
Abstract
Transboundary connectivity is a key component when conserving and managing animal species that require large areas to maintain viable population sizes. Wolves Canis lupus recolonized the Scandinavian Peninsula in the early 1980s. The population is geographically isolated and relies on immigration to not lose genetic diversity and to maintain long term viability. In this study we address (1) to what extent the genetic diversity among Scandinavian wolves has recovered during 30 years since its foundation in relation to the source populations in Finland and Russia, (2) if immigration has occurred from both Finland and Russia, two countries with very different wolf management and legislative obligations to ensure long term viability of wolves, and (3) if immigrants can be assumed to be unrelated. Using 26 microsatellite loci we found that although the genetic diversity increased among Scandinavian wolves (n = 143), it has not reached the same levels found in Finland (n = 25) or in Russia (n = 19). Low genetic differentiation between Finnish and Russian wolves, complicated our ability to determine the origin of immigrant wolves (n = 20) with respect to nationality. Nevertheless, based on differences in allelic richness and private allelic richness between the two countries, results supported the occurrence of immigration from both countries. A priori assumptions that immigrants are unrelated is non-advisable, since 5.8% of the pair-wise analyzed immigrants were closely related. To maintain long term viability of wolves in Northern Europe, this study highlights the potential and need for management actions that facilitate transboundary dispersal.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
While many large carnivore species suffer from population decline there are also species that have recolonized parts of their historic distribution, especially in Europe and North America (Ripple et al. 2014; Chapron et al. 2014). However, the conservation and management of large carnivores is challenging as most of these species require large amounts of food and extensive space, often leading to intense conflict among different interest groups in society and the management authorities (Carbone et al. 1999; Cardillo et al. 2004; Cardillo 2005). As such, it is likely that many carnivore populations will remain small and semi-isolated, which is expected to negatively affect their long-term viability (Kenney et al. 2014). From a conservation perspective it follows that population connectivity is highly important, not least when populations are small and recently founded. Immigration of unrelated individuals counteracts genetic drift, which is often accompanied with higher levels of inbreeding (Wright 1931; Nei et al. 1975; Allendorf 1986), increased genetic load, and reduced evolutionary potential (Hedrick 2001; Allendorf and Ryman 2002).
In Europe, the majority of large carnivore populations are transboundary (Linnell et al. 2008; Chapron et al. 2014). Many of these countries are obligated to ensure the long term viability of their large carnivore populations in accordance with the European Union Habitats Directive (Council Directive 92/43/EEC) and/or the Bern convention; these countries are henceforth referred to as the member states. Despite these legally binding documents it is hard or even impossible for some member states to reach and maintain population sizes that alone can be considered large enough to have long term viability. Several countries also practice lethal control to limit population sizes and distributions in order to mitigate human conflicts around carnivores, which in turn may reduce connectivity with neighboring populations and the potential for viability (Linnell et al. 2008). This gives member states the incentive to facilitate large carnivore transboundary movement and thus ensuring the connectivity between populations (Hindrikson et al. 2017). Moreover, member states also need to consider how population viability is affected by populations in non-member states (i.e. countries that are not obliged under the Bern convention or European Union Habitats Directive) or in member states where the carnivores have different legal status (Trouwborst 2018).
Wolf populations in Northern Europe currently consist of 450 individuals on the Scandinavian Peninsula, i.e. Norway and Sweden (Wabakken et al. 2020) and approximately 750 wolves in Finland and north-western Russia, including the Russian oblasts of Murmansk and Karelia in Russia, hereafter called the Finnish-Karelian wolf population (Linnell et al. 2008; Laikre et al. 2016). These two populations are separated by a 350 km wide land bridge, where the national policies of Norway, Sweden and Finland for wolf presence are restricted, in favor of human semi-domesticated reindeer herding. The population in Finland and north-western Russia is connected to a more or less continuous population of about 3600 wolves covering Estonia, Latvia, Lithuania, Poland and the Russian oblasts of Leningrad, Novgorod, Pskov, Tver, Smolensk, Bryansk, Moscow, Kaliningrad, Kaluzh, Tula, Kursk, Belgorod and Orel, which in turn are part of Russia’s 52,000 wolves (Linnell et al. 2008; Bragina et al. 2015; Hindrikson et al. 2017).
Although wolves are capable to disperse distances over 1000 km (see Wabakken et al. 2007), and individual wolves are observed almost annually in northernmost Scandinavia (Seddon et al. 2006, Fig. 1), the genetic exchange between the Scandinavian and the Finnish-Karelian populations is limited, mainly due to a high rate of human-caused mortality of wolves in northern Sweden and Finland (Kojola et al. 2009; Liberg et al. 2012b). Recent studies also show indications of decreasing connectivity between Finland and Russia as well as a reduced effective population size in Finland (Aspi et al. 2006, 2009; Jansson et al. 2012), which in turn may have a negative effect on the immigration rate to Scandinavia (Kojola et al. 2009).

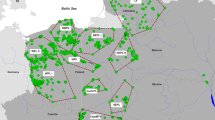
Last known position and individual identity of 20 Scandinavian immigrants (blue dots) and reference individuals (blue triangles). For some Russian samples (with number within brackets), the average location of the site for sampling is given. The grey squares indicated areas of permanent wolf occurrence (excluding the hatched area) in accordance with Chapron et al. (2014)
In Scandinavia wolves were considered functionally extinct by 1966 and the present population is traced back to a founder event in south central Scandinavia in 1983, including only a single pair of Finnish-Russian immigrants (Wabakken et al. 2001; Vilá et al. 2003). Since then and until 2020, seven more immigrants have managed to reproduce, and four of these have produced offspring that subsequently have reproduced successfully in Scandinavia (Seddon et al. 2006; Åkesson et al. 2016; Åkesson and Svensson 2020). Consequently, the Scandinavian wolf population is highly inbred (Åkesson et al. 2016; Kardos et al. 2018) and has shown several signs of inbreeding depression (Liberg et al. 2005; Bensch et al. 2006; Åkesson et al. 2016).
During the last two decades, the Swedish and Norwegian governments have commissioned several investigations focusing on the viability of the Scandinavian wolf population and the genetic effect of immigration (e.g. Hansen et al. 2011; Bruford 2015; Naturvårdsverket 2015, 2016; Laikre et al. 2016; Eriksen et al. 2020). These investigations have clarified that a prerequisite for the Scandinavian wolf population to maintain long-term viability is that it needs to be a part of a larger ‘functional metapopulation’ in North Europe (Mills and Feltner 2015; Laikre et al. 2016). This resulted in a management goal for the population to have at least one reproducing immigrant every 5-year period, regarded as the generation time of Scandinavian wolves (Naturvårdsverket 2016). The range of the larger ‘functional metapopulation’ is however less clearly described, and although Scandinavian immigrants have been studied with regard to population origin (Ellegren et al. 1996; Sundqvist et al. 2001; Flagstad et al. 2003; Vilá et al. 2003; Seddon et al. 2006; Smeds et al. 2019), little is known about the immigration from specific member states, like Finland and non-member states, like Russia. In order to meet the one-immigrant-per-generation goal, the cooperation to monitor and facilitate gene flow of wolves with neighboring countries has intensified during the last years. This includes the ambition to learn more about the movements of wolves between Scandinavia, Finland and Russia.
In this study we address three major hypotheses: (1) The temporal variation in the genetic diversity among wolves in Scandinavia between 1983 and 2014 is explained by the known founder events in the population and by genetic drift during the first decade of the population history when the population consisted of < 30 individuals (Wabakken et al. 2001). (2) The immigrants to Scandinavia are likely to originate from either Finland or the western part of Russia (see Jansson et al. 2012). (3) Under an assumption that the source population of immigrants to Scandinavia is large and panmictic, the immigrants should be largely unrelated to each other. These hypotheses are tested using 26 autosomal microsatellite loci from wolves born in Scandinavia in different cohorts, wolves from Finland and the Russian Republic of Karelia (henceforth called Russia) and wolves that were genetically identified as immigrants in Scandinavia between 1977 and 2012.
Methods
Population and study site
The wolf population in Norway and Sweden (hereafter referred to as Scandinavia) has been monitored since 1978; first based on ground tracking on snow until 1998, and thereafter also by radio telemetry and DNA analysis (Wabakken et al. 2001; Liberg et al. 2012a).
The Scandinavian wolf population is geographically separated from the Finnish-Karelian wolf breeding range (Chapron et al. 2014, Fig. 1) and, until 2015, with a minimum of 800 km distance of land travelling between the two breeding areas (Wabakken et al. 2007). This area largely matches the Fennoscandian reindeer herding ground, where DNA-identified wolves typically have been legally killed after conflict with the reindeer husbandry, poached or disappeared for unknown reasons without being identified again. In order to facilitate gene flow, the Scandinavian management authorities captured and translocated three immigrant wolves during the study period (1977–2014) from reindeer herding grounds southwards to the Scandinavian wolf breeding range.
Sampling and extraction
From 1977 to 2014, the genetic monitoring of the wolves on the Scandinavian peninsula involved the DNA-sampling of wolves, where Scandinavian origin was originally tested using Bayesian individual assignment (Rannala and Mountain 1997) and parental assignment (see Åkesson et al. 2016 for detailed description).
The study was based on DNA-samples from 20 immigrant wolves (thus wolves detected in Scandinavia that did not originate from the Scandinavian population), 143 invasively collected Scandinavian born wolves, 64 invasive tissue samples from Finnish wolves, skin tissue from 19 unrelated Russian wolves (see below) and 27 buccal samples from dogs C. familiaris. The Scandinavian reference wolves were grouped into nine cohort classes [1983–1990 (n = 6), 1991–1993 (n = 8), 1994–1996 (n = 9), 1997–1999 (n = 20), 2000–2002 (n = 20), 2003–2005 (n = 20), 2006–2008 (n = 20), 2009–2011 (n = 20), 2012–2014 (n = 20)].
Microsatellite genotyping
DNA samples were genotyped for 26 microsatellite loci (Online resource 2), located in the autosomal genome: CXX.20, CXX.123, CXX.204, CXX.225, CXX.250, CXX.253 (Ostrander et al. 1993), 2001, 2006, 2010, 2054, 2079, 2096, 2137, 2140, 2168, 2201 (Francisco et al. 1996), (AHT)002, (AHT)004, (AHT)101 (Holmes et al. 1993), AHT103, AHT119, AHT121, AHT138 (Holmes et al. 1995), PEZ03, PEZ06 (Neff et al. 1999), vWF (Shibuya et al. 1994). The markers were amplified using PCR followed by capillary electrophoresis in accordance with Åkesson et al. (2016), and using the same precautions when genotyping non-invasive samples. The microsatellite loci were regularly used to monitor of the wolves in Scandinavia, to reconstruct the pedigree of the population and study how the variation on these markers are associated with individual fitness (Liberg et al. 2005; Bensch et al. 2006; Åkesson et al. 2016).
Genetic analyses
Immigrant detection
Immigrants were detected using Bayesian individual assignment in the program Geneclass 2 (Piry et al. 2004) using nine cohort classes of Scandinavian wolves, unrelated Finnish wolves (see below), unrelated Russian wolves (see below) and dogs as reference populations (Online resource 1: Table 1, Table 2). This involved likelihood estimation of assignment using Bayesian criteria (Rannala and Mountain 1997) and an exclusion test (with α < 0.05) of the likelihood values based on Monte Carlo resampling to approximate the distribution of genotype likelihoods that would be found in the reference populations (Paetkau et al. 2004). The exclusion of Scandinavian origin were based on the cohort class of Scandinavian wolves prior to the year when the immigrant was first observed.
Apart from the DNA-sampled immigrants, two more founders (G1–83 and G1–91) of the Scandinavian population were breeding during the study period (Vilá et al. 2003; Liberg et al. 2005). The genotypes from these first two males of the Scandinavian wolf population was partly reconstructed from known offspring (see Liberg et al. 2005 for more details) but was not used in this study because reconstructed genotypes are heavily biased towards heterozygote loci.
Genetic relatedness
In order to test if wolves were related, we calculated the maximum likelihood estimates of relatedness using the program ML-RELATE (Kalinowski et al. 2006). The existence of close relatives among the immigrants was tested by likelihood ratio tests using the maximum likelihood estimates of a pair being unrelated or being close relatives (i.e. half-sibs, full sibs or parent-offspring). The possible relationships with likelihood values within the 95% confidence interval were calculated in ML-RELATE using 10,000 randomizations to get the sample distributions for the null hypothesis.
Genetic relatedness was tested among immigrants and correlated against the absolute difference in years between observations. The correlation was tested with a Mantel-test using PASSaGE v2 (Rosenberg and Anderson 2011).
To avoid inflated genetic structure due to biased sampling of close relatives among the Finnish and Russian wolves we preceded the genetic analyses with estimating genetic relatedness and randomly omitting individuals from pairs of close relatives (full sibs or parent-offspring) with 95% confidence until none of the individuals were closely related. The average relatedness was 0.045 (range between 0 and 0.65) and 86 of 3741 pairs (2.3%) were found to be close relatives. After randomly omitting individuals with close relatives, the data set was composed of 44 unrelated wolves, including 25 from Finland and 19 from Russia.
Genetic diversity and linkage disequilibrium
Expected (HE) and observed (HO) heterozygosity among immigrants, Finnish, Russian and temporally divided Scandinavian wolves was calculated in Genetix 4.05.2 (Belkhir et al. 2004). Number of alleles (A), allelic richness (AR), and private allelic richness (Π) were retrieved from HP-RARE 1.0, where AR and Π was estimated using rarefaction assuming the minimum number of genes (i.e. two times the number of sampled individuals) from the groups used in the analysis (Kalinowski 2005). When including Scandinavian wolves in the analysis, AR was based on the rarefaction of 12 genes, because sample size in one of the cohort classes was 6 individuals. When comparing immigrant, Finnish and Russian wolves, we based the rarefaction on 26 genes because the lowest sample size at any locus was 13 individuals. The statistical difference of AR and Π between groups was tested using the sign test (see Sokal and Rohlf 1995).
The presence of inbred individuals and close relatives in a sample may cause the estimators of HE to be downwardly biased (DeGiorgio and Rosenberg 2009). To account for this when comparing the temporal samples of the Scandinavian wolves we used an unbiased estimator for heterozygosity (\({\stackrel{\sim }{\mathrm{H}}}_{\mathrm{E}}\)) corrected for the average pedigree-based relatedness (\({\overline{\mathrm{R}} }_{\mathrm{P}}\)) between the individuals in each sample in accordance with DeGiorgio and Rosenberg (2009). The pedigree-based relatedness (\({\mathrm{R}}_{\mathrm{P}}\)) between individuals was calculated using CFC v1.0 (Sargolzaei et al. 2005) based on the reconstructed pedigree of the Scandinavian wolves (Åkesson et al. 2016). For one of the 143 Scandinavian born wolves knowledge of parental origin was missing and we chose to use the average relatedness in the cohort class for all pairs including this individual.
The difference in HE and \({\stackrel{\sim }{\mathrm{H}}}_{\mathrm{E}}\) among immigrants, Finnish wolves, Russian wolves and the temporal samples of Scandinavian wolves were tested using paired t-tests. Inbreeding coefficient (FIS) with 95% confidence intervals from bootstrapping over all loci 1000 times was calculated using Genetix 4.05.2 (Belkhir et al. 2004). Test of linkage disequilibrium between all pairs of loci and overall populations were calculated using 10,000 permutations and 2 randomized initial conditions for the expectation–maximization (EM) algorithm in Arlequin 3.5.1. 2 (Excoffier and Lischer 2010).
We calculated standardized multilocus heterozygosity (stMLH) for all the genetically sampled immigrants as proportion of heterozygous loci divided by mean heterozygosity of typed loci, thus accounting for the variation in the typed loci (Slate et al. 2004).
Genetic differentiation and migration
Pairwise differentiation between populations and between cohort classes of the Scandinavian wolf population was based on θ (Weir and Cockerham 1984), an FST-analogue calculated in FSTAT 2.9.3.2 (Goudet 1995). To account for within-population diversity (Charlesworth 1998; Hedrick 1999) we also calculated θʹ, where θ was divided by θmax, calculated by recoding the alleles using RECODE v. 0.1 so that all populations contains unique alleles and still have the same within population diversity (Meirmans 2012). Pairwise tests of θ were conducted in FSTAT 2.9.3.2 and were based on 1000 randomizations of genotypes and significances after strict Bonferroni corrections. The temporal variation in genetic differentiation between temporal samples in Scandinavia was analyzed by correlating the pairwise θ and θʹ between cohort classes against the temporal distance matrix (i.e. the difference in years between cohort classes) using the average year of the cohorts. The correlation was tested with a Mantel-test based on 10,000 randomizations using PASSaGE v2 (Rosenberg and Anderson 2011).
To estimate the average number of migrants (Nm) between the Finnish, Russian and “immigrant” populations, we used the method of Slatkin (1985) in Genepop (Raymond and Rousset 1995). With this method the frequency of alleles found in only one of the populations, so called private alleles, is used can be used to quantify Nm.
Microsatellite clustering analysis and assignment
Bayesian clustering analysis was conducted using STRUCTURE v2.3.4 (Pritchard et al. 2000) assuming admixture and correlated allele frequencies. No prior information about population origin was used and only reference individuals from Finland and Russia were used to update the allele frequencies. We ran models where the number of assumed populations (K) ranged between 1 and 10, each replicated 10 times. To detect the most likely genetic structure for each K, each run consisted of 200,000 Markov chain Monte Carlo iterations following 100,000 burn-in iterations. The optimal number of clusters (K) was inferred based on the rate of change in the log-likelihood (LnP(D)) and the standard deviation between replicates for each K (Evanno et al. 2005). Individual membership coefficients (q) for the optimal number of clusters (when > 1) were inferred from the 10 replicate runs using the FullSearch algortim in the program CLUMPP v1.1.2 (Jakobsson and Rosenberg 2007).
In order to account for the presence of unsampled populations, we conducted an individual-based population assignment in Geneclass 2, using a similar procedure to that described above but with only unrelated Finnish and unrelated Russian wolves as reference populations, separately and pooled.
To visualize the assignment of immigrants and the reference individuals while accounting for missing data we used Geneplot (McMillan and Fewster 2017). The sample sizes were small in relation to the population sizes, and as recommended we used a prior on allele frequencies defined in Baudouin & Lebrun (2001) and a leave-one-out approach (McMillan and Fewster 2017), where the genotype probability for each individual in the reference populations is based on the posterior distribution of allele frequencies, after the individual has been excluded in the calculation (McMillan and Fewster 2017).
The immigrant sharing of alleles that were private to the Finnish or Russian population, as well as alleles present only among immigrants was extracted from GenAlEx 6.5 (Peakall and Smouse 2012).
Results
Genetic diversity and temporal variation in Scandinavia
The genetic diversity significantly increased after the founder event in 1983 in Scandinavia. Between cohort classes 1983–1990 and 2012–2014 (Table 1) the number of alleles (A) increased at 23 loci while remaining unchanged at 3 loci (sign test, n = 26, p < 0.001). Also, the allelic richness (AR) increased at 19 of 26 loci (sign test, n = 26, p = 0.01) and average heterozygosity (HE) increased from 0.533 to 0.605 (t = 2.23, df = 25, p < 0.001, Fig. 2). During the same period \({\stackrel{\sim }{\mathrm{H}}}_{\mathrm{E}}\) (corrected for average relatedness) did not show any significant change from the first to the last cohort class (t = 0.21, df = 25, p = 0.84, Fig. 2). During the period of no effective immigration, AR decreased, with lower AR at 18 of 26 loci among individuals sampled 2003–2005 compared to 1994–1996 (sign test, n = 26, p = 0.02). Recent immigration of four wolves (2008–2013) led to increased genetic diversity, with a 10% increase in AR and with increasing AR in 22 of 26 loci (sign test, n = 26, p < 0.001). However, no significant change in HE or \({\stackrel{\sim }{\mathrm{H}}}_{\mathrm{E}}\) could be observed between the two last cohort classes (HE: t = 1.61, df = 25, p = 0.12; \({\stackrel{\sim }{\mathrm{H}}}_{\mathrm{E}}\): t = − 1.86, df = 25, p = 0.07).

Mean expected heterozygosity (HE), observed heterozygosity (HO) and average pedigree-based relatedness (\({\overline{\mathrm{R}} }_{\mathrm{P}}\)) and heterozygosity (\({\stackrel{\sim }{\mathrm{H}}}_{\mathrm{E}}\)) corrected for \({\overline{\mathrm{R}} }_{\mathrm{P}}\) among cohort classes in the Scandinavian wolf population
In Scandinavia, several cohort classes were genetically differentiated (Table 2) and the time difference between cohort classes was correlated with both θ (r = 0.58, p = 0.008) and θ’ (r = 0.66, p = 0.007). Even though the genetic diversity increased over time in Scandinavia, the latest cohort class (2012–2014) still had lower genetic diversity than Finland and Russia. Scandinavian wolves in the latest cohort carried on average 2.5 (37%) fewer alleles per locus compared to wolves from Finland (t = 6.01, df = 25, p < 0.001) and 3.15 (42%) fewer than Russian wolves (t = 6.96, df = 25, p < 0.001). The average AR in the latest Scandinavian cohort was 28% and 34% lower than Finland and Russia respectively, with lower AR at all 26 loci compared to Finland (sign test, n = 26, p < 0.001) and for 25 of 26 loci compared to Russia (sign test, n = 26, p < 0.001). Also, HE was substantially lower among Scandinavian wolves (0.605 ± 0.107 S.D.) compared to Finnish (mean difference = − 0.129, t = − 5.57, p < 0.001) and Russian wolves (mean difference = − 0.151, t = − 6.51, p < 0.001).
Incidentally, both founder events in 1991 and 2008 were followed by a decrease in FIS (Table 1), which is in agreement with previous findings of heterozygote advantage in the population after founder events (Bensch et al. 2006; Åkesson et al. 2016).
Also the incidence of linkage disequilibrium (LD) between loci differed between cohort classes (Table 1), where the proportion of locus pairs in linkage disequilibrium among cohort classes increased with decreasing average pedigree based relatedness RP (Pearson r = − 0.86, p = 0.003, n = 9, Table 1).
Genetic structure among Finnish and Russian wolves
Overall, we found very weak signs of genetic differentiation between Finnish and Russian wolves (Table 2). The genetic composition differed somewhat though, with 21 of 26 loci having higher AR in Russia than in Finland (sign test, n = 26, p < 0.001, Table 1). The incidence of LD among the 365 locus pairs was slightly more pronounced in Finland (7.1%) compared to Russia (0.3%). Heterozygosity among Finnish (HE = 0.734 ± 0.018 S.E.) and Russian wolves (HE = 0.756 ± 0.019 S.E.) was not significantly different (t = 1.45, df = 25, p = 0.16) and the genetic differentiation between Finnish and Russian wolves was low and non-significant (θ = 0.02), indicating considerable gene flow between the two countries (Table 2). With a mean frequency of private alleles of 0.045 between Finland and Russia, the estimated average number of reproducing migrants per generation was 2.92. FIS of Finnish and Russian wolves was non-significant, indicating random mating between wolves with respect to relatedness (Table 1). With STRUCTURE assignment, the most parsimonious model with Finnish and Russian wolves consisted of one cluster (K = 1), suggesting no cryptic population structure (Online resource 1: Table 3).
Immigrant wolves to Scandinavia
During the study period (1977–2014) we found DNA in Scandinavia from 20 wolves that was not born in the Scandinavian population (Online resource 1: Table 1). Using Bayesian individual assignment, the probability of assignment to Scandinavian wolves and dogs were < 0.01, demonstrating that they were wolves with non-Scandinavian origin (Online resource 1: Table 1). Invasive samples (blood or tissue) were collected from 14 of the 20 immigrant wolves, while six wolves were sampled 1–9 times non-invasively from fecal material found during snow tracking.
Among the Scandinavian immigrants, the average AR was 5.12, which was higher in 17 of 26 loci (sign test, n = 26, p = 0.047) than Finnish wolves (average AR = 4.82) and non-significantly different (sign test, n = 26, p = 0.12) from Russian wolves (average AR = 5.23). The proportion of loci in LD among immigrants was 18.1%, thus higher than both Russia and Finland (Table 1). The LD was partly explained by the relatedness between some of the immigrants (see below) as the proportion of loci in LD was reduced to 6.4% when using only immigrants that was not closely related (n = 15). The average heterozygosity among immigrants was 0.756 ± 0.016 (S.E.) with FIS being non-significantly different from zero (Table 1). The individual standardized multilocus heterozygosity (stMLH) of immigrants ranged between 0.53 and 1.24 and was not dependent on the year of detection (linear regression; t = − 1.31, df = 19, p = 0.21).
From an individual-based population assignment with a priori determined source populations, we found that immigrants assigned with the Russian population in 8 of 20 cases (Table 3). In three of these cases the probability of Finnish origin was < 0.01. In 10 of 20 cases immigrants assigned better with the Finnish population, but none of these cases with < 0.01 probability of Russian origin. For two individuals (M-09-03 and G52-09) both Finnish and Russian origin was excluded statistically. These two individuals maintained < 0.01 assignment probability when Finnish and Russian wolves were pooled (Online resource 1:Table 4). When visualizing the log10 genotype probabilities (LGP) of assignment to Finland and Russia, the same two immigrants showed profiles below the 1% quantile of both populations (Fig. 3).

Genotype probability for 20 immigrant wolves (green diamonds) to originate from a population in Finland (blue circles) and Russia (red squares) respectively, where the probabilities of the reference individuals are based on the estimation of allele frequencies from leaving the one individual out. Genotypes with missing data on any of the 26 microsatellites are indicated with an asterisk. Equal probability to belong to either population is illustrated from the thick diagonal line, while 9 times the probability to belong to one of the populations is illustrated from the thin diagonal lines. The dashed lines correspond to the 1% and 99% quantiles of Log10 genotype probabilities for the Russian (horizontal lines) and Finnish (vertical lines) wolves respectively
To test if immigrants had admixed origin with regard to the Scandinavian population we extended the STRUCTURE-analysis by including Scandinavian wolves from the cohort class 2003–2005, thus representing the later part of the period before the last two founder events. Two genetic signatures were evident, where the Scandinavian born wolves defined one distinct cluster, and the Finnish and Russian wolves defined the other (Fig. 4A, B, Online resource 1: Table 5). One Russian wolf (V39_RU) showed an admixed genotype, as did some of the immigrants, including the founding female in 1983 (D-85-01) as well as an immigrant (D-77-01) observed before the population establishment. The two latter wolves could not have been truly admixed, as they were sampled before the reestablishment of the Scandinavian population, but carried alleles that later became common in Scandinavia. Also after 2003 some level of admixture with Scandinavian wolves were evident among the immigrants where M-09-03 and G13-04 showed the highest values (Fig. 4C).

Population structure based on analysis in STRUCTURE using 26 microsatellite loci. Based on the replicated modelling of ten scenarios were the assumed number of clusters (K) was set to values 1 to 10, the average likelihood LnP(D) (A) and its standard deviation was used to decide the most parsimonious number of clusters (K = 2). The ancestry of wolves from Finland, Russia and Scandinavian cohort class 2003–2005 when assuming K = 2 is illustrated (B) as well as the ancestry of wolves immigrating to the Scandinavian Peninsula (C). The immigrants are presented chronologically and the dashed line divides immigrants that was first observed before and after 2003
Several alleles carried by the immigrants were private to either Finnish (n = 6) or Russian (n = 15) wolves (Table 3). Noteworthy, 11 alleles (carried by eight immigrants) were present neither in Finland nor Russia. When accounting for sample size, based on the minimum sample size of 26 genes, private allelic richness (Π) was higher in Russia (average Π of 1.51) than in Finland (average Π of 0.77) on 21 of 26 loci (sign test, n = 26, p < 0.001). When including immigrants, the average Π was reduced by 55% in Russia (average Π = 0.83) and 52% in Finland (average Π = 0.41), indicating that the immigrants carried alleles that where private for both Russian and Finnish wolves. In a scenario where immigrants would originate also from other non-sampled and differentiated populations, we would expect that Π would be higher among immigrants compared to Finnish and Russian wolves. The average Π among immigrants was 1.08 and not significantly different among loci (sign test, n = 26, p = 0.15) from Finnish and Russian wolves combined with average Π of 1.06. When treating Finnish and Russian wolves separately, immigrants with average Π of 1.24 had higher Π among loci (sign test, n = 26, p = 0.047) than Finnish wolves while no difference in Π among loci was found between immigrants (average Π of 0.92) and Russian wolves (average Π of 1.27).
The average relatedness among the immigrants was 0.043 (range between 0 and 0.75) and for 11 out of 190 pairs (5.8%) we found indications of close relationships (including half-sib, full-sib and parent–offspring relationships, Online resource 1: Table 6). The pairwise difference in the years of first observation and the pairwise relatedness between immigrant individuals were negatively correlated (r = − 0.246, t = − 3.37, p < 0.001). When close relationship was confirmed statistically the time between first-time observations of the two involved individuals was never more than 3 years (Fig. 5).

Association between pairwise relatedness and the absolute difference in year of first observation between immigrants. Filled circles represent pairs where the most likely relationship was significantly different from the likelihood of the pair being unrelated. The correlation coefficient (− 0.246) was significant using a Mantel test (t = − 3.378, P < 0.001)
Discussion
We analyzed the genetic effect of immigrants in the recently founded wolf population in Scandinavia, where gene flow is central for the population viability (Bruford 2015; Mills and Feltner 2015; Laikre et al. 2016). Overall the genetic diversity increased since recolonization in the early 1980s as a response to the successful reproduction of seven immigrants during the study period (Vilá et al. 2003; Bensch et al. 2006; Åkesson et al. 2016). However, after more than three decades since the first reproduction in 1983, the population had still not reached the levels observed among wolves in the source population of Finland and western Russia. Moreover, there were also indications of allelic loss occurring during a period when there was no immigration. We found no strong indications that immigrants originated from other differentiated populations than those represented by our sampled wolves from Finland and north-western Russia and several immigrants were closely related. Although it may be desirable to distinguish between immigrants originating from Finland and Russia respectively, an accurate assignment of immigrants to either country proved difficult to achieve with 26 microsatellite loci, due to a near absence of genetic differentiation.
Genetic diversity and temporal variation in Scandinavia
Even though at least 20 wolves crossed the border to Scandinavia during the 30-year study period, only seven individuals, including a pair that was translocated together, reproduced in Scandinavia. Supporting our first hypothesis, the genetic diversity increased during this period with increasing heterozygosity and allelic diversity, especially after founder events in 1991 and 2008 (Table 1, Fig. 2).
The genetic differentiation between several cohort classes (Table 2) and the strong correlation with time differences between cohort classes indicated a gradual change in genetic variation in the Scandinavian wolf population, emphasizing the importance of genetic drift. Indeed, during periods of small population size without gene flow from neighboring populations, there was a significant loss of genetic diversity (e.g. 1997–2007; Åkesson et al. 2016). Founder effects were also important to explain the observed patterns. After the founder events in 1991 and 2008, new alleles arrived in the population, followed by an increase in founder representation, leading to higher frequencies of the immigrant alleles (Åkesson et al. 2016). The negative FIS-values on the time periods following the founder events, indicate an excess of observed heterozygote genotypes (Wright 1965), likely resulting from the founder events, but also from the higher breeding success of early descendants to the founders (Bensch et al. 2006; Åkesson et al. 2016). A longer time period passed before Hardy Weinberg (HW) equilibrium was reached after the 1991 founder event compared to the 2008 events. This was likely due to different levels of genetic drift as the population size in 2008 were 15-fold larger than 1991 (Wabakken et al. 2001; Åkesson et al. 2016). The time to reach HW equilibrium may also have been affected by a change in generation time, e.g. due to the increased culling rate and faster turnover of breeding pairs during the study period (Liberg et al. 2020; see also Wikenros et al. 2021).
The founders also seemed to affect the proportion of loci in linkage disequilibrium (LD), as indicated by the strong negative correlation between LD and average relatedness (RP) among individuals in different cohort classes. Decreasing RP and increasing proportion of loci in LD coincided, as expected, with the successful immigrant reproductions in 1991, 2008 and 2013 (Slatkin 2008). Both gene flow and the relatively higher reproductive success of immigrants and their descendants (Åkesson et al 2016) may have caused the temporal variation in LD. It is also possible that other processes in the population caused LD (see Bensch et al. 2006), including genetic drift as the population grew from only one founder pair (Hill and Robertson 1968) and variation in breeding population size due to anthropogenic culling (Milleret et al. 2017; Liberg et al. 2020).
The origin of Scandinavian immigrants
We did not find any indications that Finnish and Russian wolves were genetically differentiated (Table 2). The genetic differentiation θ = 0.017 was lower than previously reported (FST = 0.151; Aspi et al. (2009); FST = 0.086; Jansson et al. (2012)), but this could be explained by the higher overall diversity for the markers used in this study (Charlesworth 1998; Hedrick 1999). The lack of differentiation was supported also from the Bayesian clustering analysis which gave support for only one cluster (Online resource 1: Table 3). Little or no genetic structure suggest considerable gene flow between the two countries. According to the private allele method (Slatkin 1985), Finland received approximately three reproducing immigrants per generation from Russia, similar to the numbers presented in Aspi et al. (2009), which should be sufficient to avoid loss of genetic variation. Still, the allelic diversity was higher among Russian wolves, possibly indicating gene flow from areas surrounding the Russian republic of Karelia.
In this study, we also found support for our second hypothesis that Scandinavian immigrants originate from either Finland or Russia. An individual based assignment test resulted in immigrants confidently assigning with either Finnish or Russian wolves, although the power to distinguish between countries with FST < 0.05 is questionable (Paetkau et al. 2004). While the immigrant group was not significantly differentiated from the Russian wolves (Table 2), Russian origin could be excluded for two (10%) immigrants. Interestingly, these two individuals (G52-09 and M-09-03) did not assign with either Finnish or Russian wolves, which may indicate that the individuals originated from a differentiated population that is not represented in the study, such as northern Karelia, the Kola Peninsula, the Baltic population, or even further east (Fig. 1). A Scandinavian-born female wolf, dispersing from the western parts of the Scandinavian breeding range to the northeastern Finnish-Russian border, travelled more than > 10,000 km during a 9-month period (Wabakken et al. 2007). This illustrates the potential for Scandinavian immigrants also to have dispersed from outside the range of the Finnish-Karelian population included in this study, which is not extending more than 760 km from the Scandinavian border. An alternative explanation to the rejected population assignments of G52-09 and M-09-03 could be that they had admixed origin with respect to the Scandinavian population, e.g. involving the breeding of Scandinavian born and Finnish-Russian wolves outside the breeding range of Scandinavia, giving descendants that immigrated to Scandinavia. We did not find significant results supporting this alternative (Fig. 3), but the power to detect admixture events more than one generation back could be low. Indeed, M-09-03 had an estimated admixture of 0.19 (credible interval: 0.00–0.46) with the cluster dominated by Scandinavian wolves (Fig. 3), supporting the possibility of partial Scandinavian ancestry. In a recent study, Smeds et al. (2020) found three wolves of Scandinavian origin among wolves sampled in Finland, demonstrating that migration between Scandinavia and the neighboring populations to the East indeed is bi-directional.
The immigrants carried alleles that were private for both Russian and Finnish wolves. Also, we found no support for immigrants having higher Π than Finland and Russia combined, as would be expected if a large proportion of the immigrants originated from an unknown differentiated population. This is in agreement with a recent study on ca 1500 Y-chromosome linked SNPs on samples from Finnish, Scandinavian and eight immigrant wolves (all immigrants also represented in this study), showing that the haplotypes found among Scandinavian and immigrant wolves were all represented among Finnish wolves (Smeds et al. 2019). Still, the higher AR and Π among immigrants compared to Finnish wolves alone, would indicate that Finland is not the only origin of immigrants to Scandinavia.
Previous studies have suggested that the immigration rate from Russia to Finland has decreased (Jansson et al. 2012). A decreasing number of immigrants from Russia to Scandinavia was also indicated in our study, where the proportion of detected immigrants that assign with the Russian wolves was 40% during the period 2001–2006 and 17% during the period 2007–2012 (Table 3). Based on a study of 35 dispersing wolves, radio-collared in Finland between 2000 and 2006, none was found to cross the border into Sweden and Norway, partly because of high mortality in the Finnish reindeer management areas (Kojola et al. 2009). However, a few years later the wolf breeding range in Finland has moved closer to the Swedish border (Heikkinen et al. 2020). It is therefore likely that the Scandinavian wolf population now rely more on immigration from Finland rather than from Russia. With lower immigration rate from Russia, we expect effective population size of wolves in the Fennoscandian region to be lower (Laikre et al. 2016). On the other hand, we did not find that stMLH of immigrants decreased significantly with year of detection in Scandinavia, as would be expected from a decreasing proportion of Russian immigrants (Jansson et al. 2012).
Genetic relatedness of Scandinavian immigrants
A long-term decrease in inbreeding among Scandinavian wolves will require immigration from less related individuals. Our present knowledge about the inbreeding status in Scandinavia is based on the assumption that the founders were unrelated (Liberg et al. 2005; Bensch et al. 2006; Åkesson et al. 2016). However, under a scenario where the North European metapopulation becomes smaller and less connected, the inbreeding level and relatedness may need to be accounted for when evaluating the genetic viability of the population (Laikre et al. 2016). If not, inbreeding among Scandinavian wolves is likely to be underestimated (see Kardos et al. 2018). Among the immigrants in this study, 15 of 20 were closely related (half sibs, full-sibs or parent-offspring) with at least one of the other immigrants, thus rejecting our third hypothesis. Expectedly, temporal proximity of first year of observation between two immigrants explained the degree of relatedness, where close relatives tended to be found within the typical generation time for wolves, i.e. < 5 years (Mech et al. 2016). The average relatedness among immigrants that was first observed within 5 years were 0.08 (0.16 S.D.), thus higher than the overall average relatedness of 0.04. In Finland, the average relatedness within cohorts has previously shown to vary between − 0.03 and 0.03 (Jansson et al. 2012). A higher average relatedness among the Scandinavian immigrants compared to Finnish wolves may suggest that some family groups in Finland or Russia have a higher probability than an average family group to contribute with offspring that disperse to Scandinavia. Geographic proximity and dispersal routes less affected by human-mediated mortality are likely to be important factors affecting this pattern.
Implications to conservation
The present and future conservation status of wolves in member states under the legislations of the European Union and/or the Bern Convention are threatened by e.g. overharvesting, low public acceptance, conflicts due to livestock depredation and poor management (Hindrikson et al. 2017). Moreover, the conservation of wolves in several of these countries also depends on immigration from countries that are not bound by the above mentioned legislations, and sometimes manage populations in a manner that does not promote the viability and connectivity of shared wolf populations. Here we present compelling evidence that immigration from the Finnish-Russian wolf population has a positive effect on the genetic diversity in the Scandinavian wolf population, a finding that also have positive fitness effects (Åkesson et al. 2016). Our results also confirm that the conservation status of the Scandinavian wolf population is dependent on the immigration from both Russia and Finland. This finding emphasizes the need for transboundary wolf management strategies guided by the connectivity among biological rather than national wolf populations (Linnell et al. 2008; Quevedo et al. 2019). However, with the assumptions of unrelated founders being violated in our study, it also means that immigration rate needed for long term viability may have to be re-evaluated. By using genetic tools to measure transboundary dispersal patterns over different legislative areas (Mason et al. 2020) our study have the potential to contribute to a more informed understanding and proactive strategy when facilitating the connectivity of wolves in Europe.
Availability of data and material
The datasets generated during and/or analysed during the current study are included in this published article and its supplementary information files.
Code availability
Not applicable.
References
Åkesson M, Svensson L (2020) Sammanställning av släktträdet över den skandinaviska vargpopulationen fram till 2019. Rapport från Viltskadecenter 2020-1
Åkesson M, Liberg O, Sand H et al (2016) Genetic rescue in a severely inbred wolf population. Mol Ecol 25:4745–4756. https://doi.org/10.1111/mec.13797
Allendorf FW (1986) Genetic drift and the loss of alleles versus heterozygosity. Zoo Biol 5:181–190. https://doi.org/10.1002/zoo.1430050212
Allendorf FW, Ryman N (2002) The role of genetics in population viability analysis. In: Population viability analysis. The University of Chicago Press, London
Aspi J, Roininen E, Ruokonen M et al (2006) Genetic diversity, population structure, effective population size and demographic history of the Finnish wolf population. Mol Ecol 15:1561–1576. https://doi.org/10.1111/j.1365-294X.2006.02877.x
Aspi J, Roininen E, Kiiskilä J et al (2009) Genetic structure of the northwestern Russian wolf populations and gene flow between Russia and Finland. Conserv Genet 10:815–826. https://doi.org/10.1007/s10592-008-9642-x
Baudouin L, Lebrun P (2001) An operational Bayesian approach for the identification of sexually reproduced cross-fertilized populations using molecular markers. Acta Hortic. https://doi.org/10.17660/ActaHortic.2001.546.5
Belkhir K, Borsa P, Chikhi L, et al (2004) GENETIX 4.0.5.2., Software under WindowsTM for the Genetics of the Populations. Laboratory Genome, Populations, Interactions. CNRS UMP 5000, University of Montpellier II, Montpellier, France
Bensch S, Andrén H, Hansson B et al (2006) Selection for heterozygosity gives hope to a wild population of inbred wolves. PLoS ONE 1:e72. https://doi.org/10.1371/journal.pone.0000072
Bragina EV, Ives AR, Pidgeon AM et al (2015) Rapid declines of large mammal populations after the collapse of the Soviet Union: Wildlife Decline after Collapse of Socialism. Conserv Biol 29:844–853. https://doi.org/10.1111/cobi.12450
Bruford MW (2015) Additional population viability analysis of the Scandinavian wolf population. Report from the Swedish Environmental Protection Agency (SEPA). Report no. 6639
Carbone C, Mace GM, Roberts SC, Macdonald DW (1999) Energetic constraints on the diet of terrestrial carnivores. Nature 402:442–442. https://doi.org/10.1038/46607
Cardillo M (2005) Multiple causes of high extinction risk in large mammal species. Science 309:1239–1241. https://doi.org/10.1126/science.1116030
Cardillo M, Purvis A, Sechrest W et al (2004) Human population density and extinction risk in the world’s carnivores. PLoS Biol 2:e197. https://doi.org/10.1371/journal.pbio.0020197
Chapron G, Kaczensky P, Linnell JDC et al (2014) Recovery of large carnivores in Europe’s modern human-dominated landscapes. Science 346:1517–1519. https://doi.org/10.1126/science.1257553
Charlesworth B (1998) Measures of divergence between populations and the effect of forces that reduce variability. Mol Biol Evol 15:538–543. https://doi.org/10.1093/oxfordjournals.molbev.a025953
DeGiorgio M, Rosenberg NA (2009) An unbiased estimator of gene diversity in samples containing related individuals. Mol Biol Evol 26:501–512. https://doi.org/10.1093/molbev/msn254
Ellegren H, Savolainen P, Rosén B (1996) The genetical history of an isolated population of the endagered grey wolf Canis lupus: a study of nuclera and mitochondrial polymorphisms. Philos Trans R Soc B Biol Sci 351:1661–1669
Eriksen A, Willebrand MH, Zimmermann B, et al (2020) Assessment of the Norwegian part of the Scandinavian wolf population, phase 1: workshop summary. Skriftserien nr. 19 2020. Høgskolen i Innlandet
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Flagstad Ø, Walker CW, Vilà C et al (2003) Two centuries of the Scandinavian wolf population: patterns of genetic variability and migration during an era of dramatic decline. Mol Ecol 12:869–880
Francisco LV, Langsten AA, Mellersh CS et al (1996) A class of highly polymorphic tetranucleotide repeats for canine genetic mapping. Mamm Genome 7:359–362. https://doi.org/10.1007/s003359900104
Goudet J (1995) FSTAT (Version 1.2): a computer program to calculate F-statistics. J Hered 86:485–486. https://doi.org/10.1093/oxfordjournals.jhered.a111627
Hansen MM, Andersen LW, Aspi J, Fredrickson R (2011) Evaluation of the conservation genetic basis of management of grey wolves in Sweden. Report from the international evaluation panel of the Swedish Large Carnivore Inquiry, Swedish Government Investigation SOU 2011:37. Statens Offentliga Utredningar (the Swedish Government’s Official Investigations)
Hedrick PW (1999) Perspective: highly variable loci and their interpretation in evolution and conservation. Evolution 53:313–318. https://doi.org/10.1111/j.1558-5646.1999.tb03767.x
Hedrick PW (2001) Conservation genetics: where are we now? Trends Ecol Evol 16:629–636. https://doi.org/10.1016/S0169-5347(01)02282-0
Heikkinen S, Kojola I, Mäntyniemi S, Härkälä A (2020) Vargstammen i Finland i mars 2020. Naturresursinstitutet
Hill WG, Robertson A (1968) Linkage disequilibrium in finite populations. Theor Appl Genet 38:226–231. https://doi.org/10.1007/BF01245622
Hindrikson M, Remm J, Pilot M et al (2017) Wolf population genetics in Europe: a systematic review, meta-analysis and suggestions for conservation and management. Biol Rev 92:1601–1629. https://doi.org/10.1111/brv.12298
Holmes NG, Humphreys SJ, Binns MM et al (1993) Isolation and characterization of microsatellites from the canine genome. Anim Genet 24:289–292. https://doi.org/10.1111/j.1365-2052.1993.tb00313.x
Holmes NG, Dickens HF, Parker HL et al (1995) Eighteen canine microsatellites. Anim Genet 26:132–133. https://doi.org/10.1111/j.1365-2052.1995.tb02659.x
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806. https://doi.org/10.1093/bioinformatics/btm233
Jansson E, Ruokonen M, Kojola I, Aspi J (2012) Rise and fall of a wolf population: genetic diversity and structure during recovery, rapid expansion and drastic decline. Mol Ecol 21:5178–5193. https://doi.org/10.1111/mec.12010
Kalinowski ST (2005) hp-rare 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5:187–189. https://doi.org/10.1111/j.1471-8286.2004.00845.x
Kalinowski ST, Wagner AP, Taper ML (2006) ml-relate: a computer program for maximum likelihood estimation of relatedness and relationship. Mol Ecol Notes 6:576–579. https://doi.org/10.1111/j.1471-8286.2006.01256.x
Kardos M, Åkesson M, Fountain T et al (2018) Genomic consequences of intensive inbreeding in an isolated wolf population. Nat Ecol Evol 2:124–131. https://doi.org/10.1038/s41559-017-0375-4
Kenney J, Allendorf FW, McDougal C, Smith JLD (2014) How much gene flow is needed to avoid inbreeding depression in wild tiger populations? Proc R Soc B Biol Sci 281:20133337. https://doi.org/10.1098/rspb.2013.3337
Kojola I, Kaartinen S, Hakala A et al (2009) Dispersal behavior and the connectivity between wolf populations in northern Europe. J Wildl Manag 73:309–313. https://doi.org/10.2193/2007-539
Laikre L, Olsson F, Jansson E et al (2016) Metapopulation effective size and conservation genetic goals for the Fennoscandian wolf (Canis lupus) population. Heredity 117:279–289. https://doi.org/10.1038/hdy.2016.44
Liberg O, Andrén H, Pedersen H-C et al (2005) Severe inbreeding depression in a wild wolf (Canis lupus) population. Biol Lett 1:17–20. https://doi.org/10.1098/rsbl.2004.0266
Liberg O, Aronson Å, Sand H et al (2012a) Monitoring of wolves in Scandinavia. Hystrix Ital J Mammal. https://doi.org/10.4404/hystrix-23.1-4670
Liberg O, Chapron G, Wabakken P et al (2012b) Shoot, shovel and shut up: cryptic poaching slows restoration of a large carnivore in Europe. Proc R Soc B Biol Sci 279:910–915. https://doi.org/10.1098/rspb.2011.1275
Liberg O, Suutarinen J, Åkesson M et al (2020) Poaching-related disappearance rate of wolves in Sweden was positively related to population size and negatively to legal culling. Biol Conserv 243:108456. https://doi.org/10.1016/j.biocon.2020.108456
Linnell JDC, Salvatori V, Boitani L (2008) Guidelines for population level management plans for large carnivores in Europe. A Large Carnivore Initiative for Europe report prepared for the European Commission (contract 070501/2005/424162/MAR/B2)
Mason N, Ward M, Watson JEM et al (2020) Global opportunities and challenges for transboundary conservation. Nat Ecol Evol 4:694–701. https://doi.org/10.1038/s41559-020-1160-3
McMillan LF, Fewster RM (2017) Visualizations for genetic assignment analyses using the saddlepoint approximation method: visualizations for genetic assignment analyses. Biometrics 73:1029–1041. https://doi.org/10.1111/biom.12667
Mech LD, Barber-Meyer SM, Erb J (2016) Wolf (Canis lupus) generation time and proportion of current breeding females by age. PLoS ONE 11:e0156682. https://doi.org/10.1371/journal.pone.0156682
Meirmans PG (2012) AMOVA-based clustering of population genetic data. J Hered 103:744–750. https://doi.org/10.1093/jhered/ess047
Milleret C, Wabakken P, Liberg O et al (2017) Let’s stay together? Intrinsic and extrinsic factors involved in pair bond dissolution in a recolonizing wolf population. J Anim Ecol 86:43–54. https://doi.org/10.1111/1365-2656.12587
Mills LS, Feltner J (2015) An updated synthesis on appropriate science-based criteria for “favorable reference population” of the Swedish wolf (Canis lupus) population. Report to the Swedish Environmental Protection Agency NV-02945-15
Naturvårdsverket (2015) Delredovisning av regeringsuppdraget att utreda gynnsam bevarandestatus för varg (M2015/1573/Nm)
Naturvårdsverket (2016) Nationell förvaltningsplan för varg: Förvaltningsperioden 2014–2019. Swedish Environmental Protection Agency
Neff MW, Broman KW, Mellersh CS et al (1999) A second-generation genetic linkage map of the domestic dog, Canis familiaris. Genetics 151:803–820
Nei M, Maruyama T, Chakraborty R (1975) The bottleneck effect and genetic variability in populations. Evolution 29:1–10. https://doi.org/10.2307/2407137
Ostrander EA, Sprague GF, Rine J (1993) Identification and characterization of dinucleotide repeat (CA)n markers for genetic mapping in dog. Genomics 16:207–213. https://doi.org/10.1006/geno.1993.1160
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research: an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Paetkau D, Slade R, Burden M, Estoup A (2004) Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol Ecol 13:55–65. https://doi.org/10.1046/j.1365-294X.2004.02008.x
Piry S, Alapetite A, Cornuet J-M et al (2004) GENECLASS2: a software for genetic assignment and first-generation migrant detection. J Hered 95:536–539. https://doi.org/10.1093/jhered/esh074
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Quevedo M, Echegaray J, Fernández-Gil A et al (2019) Lethal management may hinder population recovery in Iberian wolves. Biodivers Conserv 28:415–432. https://doi.org/10.1007/s10531-018-1668-x
Rannala B, Mountain JL (1997) Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci 94:9197–9201. https://doi.org/10.1073/pnas.94.17.9197
Raymond M, Rousset F (1995) GENEPOP (Version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249. https://doi.org/10.1093/oxfordjournals.jhered.a111573
Ripple WJ, Estes JA, Beschta RL et al (2014) Status and ecological effects of the world’s largest carnivores. Science 343:1241484–1241484. https://doi.org/10.1126/science.1241484
Rosenberg MS, Anderson CD (2011) PASSaGE: pattern analysis, spatial statistics and geographic exegesis. Version 2. Methods Ecol Evol 2:229–232. https://doi.org/10.1111/j.2041-210X.2010.00081.x
Sargolzaei M, Iwaisaki H, Colleau J-J (2005) A fast algorithm for computing inbreeding coefficients in large populations. J Anim Breed Genet 122:325–331. https://doi.org/10.1111/j.1439-0388.2005.00538.x
Seddon JM, Sundqvist A-K, Björnerfeldt S, Ellegren H (2006) Genetic identification of immigrants to the Scandinavian wolf population. Conserv Genet 7:225–230. https://doi.org/10.1007/s10592-005-9001-0
Shibuya H, Collins BK, Huang THM, Johnson GS (1994) A polymorphic (AGGAAT), tandem repeat in an intron of the canine von Willebrand factor gene. Anim Genet 25:122. https://doi.org/10.1111/j.1365-2052.1994.tb00094.x
Slate J, David P, Dodds KG et al (2004) Understanding the relationship between the inbreeding coefficient and multilocus heterozygosity: theoretical expectations and empirical data. Heredity 93:255–265. https://doi.org/10.1038/sj.hdy.6800485
Slatkin M (1985) Rare alleles as indicators of gene flow. Evolution 39:53–65. https://doi.org/10.1111/j.1558-5646.1985.tb04079.x
Slatkin M (2008) Linkage disequilibrium—understanding the evolutionary past and mapping the medical future. Nat Rev Genet 9:477–485. https://doi.org/10.1038/nrg2361
Smeds L, Kojola I, Ellegren H (2019) The evolutionary history of grey wolf Y chromosomes. Mol Ecol 28:2173–2191. https://doi.org/10.1111/mec.15054
Smeds L, Aspi J, Berglund J et al (2020) Whole-genome analyses provide no evidence for dog introgression in Fennoscandian wolf populations. Evol Appl. https://doi.org/10.1111/eva.13151
Sokal RR, Rohlf FJ (1995) Biometry, 3rd edn. W.H. Freeman and Company, New York
Sundqvist A-K, Ellegren H, Olivier M, Vila C (2001) Y chromosome haplotyping in Scandinavian wolves (Canis lupus) based on microsatellite markers. Mol Ecol 10:1959–1966
Trouwborst A (2018) Wolves not welcome? Zoning for large carnivore conservation and management under the Bern Convention and EU Habitats Directive. Rev Eur Comp Int Environ Law 27:306–319. https://doi.org/10.1111/reel.12249
Vilá C, Sundqvist A-K, Flagstad O et al (2003) Rescue of a severely bottlenecked wolf (Canis lupus) population by a single immigrant. Proc R Soc B Biol Sci 270:91–97. https://doi.org/10.1098/rspb.2002.2184
Wabakken P, Sand H, Liberg O, Bjärvall A (2001) The recovery, distribution, and population dynamics of wolves on the Scandinavian peninsula, 1978–1998. Can J Zool 79:710–725. https://doi.org/10.1139/cjz-79-4-710
Wabakken P, Sand H, Kojola I et al (2007) Multistage, long-range natal dispersal by a global positioning system–collared Scandinavian wolf. J Wildl Manag 71:1631–1634. https://doi.org/10.2193/2006-222
Wabakken P, Svensson L, Maartmann E, et al (2020) Bestandsovervåking av ulv vinteren 2019–2020. Inventering av varg vintern 2019–2020. Bestandsstatus for store rovdyr i Skandinavia. Beståndsstatus för stora rovdjur i Skandinavien 1–2020.
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370. https://doi.org/10.2307/2408641
Wikenros C, Gicquel M, Zimmermann B et al (2021) Age at first reproduction in wolves: different patterns of density dependence for females and males. Proc R Soc Lond B Biol Sci 288:20210207. https://doi.org/10.1098/rspb.2021.0207
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–159
Wright S (1965) The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution 19:395–420. https://doi.org/10.1111/j.1558-5646.1965.tb01731.x
Acknowledgements
This work was funded by the Swedish Research Council Formas, the Swedish Environmental Protection Agency (SEPA), the Norwegian Research Council, the Norwegian Environment Agency, the Swedish University of Agricultural Sciences (SLU), Norwegian Institute for Nature Research, Inland Norway University of Applied Sciences, County Governor of Hedmark and Marie Claire Cronstedts Foundation. The Swedish County Administration boards and Wildlife Damage Center, Sweden are thanked for coordinating the field work together with HUC, Norway. Camilla Wikenros, SLU, was part in securing the funding from SEPA. The genetic laboratory work was conducted by Anna Danielsson and Eva Hedmark, SLU, the Science for Life Laboratory (SciLifeLab, Sweden), and Torveig Balstad, Line Birkeland Eriksen, Merethe Hagen Spets, Line Syslak and Caroline Danielsen Søgaard at NINA, Norway. The data reported in the paper are presented in the Supplementary Material.
Funding
Open access funding provided by Swedish University of Agricultural Sciences. This work was funded by funded by the Swedish Research Council Formas (2011-686), the Swedish Environmental Protection Agency (nr 802-0193-18), the Norwegian Research Council, the Norwegian Environment Agency, the Swedish University of Agricultural Sciences (SLU), Norwegian Institute for Nature Research, Inland Norway University of Applied Sciences, County Governor of Hedmark and Marie Claire Cronstedts Foundation.
Author information
Authors and Affiliations
Contributions
MÅ and HS secured funding. MÅ conceived and designed the study, carried out the statistical analyses and drafted the manuscript; MÅ and ØF was involved in the laboratory work and compiled data; HS, PW, OL, IK and ØF provided samples. ØF, JA, IK, OL, PW and HS revised the manuscript. All authors gave final approval for publication and agree to be held accountable for the work performed therein.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
All procedures including capture and handling of wolves, fulfilled ethical requirements and have been approved by the Swedish Animal Experiment Ethics Board (Permit Number: C 281/6), the Norwegian Experimental Animal Ethics Committee (Permit Number 2014/284738–1) and the Animal Care and Use Committee at the University of Oulu (OYEKT-6–99), the provincial government of Oulu (OLH-01951/Ym-23).
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Åkesson, M., Flagstad, Ø., Aspi, J. et al. Genetic signature of immigrants and their effect on genetic diversity in the recently established Scandinavian wolf population. Conserv Genet 23, 359–373 (2022). https://doi.org/10.1007/s10592-021-01423-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-021-01423-5