataract Dam individuals were tested for a fit to two genotypic clusters (K = 2, Cataract Dam or Cataract River) using the 370 potentially more-discriminating loci and also the full 2777 loci. Thirty replicates were run using 1 million burn-in and MCMC runs, summarised using CLUMPAK (Kopelman et al. 2015). To test for hybrid classes that are feasible to identify using available software and the present data set, individuals were assigned to six ancestry classes (two sources, F1, F2, and both backcross types) using 370 loci in NEWHYBRIDS (10 runs with 1 million burn-in and 1 million sweeps each; Jeffrey’s prior for π and θ) with a probability of P > 0.5. Cataract Dam and Cataract River individuals were preassigned to their respective genotype frequency category using the z option in NEWHYBRIDS (z0 and z1 respectively). If Cataract Dam and Cataract River fish interbred approximately freely in the Cataract River and similarly in both directions with respect to matrilines and patrilines, we expected MDB and HNB mitochondrial DNA to be found proportionately among individuals identified as hybrids.
Results
Population structure
There was clear genotypic separation of individuals sampled from Cataract River vs those from the Dam, according to principal coordinates (PC) analysis (Fig. 2). Using the 2777 loci, PC1 explained 27.9% of the variance in the data, and PC2 1.8%. The PCA of the 370 potentially more-discriminating loci explained more variance: 55.5% and 3.8%, respectively. Most of the variation was distributed amongst Cataract River individuals, as expected given that the Cataract River population is admixed whereas the Cataract Dam population is not. Cataract River fish bearing the MDB mitolineage were interspersed within the Cataract River population along both PC1 and PC2, indicating approximately free admixture. No fish bearing the HNB mitolineage were identified amongst the fish sampled from Cataract Dam, consistent with the expectation that fish might move unassisted downstream but not upstream (Fig. 2).

PCoA analysis of genotypes for Cataract Dam (N = 56) and Cataract River (N = 51) using a 370 loci, and b 2777 loci, respectively. Pie charts on scatterplot indicate assignment to HNB nuclear lineage (black) and MDB nuclear lineage (white) by STRUCTURE. Cataract River fish assigned to the MDB mitolineage have a blue outline (N = 20) and fish assigned to the HNB mitolineage have a red outline (N = 31). Cataract Dam fish have a black outline as their mitolineage was not assigned. MDB Murray-Darling Basin; HNB Hawkesbury-Nepean Basin
Fish sampled from Cataract River spanned much of the possible range of genotypic membership to the Cataract River STRUCTURE cluster (QR): values ranged from 0.056 to 1 (mean = 0.842, SD = 0.222) for 370 loci, and 0.019 to 0.696 (mean = 0.437, SD = 0.126) for 2777 loci (Fig. 3). Similar distributions were seen for fish bearing each mitolineage: for the MDB mitolineage QR values were 0.056–0.998 (mean = 0.781, SD = 0.299) for 370 loci (0.019–0.546, mean = 0.396, SD = 0.155 for 2777), and for fish bearing the HNB mitolineage QR values were 0.494–1 (mean = 0.882, SD = 0.145) for 370 loci (0.238–0.696, mean = 0.464, SD = 0.098 for 2777). These results strongly support that escaped Cataract Dam fish have admixed approximately freely with indigenous Cataract River fish in the Cataract River, with hybridization similarly bidirectional with respect to matrilines and patrilines of mating fish. Two fish sampled in Cataract River belonged strongly to the MDB lineage, with MDB mitolineage and Cataract Dam STRUCTURE cluster (QD) = 0.933 and 0.944 for the 370 loci (0.979 and 0.981 for the 2777 loci) suggesting that these might be the product of extensive backcrossing to the MDB lineage and one was indeed identified as a F1 × Cataract Dam backcross by NEWHYBRIDS (see results below; Fig. 3). It is unlikely that these two individuals are Dam escapees because all Cataract Dam fish were assigned completely to the Cataract Dam cluster (Online Appendix Table 1).

Results of STRUCTURE analysis, built from a 370 loci most useful at distinguishing between Cataract Dam (N = 56) and Cataract River (N = 51) populations and b 2777 loci (Cataract Dam N = 56 and Cataract River N = 51). Membership of each individual to River (black, QR) cluster is indicated by histogram bars. River individuals identified as being part of the MDB mitolineage are highlighted in blue (N = 14), fish with HNB mitolineage are highlighted in red (N = 26). Fish assigned as F1 × Cataract Dam, F1 × Cataract River and F2 in NEWHYBRIDS are shown in detail in inset graphs (QR-values and mitolineage (MDB = blue; HNB = red; N = 10). MDB Murray-Darling Basin; HNB Hawkesbury-Nepean Basin; Dam Cataract Dam; River Cataract River
Hybrid categorization
Among the River individuals, two fish (one of each mitolineage) were identified with quantified support in NEWHYBRIDS as F2, one as F1 × Cataract Dam backcross and seven as F1 × River backcrosses (Fig. 3, Online Appendix Table 1). Of these ten fish, five were of the MDB and five of the HNB mitolineage. The detection of the F2s of both mitolineages, the presence of F1 × Cataract Dam and F1 × Cataract River backcrossed individuals, and the fact that the latter were similarly distributed between the two mitolineages are all consistent with the STRUCTURE results that F1 offspring can successfully breed with other MDB- and HNB-mitolineage fish with little apparent sex-asymmetry.
Discussion
The serendipitous field experiment of Macquarie perch in the Cataract River offered a rare opportunity to infer multigenerational genetic mixing in the wild between two lineages of a threatened species separated for > 100,000 years. Despite the evolutionary separation of the parental lineages well in excess of precautionary guidelines for management for gene-pool mixing of lineages evolved in different environments (Frankham et al. 2011), Cataract Dam fish were shown to have admixed relatively freely with indigenous Cataract River fish in the Cataract River. We found no evidence for genomic barriers to interbreeding between the MDB and HNB lineages of Macquarie perch in the Cataract River. Bearers of both mitolineages in the Cataract River showed quite continuous distributions of genome-wide membership to the nuclear genotypic lineage characteristic of Cataract Dam. Specifically, hybridisation beyond F1 was supported by the inference of an array of backcrossed and F2 individuals. Taken together, these data suggest that hybridisation between the two lineages has been extensive, without obvious impediment, and with little or no sex-asymmetry with respect to membership of parental lineages. While successful interbreeding has been recorded for fishes with longer times of divergence than the two lineages investigated in our study (e.g., viable F2 hybrids were produced between cichlid species that diverged > 2 million years ago; Stelkens et al. 2015), few studies have been conducted to investigate the effects of multigenerational mixing between divergent lineages of threatened fish species of well-estimated divergence times, and the associated implications for conservation. In the present test case, lineages diverged in contrasting environments for as much as 385,000 years, showing no evidence of strong restriction to inbreeding in either direction. This far exceeds Frankham et al. (2011)’s precautionary guideline of safe separation (< 500 years), highlighting the value of applying evidence-based risk–benefit analysis when considering genetic mixing of lineages.
Our analyses showed that mixing populations between the two long-separated lineages (MDB and HNB) did not lead to noticeable outbreeding depression, because we detected successful interbreeding across multiple generations with no indication of sex-asymmetry in direction. This is in line with assertions that the risks of outbreeding depression might often be overestimated, given that it is uncommon, often transient or mild when it occurs, and can often be anticipated (Frankham et al. 2011; Ralls et al. 2018). Numerous studies have shown increased genetic diversity, improved fitness or increased population growth after mixing of populations (e.g., Johnson et al. 2010; Robinson et al. 2017). Inbreeding depression is virtually ubiquitous in species that usually outbreed, and the levels of inbreeding at which it occurs are relatively well understood (Frankham et al. 2019). Outbreeding depression is less well-studied and is more challenging to directly assess so is typically assessed via proxies, and is rarely evaluated even when data are available (Liddell et al. 2021). Despite strong evidence for the benefits of gene-pool mixing, it has been relatively underutilized in conservation management, owing to fears of negative consequences of genetic admixture (Frankham et al. 2019). While these risks need to be weighed against potential benefits, this is rarely done; instead the default position is often to design management regimes for each separately, which may inadvertently result in the loss of potential benefits and increase the extinction risk for threatened populations (Liddell et al. 2021).
For the Macquarie perch, genetically-informed population viability models suggested that the risk of demographically harmful inbreeding depression was high for many populations, in the absence of management actions to increase genetic variation (Pavlova et al. 2017a). Based on a thorough risk assessment, the risk of outbreeding depression was estimated to be low if populations within the MDB were mixed. Accordingly, attempts at genetic augmentation for conservation of this threatened species are underway. One such attempt has yielded an empirical test of the effects of genetic augmentation in the wild, finding that admixture of multiple MDB source populations of Macquarie perch improved survival and reproduction when reintroducing an extinct population (Lutz et al. 2021). In that case, non-risky source populations, i.e. two MDB populations, were available, and so the favourable outcomes are perhaps unsurprising.
In the present study, we used time of isolated evolution in different environments as a proxy to estimate the risk of outbreeding depression experienced by admixed populations (Frankham et al. 2011). Our results suggest that for Macquarie perch, even divergence > 100,000 years did not lead to obvious indications of harmful outbreeding depression following secondary contact. This result is consistent with meta-analysed outcomes of human-induced interspecific hybridization showing that strongly negative effects were rare (Ottenburghs 2021). Accumulating empirical cases such as the present one will be helpful for validating the assumptions underlying the risk assessment process for outbreeding depression initiated by Frankham et al. (2011). However, we surveyed only a small part of the whole genome of the Macquarie perch and might therefore have missed some signals of genomic incompatibilities between the two lineages. We tried to offset these limitations by using a reasonable number of SNPs and a selective set of loci chosen to differentiate River and Dam fish, furthermore ensuring that the sample was balanced to avoid biasing our results when investigating the genomic structure of the populations (Toyama et al. 2020).
Concerns are sometimes raised in the literature that assessment of genetic benefits of crossing populations tend to be over a limited number of generations, and that negative fitness consequences might take some generations to accrue (Frankham et al. 2011; Frankham 2016). In the present case, Macquarie perch separated for > 100,000 years are likely to have been interbreeding for ~ 7 to 14 generations—there is little reason to suppose that overtopping and releases of water from Cataract Dam did not occur from shortly after the introduction of the MBD fish in 1915. We were able to statistically identify individuals as F2s and backcrosses; in addition, the continuous distributions of STRUCTURE memberships across most of the range of possible values, and the identification of two highly-backcrossed individuals, suggest that admixture is many generations deep. This concurs with a meta-analysis showing that genetic admixture effects are usually enduringly beneficial beyond the F3 generation (Frankham 2016), several experimental studies, and observations from hybridization studies including of well-diverged species adapted to different environments (Mitchell et al. 2019; Lamichhaney et al. 2020; Ottenburghs 2021).
It is possible that the prevalence of Dam fish and their offspring in the Cataract River is partially due to the less extreme environmental and climatic conditions in the HNB compared to the harsher conditions the MDB fish have been adapted to (Byrne et al. 2011; Pavlova et al. 2017b). Due to these warmer and more stable conditions, HNB Macquarie perch may experience relaxed purifying selection and accumulate slightly deleterious mutations at a higher rate than do MDB fish, potentially putting HNB fish at a competitive disadvantage (Pavlova et al. 2017b), as is thought to be the case with Lake Dartmouth fish in the Ovens River (Lutz et al. 2021). It is likely the HNB lineage Macquarie perch inhabited the upper Cataract River before the construction of Cataract Dam, but has either gone extinct or been outcompeted by MDB fish after translocations, given no HNB alleles were detected in the Cataract Dam fish. We were unable to source any ‘pure’ Cataract River fish for our study because none are known to be isolated from the likely mixing of the Cataract Dam and Cataract River populations in the Cataract River following the release of Macquarie perch into Cataract Dam in the early twentieth century, and other coastal basin populations would not be suitable proxies because they are differentiated strongly by genetic drift (Pavlova et al. 2017a). The hybridisation seems to be confined to the Cataract River, given that no introgression of MDB alleles has been detected in Macquarie perch in Cordeaux Dam and Wongawilli Creek, other tributaries of the upper Nepean River, connected with the lower Cataract River by fish passages (Faulks et al. 2010, 2011).
Conclusions
This study adds to the growing body of evidence that mixing populations adapted to different environments that have been separated for many generations may not be detrimental and can be an effective conservation management tool (Weeks et al. 2017; Fitzpatrick et al. 2020; Ottenburghs 2021). Careful planning and monitoring of genetic conservation actions can alleviate many of the risks thought to be associated with augmented gene flow even between long-separated populations (Frankham et al. 2017; Love Stowell et al. 2017). While we do not currently advocate crossing Macquarie perch between basins, since sufficient sources are available for within-basin crosses, future conditions such as continuing loss of populations and considering climate-preparedness may call for different recommendations. Instead, this study presents a useful calibration point: two morphologically different lineages estimated to have evolved in different habitats for 119,000–385,000 years can successfully interbreed.
Data availability
Genotypic data for this study are available at Bridges research repository at https://doi.org/10.26180/16553151.
Code availability
Not applicable.
References
Anderson E, Thompson E (2002) A model-based method for identifying species hybrids using multilocus genetic data. Genetics 160:1217–1229. https://doi.org/10.1093/genetics/160.3.1217
Byrne M, Steane DA, Joseph L, Yeates DK, Jordan GJ, Crayn D, Aplin K, Cantrill DJ, Cook LG, Crisp MD (2011) Decline of a biome: evolution, contraction, fragmentation, extinction and invasion of the Australian mesic zone biota. J Biogeogr 38:1635–1656. https://doi.org/10.1111/j.1365-2699.2011.02535.x
Cadwallader PL (1978) Some causes of the decline in range and abundance of native fish in the Murray-Darling river system. Proc R Soc Vic 90:211–224
Cadwallader P (1981) Past and present distributions and translocations of Macquarie perch Macquaria australasica (Pisces: Percichthyidae), with particular reference to Victoria. Proc R Soc Vic 93:23–30
Chan WY, Hoffmann AA, van Oppen MJ (2019) Hybridization as a conservation management tool. Conserv Lett 12:e12652. https://doi.org/10.1111/conl.12652
Dufty S (1986) Genetic and morphological divergence between populations of Macquarie perch (Macquaria australasica) east and west of the Great Dividing Range. Unpublished B.Sc.(Hons) Thesis, University of New South Wales, Kensington
Faulks LK, Gilligan DM, Beheregaray LB (2010) Evolution and maintenance of divergent lineages in an endangered freshwater fish, Macquaria australasica. Conserv Genet 11:921–934. https://doi.org/10.1007/s10592-009-9936-7
Faulks LK, Gilligan DM, Beheregaray LB (2011) The role of anthropogenic vs. natural in-stream structures in determining connectivity and genetic diversity in an endangered freshwater fish, Macquarie perch (Macquaria australasica). Evol Appl 4:589–601. https://doi.org/10.1111/j.1752-4571.2011.00183.x
Fitzpatrick SW, Bradburd GS, Kremer CT, Salerno PE, Angeloni LM, Funk WC (2020) Genomic and fitness consequences of genetic rescue in wild populations. Curr Biol 30:517-522.e515. https://doi.org/10.1016/j.cub.2019.11.062
Frankham R (2015) Genetic rescue of small inbred populations: meta-analysis reveals large and consistent benefits of gene flow. Mol Ecol 24:2610–2618. https://doi.org/10.1111/mec.13139
Frankham R (2016) Genetic rescue benefits persist to at least the F3 generation, based on a meta-analysis. Biol Conserv 195:33–36. https://doi.org/10.1016/j.biocon.2015.12.038
Frankham R, Ballou JD, Eldridge MDB, Lacy RC, Ralls K, Dudash MR, Fenster CB (2011) Predicting the probability of outbreeding depression. Conserv Biol 25:465–475. https://doi.org/10.1111/j.1523-1739.2011.01662.x
Frankham R, Ballou JD, Ralls K, Eldridge MDB, Dudash MR, Fenster CB, Lacy RC, Sunnucks P (2017) Genetic management of fragmented animal and plant populations. Oxford University Press, Oxford
Frankham R, Ballou JD, Ralls K, Eldridge MDB, Dudash MR, Fenster CB, Lacy RC, Sunnucks P (2019) A practical guide for genetic management of fragmented animal and plant populations. Oxford University Press, Oxford
Georges A (2018) gl.pcoa: PCoA ordination (glPca) In: Gruber B, Georges A, Unmack PJ, Clark LV, Berry O (eds) Importing and analysing SNP and silicodart data generated by genome-wide restriction fragment analysis. pp 68–70. https://cran.r-project.org/web/packages/dartR/dartR.pdf
Gruber B, Unmack PJ, Berry OF, Georges A (2018) dartr: an r package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Mol Ecol Resour 18:691–699. https://doi.org/10.1111/1755-0998.12745
Harrisson KA, Pavlova A, Telonis-Scott M, Sunnucks P (2014) Using genomics to characterize evolutionary potential for conservation of wild populations. Evol Appl 7:1008–1025. https://doi.org/10.1111/eva.12149
Hoffmann AA, Miller AD, Weeks AR (2021) Genetic mixing for population management: from genetic rescue to provenancing. Evol Appl 14:634–652. https://doi.org/10.1111/eva.13154
Ingram B, Barlow C, Burchmore J, Gooley G, Rowland S, Sanger A (1990) Threatened native freshwater fishes in Australia—some case histories. J Fish Biol 37:175–182. https://doi.org/10.1111/j.1095-8649.1990.tb05033.x
Ingram BA, Douglas JW, Lintermans M (2000) Threatened fishes of the world: Macquaria australasica Cuvier, 1830 (Percichthyidae). Environ Biol Fish 59:68–68. https://doi.org/10.1023/A:1007669423532
Johnson WE, Onorato DP, Roelke ME, Land ED, Cunningham M, Belden RC, McBride R, Jansen D, Lotz M, Shindle D, Howard J, Wildt DE, Penfold LM, Hostetler JA, Oli MK, O’Brien SJ (2010) Genetic restoration of the Florida panther. Science 329:1641–1645. https://doi.org/10.1126/science.1192891
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191. https://doi.org/10.1111/1755-0998.12387
Kronenberger J, Funk W, Smith J, Fitzpatrick S, Angeloni L, Broder E, Ruell E (2017) Testing the demographic effects of divergent immigrants on small populations of Trinidadian guppies. Anim Conserv 20:3–11. https://doi.org/10.1111/acv.12286
Lamichhaney S, Han F, Webster MT, Grant BR, Grant PR, Andersson L (2020) Female-biased gene flow between two species of Darwin’s finches. Nat Ecol Evol 4:979–986. https://doi.org/10.1038/s41559-020-1183-9
Legislative Assembly of New South Wales (1916) Report on the fisheries of New South Wales for the year 1915
Liddell E, Sunnucks P, Cook CN (2021) To mix or not to mix gene pools for threatened species management? Few studies use genetic data to examine the risks of both actions, but failing to do so leads disproportionately to recommendations for separate management. Biol Conserv 256:109072. https://doi.org/10.1016/j.biocon.2021.109072
Lintermans M (2007) Fishes of the Murray-Darling Basin: an introductory guide. Murray-Darling Basin Commission, Canberra
Lintermans M (2012) Managing potential impacts of reservoir enlargement on threatened Macquaria australasica and Gadopsis bispinosus in southeastern Australia. Endanger Species Res 16:1–16. https://doi.org/10.3354/esr00382
Lintermans M, Pearce L, Tonkin Z, Bruce A, Gilligan D (2019) Macquaria australasica (errata version published in 2020). The IUCN Red List of Threatened Species 2019:e.T12581A167651778. Accessed 9 Aug 2021
Love Stowell SM, Pinzone CA, Martin AP (2017) Overcoming barriers to active interventions for genetic diversity. Biodivers Conserv 26:1753–1765. https://doi.org/10.1007/s10531-017-1330-z
Lutz ML, Tonkin Z, Yen JD, Johnson G, Ingram BA, Sharley J, Lyon J, Chapple DG, Sunnucks P, Pavlova A (2021) Using multiple sources during reintroduction of a locally extinct population benefits survival and reproduction of an endangered freshwater fish. Evol Appl 14:950–964. https://doi.org/10.1111/eva.13173
Mitchell N, Owens GL, Hovick SM, Rieseberg LH, Whitney KD (2019) Hybridization speeds adaptive evolution in an eight-year field experiment. Sci Rep-UK 9:1–12. https://doi.org/10.1038/s41598-019-43119-4
Ottenburghs J (2021) The genic view of hybridization in the anthropocene. Evol Appl. https://doi.org/10.1111/eva.13223
Pavlova A, Beheregaray LB, Coleman R, Gilligan D, Harrisson KA, Ingram BA, Kearns J, Lamb AM, Lintermans M, Lyon JP, Nguyen T, Sasaki M, Tonkin Z, Yen JDL, Sunnucks P (2017a) Severe consequences of habitat fragmentation on genetic diversity of an endangered Australian freshwater fish: a call for assisted gene flow. Evol Appl 10:531–550. https://doi.org/10.1111/eva.12484
Pavlova A, Gan HM, Lee YP, Austin CM, Gilligan D, Lintermans M, Sunnucks P (2017b) Purifying selection and genetic drift shaped Pleistocene evolution of the mitochondrial genome in an endangered Australian freshwater fish. Heredity 118:466–476. https://doi.org/10.1038/hdy.2016.120
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1093/genetics/155.2.945
R Core Team (2019) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/
Ralls K, Ballou JD, Dudash MR, Eldridge MD, Fenster CB, Lacy RC, Sunnucks P, Frankham R (2018) Call for a paradigm shift in the genetic management of fragmented populations. Conserv Lett 11:e12412. https://doi.org/10.1111/conl.12412
Ralls K, Sunnucks P, Lacy RC, Frankham R (2020) Genetic rescue: a critique of the evidence supports maximizing genetic diversity rather than minimizing the introduction of putatively harmful genetic variation. Biol Conserv 251:108784. https://doi.org/10.1016/j.biocon.2020.108784
Robinson ZL, Coombs JA, Hudy M, Nislow KH, Letcher BH, Whiteley AR (2017) Experimental test of genetic rescue in isolated populations of brook trout. Mol Ecol 26:4418–4433. https://doi.org/10.1111/mec.14225
Stelkens RB, Schmid C, Seehausen O (2015) Hybrid breakdown in cichlid fish. PLoS ONE 10(5):e0127207
Toyama KS, Crochet PA, Leblois R (2020) Sampling schemes and drift can bias admixture proportions inferred by structure. Mol Ecol Resour 20:1769–1785. https://doi.org/10.1111/1755-0998.13234
Trueman WT (2011) True tales of the trout cod: river histories of the Murray-Darling Basin. MDBA Publication No. 215/11
Weeks AR, Sgro CM, Young AG, Frankham R, Mitchell NJ, Miller KA, Byrne M, Coates DJ, Eldridge MD, Sunnucks P, Breed MF, James EA, Hoffmann AA (2011) Assessing the benefits and risks of translocations in changing environments: a genetic perspective. Evol Appl 4:709–725. https://doi.org/10.1111/j.1752-4571.2011.00192.x
Weeks AR, Moro D, Thavornkanlapachai R, Taylor HR, White NE, Weiser EL, Heinze D (2015) Conserving and enhancing genetic diversity in translocation programs. In: Armstrong D, Hayward M, Moro D, Seddon PJ (eds) Advances in reintroduction biology of Australian and New Zealand fauna. CSIRO Publishing, Melbourne, pp 127–140
Weeks AR, Heinze D, Perrin L, Stoklosa J, Hoffmann AA, Rooyen A, Kelly T, Mansergh I (2017) Genetic rescue increases fitness and aids rapid recovery of an endangered marsupial population. Nat Commun 8:1071. https://doi.org/10.1038/s41467-017-01182-3
Wells ZR, Bernos TA, Yates MC, Fraser DJ (2019) Genetic rescue insights from population-and family-level hybridization effects in brook trout. Conserv Genet 20:851–863. https://doi.org/10.1007/s10592-019-01179-z
Acknowledgements
MLL was supported by Monash Graduate and International Postgraduate Research Scholarships. This work was carried out under the Research Permit P01/0059 and Animal Ethics authority NSW Fisheries ACEC 05/06 and Ethics. We thank Maria Roitman, Anna Polesskiy and Alice Sunnucks for laboratory assistance.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. This work was supported by the Australian Research Council Grants LP110200017 and LP160100482 with Partner Organizations Department of Environment, Land, Water and Planning, State Government of Victoria; Environment, Planning and Sustainable Development Directorate (ACT Government); Department of Biodiversity, Conservation and Attractions; Zoos Victoria; Diversity Arrays Technology, Icon Water (formerly ACTEW Corporation), Melbourne Water, and Fisheries Victoria (now within DEDJTR—Department of Economic Development, Jobs, Transport and Resources). Additional funding was provided by Holsworth Wildlife Research Endowment—Equity Trustees Charitable Foundation & the Ecological Society of Australia (to MLL) and NSW DPI-Fisheries (to DG). MLL was supported by Monash Graduate and International Postgraduate Research Scholarships.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors have not disclosed any competing interests.
Ethical approval
This work was carried out under the Research Permit P01/0059 and Animal Ethics authority NSW Fisheries ACEC 05/06 and Ethics.
Consent to participate
Not applicable,
Consent for publication
All authors have read, commented on and approved the manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
10592_2022_1476_MOESM1_ESM.docx
Supplementary file1 (DOCX 45 kb)—Online Appendix Table 1: Results of PCoA and STRUCTURE analysis built from 370 loci thought to be most useful at distinguishing between Cataract Dam (N = 56) and Cataract River (N = 51) populations and 2777 loci. Values shown are for QRiver only. Results of NEWHYBRIDS analysis built from 370 loci potentially most useful at distinguishing between genotypic groups characteristic of Cataract Dam (N = 56) vs Cataract River (N = 51). F2, F1 × Cataract Dam and F1 × Cataract River values are highlighted in bold for individuals that were assigned to a hybrid class with probability > 0.5. HNB Hawkesbury-Nepean Basin; MDB Murray-Darling Basin; Dam Cataract Dam; River Cataract River.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lutz, M.L., Sunnucks, P., Chapple, D.G. et al. Strong bidirectional gene flow between fish lineages separated for over 100,000 years. Conserv Genet 23, 1105–1113 (2022). https://doi.org/10.1007/s10592-022-01476-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-022-01476-0