
Abstract
Despite increased use of species translocations, controversy remains regarding the efficacy and efficiency of the strategy in obtaining conservation goals. Much of this controversy results from vague program objectives, unclear definitions of success, and lack of follow-up monitoring. We used the translocation program initiated by Zoo Knoxville for the federally threatened Bog Turtle (Glyptemys muhlenbergii) as a case study to demonstrate how genomic assessments not only assess the success of program objectives, but also allow managers to quickly obtain baseline data from which program objectives and explicit definitions of ‘success’ can be determined. Here we used 7030 SNP markers derived from RADseq data to confirm the premise that different source populations are genetically differentiated. Then we tested whether the release population has enhanced genetic diversity, as expected from a deliberate admixture. Although the release population had greater diversity than any source population, variation was lower than expected from modeling admixture with equal source contribution. Our results support the premise that genetic diversity can be maximized by including representatives from as many natural populations as possible. But failure to achieve the expected level of diversity could result from nonrandom success of founders from different sources or unrecorded bias in the implementation of the release program. Many existing and future translocation programs would benefit from genetic assessment similar to that conducted here with Bog Turtles.
Similar content being viewed by others
Introduction
Habitat destruction, degradation, and fragmentation are shrinking population sizes and altering historic patterns of gene flow in numerous species (Andrén 1994; Bender et al. 1998; Cushman 2006; Swift and Hannon 2009; Quesnelle et al. 2013). Without human intervention, many of these populations would face an early extinction. Such interventions are often outlined in species action plans. One intervention in particular has become increasingly common since the 1970s: human-mediated migration (i.e. translocation—including introduction, relocation, reintroduction, and supplementation). Despite increased use of species translocations, less than 50% have been formally assessed and only a small fraction of those have been deemed successful (Fischer and Lindenmayer 2000; Tarszisz et al. 2014). Lack of assessment (i.e. poor follow-up monitoring) has likely contributed to uncertainty regarding the efficacy and efficiency of translocations: numerous studies advocate for translocations (Marsh and Trenham 2001; Strum 2005; Parker 2008; Decesare et al. 2011; Estrada 2014; Watson and Watson 2015), but many others advocate against them (Dodd and Seigel 1991; Struhsaker and Siex 1998; Ricciardi and Simberloff 2009; Godefroid et al. 2011; Oro et al. 2011). And those studies that do not explicitly advocate for or against translocations emphasize that extreme caution should be taken when considering translocations as a conservation strategy (Cope and Waller 1995; Menges 2008; Schwartz et al. 2012). One factor likely contributing to the lack of consensus regarding translocations is that program objectives are often unclear or nonexistent, resulting in vague criteria for success (Weeks et al. 2011; Ewen et al. 2014).
Genomic assessments offer an objective means to monitor translocation programs and establish specific criteria to characterize success, yet such assessments are rarely incorporated in program evaluations (Frankham et al. 2014). This is particularly surprising considering the growing realization that genetic factors often impact populations prior to their extinction (Spielman et al. 2004). Historically, and to a lesser extent presently, probability of persistence was inferred by estimating census population sizes using mark-recapture techniques (Seber 1982), management units were based solely on political boundaries, translocations were implemented based on expert opinion, and taxonomic uncertainties were resolved using morphology. However, the emergence of new genomic techniques now allows thousands of markers to be examined with relative ease, making previously unattainable information accessible and previously accessible information more reliable and objective (Allendorf et al. 2010). Such advances are changing the way populations are monitored, how they are managed, and how uncertainty is addressed.
Here we illustrate how genetic assessment can be used to evaluate a translocation program using Tennessee’s Bog Turtle (Glyptemys muhlenbergii) captive breeding and release program. This program presents itself as a useful case study because several aspects of the program mirror those of many extant translocation programs, namely the lack of clear objectives when the program was first initiated, subjective success criteria, and missing breeding and release records for a species notoriously difficult to monitor using conventional survey methods.
The Tennessee Bog Turtle captive breeding and release program was initiated nearly 30 years ago by Zoo Knoxville to aid in the conservation of this federally threated species. The goal was to successfully breed Bog Turtles in captivity and release the offspring in the wild. Bog Turtles from North Carolina and Tennessee were successfully bred in captivity and over 100 turtles were released to a single, experimental release site between 1991 and 2015 with an 84% annual survival rate. Was this program successful? Well, that depends on what the program objectives were. If the only objectives were to successfully breed Bog Turtles in captivity and that the released offspring survived, then the program successfully met those objectives. However, if the objective was to create a self-sustaining population, then the success of the program has not yet been determined. Specifically, if the objective was to maximize the long-term sustainability of the introduced population by maximizing genetic variation, then records documenting the release and survival of an equal number of offspring (particularly females) from each source population (assuming each source was a genetically distinct population) would suggest this objective was met. However, some breeding and release records were missing or undocumented and all but two founding captive individuals were predated by raccoons, which prohibited retroactive genetic analysis to assign missing parentage records (see Online Resource 1 for details).
Another motivating factor, one of primary importance to agencies and personnel managing Bog Turtle populations for conducting a genetic assessment on the Tennessee captive breeding and release program is the fact that the Species Recovery Plan (USFWS 2001) mandates the “investigation of the genetic impacts of reintroduction”. Many captive breeding programs, including the Bog Turtle program are intentionally managed to avoid breeding close relatives (i.e. inbreeding avoidance) to minimize loss of genetic variation due to inbreeding. A formal genetic assessment would indicate whether the implemented breeding strategy achieved said goal or if adjustments are necessary in the future. Such evaluation is timely as conservation options are dwindling as Bog Turtle populations continue to plummet and managing partners are expressing interest in implementing similar translocation programs in other parts of the Bog Turtle range. Since most interested partners will face implementation challenges associated with limited time, money, and personnel, our approach also demonstrates how managers could determine whether or not female turtles from all wild populations are necessary for a sustainable head-start program based on the degree of genetic differentiation among populations (i.e. whether or not the inclusion of the focal population maximizes the genetic variation in the release population).
We use reduced representation next-generation sequencing (RADseq) to evaluate whether the Bog Turtle translocation program implemented by Zoo Knoxville successfully maintained genetic variation within the release population relative to neighboring wild populations and source populations. First, we tested the working assumption that different source populations are genetically differentiated, and therefore of equal importance in establishing a reservoir of genetic variation. Then we compared the estimated genetic variation in the release site to the expected level based on an assumed even admixture of source populations. Taken together, these genetic measures not only satisfy the requirements mandated in the Species Recovery Plan, but also inform current and future management programs for the federally threatened Bog Turtle. Furthermore, our genetic assessment highlights how careful record keeping can complement and even inform quantitative standards of success in regards to program objectives, bringing us one step closer to resolving the uncertainty surrounding translocation as a viable conservation strategy.
Methods
Study system
North America’s smallest semi-aquatic turtle, the Bog Turtle (Glyptemys muhlenbergii) continues to require conservation action as populations are declining throughout their range, from northern Georgia to upstate New York and Massachusetts due to habitat destruction (Gibbons et al. 2000), road mortality (Mitchell 1994), and illegal collection for the pet trade (Tesauro 2001). Long-term programs are particularly valuable considering the life history of the species; Bog Turtles typically reach reproductive age at 6–12 years old, with males maturing before females and each female only lays an average of three eggs per year. A handful of Bog Turtle head-start programs have been launched, but the Tennessee program is the largest and longest running. The program has a blended approach, combining captive breeding and head-starting: (1) captive breeding and release, where initial breeding pairs were obtained from wild populations in the southern United States and offspring were released into a wild experimental population and (2) head-starting, where local, wild females are tracked using radio-telemetry, brought into a local laying facility to deposit their eggs and immediately returned while the eggs and eventually hatchlings are raised in a secure environment for approximately 9 months before release into the wild experimental population (although early on in the program, hatchlings were raised for 22 months before release).
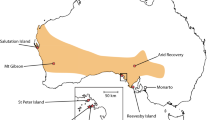
The initial captive population included one individual from western North Carolina (Site F; Fig. 1) and a few individuals from northeast Tennessee. To date, only first generation (F1) offspring from the captive breeding program have been translocated to the release site. Females for the head-start program were obtained from three sites in northeast Tennessee (Site L, O, and Q; Fig. 1). A fourth wild site exists in Tennessee (Site B; Fig. 1), but permission to access the property has been revoked for an unknown period of time and thus is no longer part of the program. Like most Bog Turtle sites, the experimental release site (Release Site; Fig. 1) is also on private land (only Site O is entirely on public land), at a pristine high-elevation bog approximately 48 km south of the nearest known wild site in Tennessee. The wild sites in Tennessee are relatively close to each other geographically, particularly Site L and Site O for which previous radio-telemetry data has shown turtles occasionally moving across the 2 km agricultural field between the two wetland sites (A. Eastin, personal communication).

Bog turtle sampling locations in Tennessee and North Carolina. Site F represents the source population for one individual used in the original captive breeding program at Zoo Knoxville (Site Z), for which offspring were introduced to the release site (Site R) located south of the wild Tennessee Bog Turtle populations (Site O, L, B, and Q). The location of the head-start facility where offspring of wild females were raised for 9 months prior to release is shown adjacent to the wild sites within the map inset
Sampling
We collected tissue samples from a total of 124 individual turtles, representing all known Tennessee populations and nearly half of all extant individuals at accessible sites. All Tennessee turtles were hand-captured using visual and tactile methods (Whitlock 2002) and augmented with trapping as needed (Somers 2000; Whitlock 2002) between April and October of 2014 and 2015. For each new capture a tissue sample was collected (<0.5 cm distal portion of the tail) and was immediately placed in 95% ethanol and stored at −20 °C until DNA extraction (Hughe 2010). We were able to confirm that each sample represented a different individual because all turtles had or were given a unique notching pattern on their marginal scutes (method modified from Cagle 1939).
Of the estimated number of extant turtles from each site, 63% of turtles from the Release Site were sampled (N = 57), 71% of Site L (N = 5), 30% of Site O (N = 9), 57% of Site Q (N = 12). Two turtles from Site F were donated by the North Carolina Wildlife Resources Commission and J. Apodaca (Warren–Wilson College), all extant captive individuals (N = 5) were donated by Zoo Knoxville, four turtles from Site B were donated by Tim King (USGS), and the remaining samples came from juveniles at the head-start facility (N = 30). This is the most extensive and complete genetic sampling ever obtained for the Bog Turtles in Tennessee, an impressive feat considering the cryptic nature of this species and the logistical constraints on the efficiency of capture methods because of habitat characteristics at some sites. Considering that only one captive breeding pair survived the raccoon predation at the outdoor exhibit at Zoo Knoxville, we supplemented the sampling of the extant captive population with samples previously collected from several turtles at the North Carolina source (Site F) and the fourth Tennessee site (Site B).
Laboratory and post-sequencing procedures
We isolated DNA from tissue using the DNeasy Blood and Tissue Kit (Qiagen Corporation, Valencia, CA), quantified extracted products using a fluorometer, and visualized DNA using gel electrophoresis to confirm that the DNA was not degraded. We digested DNA products using three enzymes (ClaI, MspI, and BamI) rather than the two enzymes typically used in double-digest restriction site associated DNA sequencing (ddRAD; Peterson et al. 2012) to reduce chimeras, increases the efficiency of adapter ligation, and minimize the occurrence of adapter dimers (3RADseq; T Glenn, unpublished). The 3RADseq protocol requires less input DNA and adapters and uses variable length quadruple-index tags to improve sequencing efficiency and allow pooling of more samples. We successfully generated 3RAD libraries for 113 individuals. Individual RADseq libraries were pooled relative to their DNA concentrations prior to size selection of 500 bp fragments using a PippenPrep system (Sage Science Corporation, Beverly, MA) and sequenced on an Illumina NextSeq for 75-bp paired end reads for approximately 2 million reads per individual.
After the inner barcode and cut site were trimmed, quality control and filtering of the genotypes for each sample were performed with the software pipeline, pyRAD (Eaton 2014). The default parameter values were used, with the following exceptions: the minimum depth of coverage was set to ten, the clustering threshold was 0.907 (allowing no more than 6 bp mismatches), the minimum number of samples required per locus was 2, and the maximum number of shared polymorphic sites allowed per locus was 3 bp. After library preparation, sequencing, and quality control of Illumina reads, data from 7030 markers and 95 individual turtles remained (2 turtles from Site F, 2 from Site Z, 22 from Site H, 3 from Site B, 4 from Site Q, 6 from Site O, 4 from Site L, and 52 from Site R).
Data analysis
To assess genetic differentiation among source populations, we estimated F ST for each pair of samples to represent the between-population fraction of genetic variation (Allendorf and Luikart 2009). Specifically, we calculated Weir and Cockerham’s FST for comparative purposes; an unbiased test statistic with respect to sample size (Weir and Cockerham 1984). We used parametric randomization to evaluate statistical significance while avoiding potential bias arising from missing data (i.e. when resampling individuals with varying amounts of missing data, the resampled distributions are not comparable because they vary in the number of usable loci). For each pair of populations, we estimated population allele frequencies and pooled allele frequencies for the subset of markers with shared data for the particular pair of populations. Then, for 10,000 replicates we generated two simple random samples of two alleles per locus from the pooled allele frequencies to get a distribution of F ST under the null hypothesis of no population differentiation. We also generated one random sample from each population to get a distribution of F ST under the alternative hypothesis of differentiated populations. Put simply, we used this procedure to test the hypothesis of differentiation based on the probability of identity of alleles rather than observed genotypes, by literally taking an allele at random and asking if it was different from a second allele, randomly drawn from either the same site or different site. Gene diversity is the probability that two randomly sampled alleles are different; often misleadingly called heterozygosity because it corresponds to the expected proportion of heterozygous genotypes under Hardy–Weinberg assumptions (Nei 1987; Gillespie 2004). Hardy–Weinberg assumptions are explicitly not met in the experimental release population because it still includes first generation transplants (Wahlund 1928). Therefore, we restricted our analyses to those based on allele frequencies, and made no assumptions regarding genotype frequencies. Two alleles per locus per population is the minimal sample of alleles suitable for estimating F ST , resulting in maximal sampling variance and minimal risk of Type I error. We estimated p-values as the proportion of replicates in which F ST under the null hypothesis was greater than or equal to F ST under the alternative hypothesis of distinct populations. R code for these randomizations is given in Online Resource 2.
To compare the genetic diversity within the release population versus within each natural population, we estimated gene diversity for each population. We estimated bootstrap 95% confidence intervals by resampling from the full set of 7030 loci with replacement to generate 10,000 bootstrap samples.
To specifically test whether the gene diversity in the release population (Site R) was consistent with an equal contribution from each source population, we used parametric bootstrapping to estimate the expected distribution of gene diversity in an equal mixture. The unweighted mean allele frequencies were calculated for each locus across source populations to estimate the expected allele frequencies in a source pool with equal representation from each source population. Then, 10,000 replicate random samples of two alleles per locus were generated from the pooled allele frequencies to get a distribution of sample gene diversities under the equal mixture model. We compared this distribution to the parametric bootstrap distribution of sample gene diversities generated by drawing samples from the observed allele frequencies within the release population. R code for these simulations is given in Online Resource 2.
As a follow-up to the previous analysis that compared observed versus expected gene diversity in the released population (Site R), we assessed expectations of gene diversity loss associated with genetic drift assuming a constant population of 176 individuals (which was the number of individuals released in Site R) comprised of an equal number of founders from each source population. Using standard theory for the loss of gene diversity owing to drift (Gillespie 2004) we calculated the number of generations it would take for the expected gene diversity to match the observed gene diversity. We used 10,000 stochastic simulations to assess the variability of gene diversity loss owing to drift (Online Resource 2).
Finally, to evaluate whether the release population was more genetically similar to some source populations than others, we used the pairwise F ST parametric bootstrap routine (Online Resources 2) to estimate bootstrap distributions of F ST and Jost’s D (Jost 2008) between the release site and each source.
Results
We obtained a total of 163,139,257 paired-end reads, each 150 bp across 113 individual turtles in four wild Tennessee populations, one North Carolina wild population, the captive population at Zoo Knoxville and the head-start population at the local rearing site. After filtering for a minimum coverage depth of ten and a minimum number of samples per locus of two, 7030 total markers were found across 95 turtles, for which nine loci had three alleles.
Partitioning of genetic diversity as estimated using pairwise F ST values ranged from 0.411 (Head Start, H—Release Site, R) to 0.717 (Site L—Site O) (Table 1). The null distributions of F ST values produced from parametric randomizations indicated that all pairwise F ST values were significantly higher than would be expected if the sites were parts of a single panmictic population (Table 1). The observed genetic differentiation among wild source populations did not correspond to expectations given the geographic proximity of populations. For example, Site L was geographically closest to Site O (within 2 km of each other), but the pairwise F ST value between these two sites was higher than pairwise F ST values between either site and the other, more geographically distant wild sites (Site Q and Site B).
Gene diversity measures within wild source populations ranged from 0.08 (Site L) to 0.14 (Site Q) (Fig. 2). The captive population at Zoo Knoxville (Site Z) and the head-start population at the local rearing site (Site H) had higher gene diversity values relative to the wild source populations (Fig. 2). Pertinent to our overall research question, regarding whether the release program successfully increased gene diversity in the experimental release population, we did observe a substantially higher gene diversity value in the released population relative to its source populations (Fig. 2). However, the observed population level gene diversity in the released population falls significantly short of the expected gene diversity based on the admixture model where all source populations contributed equally to the released population (Fig. 3). In fact, it would take, on average, about 47 generations (perhaps 400–500 years) for genetic drift to reduce the expected gene diversity of 0.280 to the observed gene diversity of 0.245 (Online Resource 2).

Mean gene diversity at each Bog Turtle sampling location (see Fig. 1) across 7030 SNP markers 95% bootstrap confidence intervals are shown around each mean

Observed (gray) versus expected (white) gene diversity for the experimental Bog Turtle release population (Site R) based on 1000 bootstrap replicates. Expected gene diversity was calculated assuming an equal contribution from the captive breeding population (Z) and each potential wild source population (Sites F, O, L, B, and Q in Fig. 1)
Measures of genetic differentiation were also inconsistent with equal contribution of source populations (Fig. 4). The release population had the greatest genetic similarity to populations Q and B and the Zoo population (Z), and the greatest differentiation from populations O, L, and F (Fig. 4). The head-start population was surprisingly dissimilar to the release population according to Jost’s D, while F ST between them was relatively low (likely reflecting the relatively high within-population variation seen in both samples, see Fig. 2).

Genetic differentiation between Bog Turtles sampled from the release site and other samples with 95% parametric bootstrap confidence intervals
Discussion
Nearly 30 years ago, Zoo Knoxville started a captive breeding and release program for the federally endangered Bog Turtle (G. muhlenbergii) as a preemptive conservation measure to combat the observed decline of populations throughout their range. Within 10 years, the IUCN listed the Bog Turtle as endangered, and the US government listed populations within the northern region as Threatened under the ESA. As a result, a species recovery plan was drafted (USFWS 2001), which included an objective to genetically assess the impacts of reintroductions (i.e. ‘translocations’). Although the particular long-term objectives of the Tennessee program were either vague or unwritten when the program was initiated nearly 30 years ago, implementation of the program has generally been guided by the idea that the experimental release population should be composed of individuals from many distinct natural populations to maximize genetic diversity (presumably maximizing adaptive capacity and minimizing risks associated with inbreeding depression). Here we confirmed the inferred premise that the source populations represent distinct gene pools, and showed that the experimental released population has greater genetic diversity than any of the possible source populations. However, the level of genetic diversity falls short of the predicted diversity of a truly equal mixture from all sources. Understanding the causes of this shortfall could help guide future management decisions.
Experts believed the Bog Turtle was in decline long before their addition to the IUCN Red List in 1996 (Ernst and Barbour 1972; Behler 1974; Bury 1979; Chase et al. 1989). With each passing year, the IUCN Red List grows; in fact since 2000 the number of listed threatened species has more than doubled (IUCNredlist.org). Many of these additions result from delayed assessments of less well-known groups or the eventual filling in of the knowledge gaps from insufficient preliminary assessments (e.g. marine turtles; Seminoff and Shanker 2008). Although the IUCN is often regarded as the world’s primary authority on the conservation status of species (Mrosovsky 1997), within the United States federal protections are not established until they are listed under the Endangered Species Act. For Bog Turtles, ESA listing came the following year (although only for the northern populations), but for most IUCN-listed species ESA listing has never followed (Harris et al. 2011). Unfortunately, delayed prioritization and protections can leave managers with fewer conservation options with potentially greater uncertainty regarding their outcomes. In situations such as these, translocations may be proposed to artificially restore historic gene flow or introduce a population to a more suitable habitat (e.g. freshwater mussel; Cosgrove and Hastie 2001). However, to increase the likelihood of success, translocations should be considered long before they become a last resort (Griffith et al. 1989). Clearly, endangered species conservation programs could benefit from a greater understanding of the efficacy and efficiency of controversial translocation strategies.
Genetic, or more recently genomic assessments offer a way to address the current deficiency in our ability to evaluate translocation as a possible strategy by providing a relatively quick method for collecting baseline data from which to formulate quantitative program objectives and conduct follow-up monitoring. Specifically, we used RADseq to characterize the genetic variation present in potential source populations (i.e. starting gene diversity) to serve as a baseline to compare to the translocated population, where greater gene diversity would provide an objective measure of ‘success’.
Pairwise F ST values indicated that all wild source populations are genetically distinct and thus worthy of inclusion in the program. This is immediately pertinent considering several of the populations are continually threatened by human-caused habitat degradation. Thus the offspring currently existing in the release population may soon be the only genetic representatives left from some natural populations. With continued climate change on the horizon and the unpredictable impacts of anthropogenic activities, the variants of a gene that will be beneficial in the future will be hard to predict, thus preserving genetic variation may be the most conservative strategy (Bonin et al. 2007)—in which case the Bog Turtles of Tennessee are in a better position than they were in the recent past.
Careful and complete bookkeeping of any translocation program would also reduce ambiguity in assessments of success. In our case, the absence of complete breeding and release records inhibit our ability to distinguish between two possible explanations for the lower than expected gene diversity in the translocated population; (1) unrecorded bias in the implementation of the release program and (2) nonrandom success of released individuals (including the dominance of one or a few breeding males). If more individuals were released from one source relative to another then our assumption of equal contribution from each source would yield unrealistic expected gene diversity; however, if these data were available it would be an easy assumption to update by using weighted means for each source population. If our assumption of equal mixture was incorrect, our simulations indicate that genetic variation could be increased by 25% if an equal number of offspring from each genetically distinct source population were successfully released. On the other hand, if our assumption of equal contribution was correct, then the lower than expected gene diversity is likely a result of unequal survival or reproductive success between individuals of different source populations, rather than inbreeding depression or genetic drift. Many captive populations are specifically managed to prevent inbreeding, as is true for the captive breeding program at Zoo Knoxville. Offspring of captive turtles were released rather than added to the captive breeding stock and over 170 turtles were released from five different wild source populations (either via the captive breeding program or head-start program) over the past 3 decades, minimizing breeding between close relatives. Furthermore, only a maximum of three generations of turtles are present at the release site compared to the 47 generations necessary for genetic drift to produce the observed gene diversity (assuming an equal contribution from each source population). In fact, the 99% interquantile of our simulated data under genetic drift did not overlap the 99% interquantile range of the bootstrap distribution based on the real data until 22 generations. Thus, there has not been enough time for drift to explain the discrepancy between the expected and observed gene diversity in the release population. However, a disparity in reproductive success is possible as Bog Turtles are hypothesized to have a polygamous mating system. Such a mating system was observed in their close relative, the Wood Turtle (Glyptemys insculpta) where high-ranking males were found to father more offspring (Galbraith 1991) and half of all clutches had multiple sires (Pearse and Avise 2001). An extremely skewed effective sex ratio could lower the effective population size enough to achieve the observed level of gene diversity in one or two generations (Gillespie 2004) and a milder skew could be a contributing factor. Such a diagnosis would be valuable to those implementing the program and would help guide managers in making appropriate adjustments to improve the program.
Hundreds of translocation programs have been implemented (Fischer and Lindenmayer 2000), providing a great sample size for which to implement genomic assessments to evaluate program success, at least in terms of genetically relevant objectives. Of course, other assessments will be valuable to determine other dimensions of success, such as habitat assessments (e.g. bighorn sheep; Zeigenfuss et al. 2000) and health screenings to monitor the spread of disease (Griffith et al. 1993). Broad implementation of genetic assessments would help evaluate the success of previously implemented translocations, providing managers with more concrete direction regarding whether translocation is a viable strategy in general. Moreover, genetic assessments could provide managers with a baseline from which specific program objectives could be developed and hypothetical outcomes of alternative strategies could be modeled. Conservation decisions are often challenged with uncertainty and urgency, but post hoc genetic analyses of existing translocation programs and a priori assessments of any population that may be considered in future translocation programs could remove some of the uncertainty surrounding the idea of translocations as a viable conservation strategy.
References
Allendorf FW, Luikart GH (2009) Conservation and the genetics of populations. Wiley, Hoboken
Allendorf FW, Hohenlohe PA, Luikart G (2010) Genomics and the future of conservation genetics. Nat Rev 11:697–709
Andrén H (1994) Effects of habitat fragmentation on birds and mammals in landscapes with different proportions of suitable habitat: a review. Oikos 71:355–366
Behler JL (1974) Efforts to save the bog turtle. Herpetol Rev 5:44
Bender DJ, Contretas TA, Fahrig L (1998) Habitat loss and population decline: a meta-analysis of the patch size effect. Ecology 79:517–533
Bonin A, Nicole F, Pompanon F, Miaud C, Taberlet P (2007) Population adaptive index: a new method to help measure intraspecific genetic diversity and prioritize populations for conservation. Conserv Biol 21:697–708
Bury RB (1979) Review of the ecology and conservation of the bog turtle, Clemmys muhlenbergii. In: US Fish and Wildlife Service Special Scientific Report of Wildlife. Washington DC, No 219, p 9
Cagle FR (1939) A system of making turtles for future identification. Copeia 1939:170–173
Chase JD, Dixon KR, Gates JE, Jacobs D, Taylor GJ (1989) Habitat characteristics, population size, and home range of the Bog Turtle, Clemmys muhlenbergii, in Maryland. J Herpetol 23:356–362
Cope WG, Waller DL (1995) Evaluation of freshwater mussel relocation as a conservation and management strategy. Regul Rivers Res Manage 11:147–155
Cosgrove PJ, Hastie LC (2001) Conservation of threatened freshwater pearl mussel populations: river management, mussel translocation and conflict resolution. Biol Conserv 99:183–190
Cushman SA (2006) Effects of habitat loss and fragmentation on amphibians: a review and prospectus. Biol Conserv 128:231–240
Decesare NJ, Whittington J, Hebblewhite M, Robinson H, Bradley M, Neufeld L, Musiani M (2011) The role of translocation in recovery of woodland caribou populations. Conserv Biol 25:365–373
Dodd CK, Seigel RA (1991) Relocation, repatriation, and translocation of amphibians and reptiles: are they conservation strategies that work? Herpetologica 47:336–350
Eaton DAR (2014) PyRAD: assembly of de novo RADseq loci for phylogenetic analyses. Bioinformatics doi:10.1093/bioinformatics/btu121
Ernst CH, Barbour RW (1972) Turtles of the United States. University of Kentucky Press, Lexington
Estrada J (2014) Finding correlations among successful reintroduction programs: an analysis and review of current and past mistakes. Thesis, University of Michigan
Ewen JG, Soorae PS, Canessa S (2014) Reintroduction objectives, decisions and outcomes: global perspectives from the herpetofauna. Animal Conserv 17:74–81
Fischer J, Lindenmayer DB (2000) An assessment of the published results of animal relocations. Biol Conserv 96:1–11
Frankham R, Bradshaw CJA, Brook BW (2014) Genetics in conservation management: revised recommendations for the 50/500 rules, red list criteria and population viability analysis. Biol Conserv 170:56–63
Galbraith DA (1991) Studies of mating systems in wood turtles (Clemmys insculpta) and snapping turtles (Chelydra serpentina) using DNA fingerprinting. Dissertation, Queen’s University, Ontario
Gibbons JW, Scott DE, Ryan TJ, Buhlmann KA, Tuberville TD, Metts BS, Greene JL, Mills T, Leiden Y, Poppy S, Winne CT (2000) The global decline of reptiles, déjà vu amphibians. Bioscience 50:653–666
Gillespie JH (2004) Population genetics: A concise guide, 2nd edn. Johns Hopkins University Press, Boston
Godefroid S, Piazza C et al (2011) How successful are plant species reintroductions. Biol Conserv 144:672–682
Griffith B, Scott JM, Carpenter JW, Reed C (1989) Translocation as a species conservation tool: status and strategy. Science 245:477–479
Griffith B, Scott JM, Carpenter JW, Reed C (1993) Animal translocations and potential disease transmission. J Zoo Wildlife Med 24:231–236
Harris JBC, Reid JL, Scheffers BR, Wanger TC, Sodhi NS, Fordham DA, Brook BW (2011) Conserving imperiled species: a comparison of the IUCN Red List and US Endangered Species Act. Conserv Lett 00:1–9
Hughe E (2010) Genetic Sampling Protocol: protocol for sampling and storage of painted turtle (Chrysemys picta) blood and tissue for genetic analysis. CRD Species at Risk Information and Collabative. http://www.speciesatrisk.hat.bc.ca. Accessed 8 Mar 2014
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Marsh DM, Trenham PC (2001) Metapopulation dynamics and amphibian conservation. Conserv Biol 15:40–49
Menges ES (2008) Restoration demography and genetics of plants: when is a translocation successful? Aust J Bot 56:187–196
Mitchell JC (1994) The reptiles of Virginia. Smithsonian Institution Press, Washington DC
Mrosovsky N (1997) IUCN’s credibility critically endangered. Nature 389:436
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Oro D, Martínez-Abraín, Villuendas E, Sarzo B, Mínguez E, Carda J, Genovart M (2011) Lessons from a failed translocation program with a seabird species: determinants of success and conservation value. Biol Conserv 144:851–858
Parker KA (2008) Translocations: providing outcomes for wildlife, resource managers, scientists, and the human community. Restor Ecol 16:204–209
Pearse DE, Avise JC (2001) Turtle mating systems: behavior, sperm storage, and genetic paternity. J Hered 92:206–211
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoSONE 7:e37153
Quesnelle PE, Fahrig L, Lindsay KE (2013) Effects of habitat loss, habitat configuration and matrix composition on declining wetland species. Biol Conserv 160:200–208
Ricciardi A, Simberloff D (2009) Assisted colonization is not a viable conservation strategy. Trends Ecol Evol 24:248–253
Schwartz MW, Hellmann JJ et al (2012) Managed relocation: integrating the scientific, regulatory, and ethical challenges. Bioscience 62:732–743
Seber GAF (1982) The estimation of animal abundance and related parameters. Blackburn Press, New Jersey
Seminoff JA, Shanker K (2008) Marine turtles and IUCN Red Listing: a review of the process, pitfalls, and novel assessment approaches. J Exp Mar Biol Ecol 356:52–68
Somers AB (2000) A population of Bog Turtles in the Piedmont of North Carolina: habitat preferences, capture method efficacy, conservation initiatives, and site enhancement. In: Report submitted to Natural Resources Conservation Service Wetlands Institute
Spielman D, Brook BW, Frankham R (2004) Most species are not driven to extinction before genetic factors impact them. PNAS 101:15261–15264
Struhsaker TT, Siex KS (1998) Translocation and introduction of the Zanzibar red colobus monkey: success and failure with an endangered island endemic. Oryx 32:277–284
Strum SC (2005) Measuring success in primate translocation: a baboon case study. Am J Primatol 65:117–140
Swift TL, Hannon SJ (2009) Critical thresholds associated with habitat loss: a review of the concepts, evidence, and applications. Biol Rev 85:35–53
Tarszisz E, Dickman CR, Munn AJ (2014) Physiology in conservation translocations. Conserv Physiol 2:1–19
Tesauro J (2001) Restoring wetland habitats with cows and other livestock. Conservation 2:26–31
United States Fish and Wildlife Service (USFWS) (2001) Bog Turtle (Clemmys muhlenbergii) Northern population recovery plan. Prepared by M Klemens. Pennsylvania Field Office, Hadley, Massachusetts
Wahlund S (1928) Zusammensetzung von Population und Korrelationserscheinung vom Standpunkt der Vererbungslehre aus betrachtet. Hereditas 11:65–106
Watson DM, Watson MJ (2015) Wildlife restoration: mainstream translocations to keep common species common. Biol Conserv 191:830–838
Weeks AR, Sgro CM, Young AG, Frankham R, Mitchell NJ, Miller KA, Byrne M, Coates DJ, Eldridge MDB, Sunnucks P, Breed MF, James EA, Hoffmann AA (2011) Assessing the benefits and risks of translocations in changing environments: a genetic perspective. Evol Appl 4:709–725
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolut Int J org Evolut 38:1358–1370
Whitlock AL (2002) Ecology and status of the Bog Turtle (Clemmys muhlenbergii) in New England. Dissertation, University of Massachusetts Amherst
Zeigenfuss LC, Singer FJ, Gudorf MA (2000) Test of a modified habitat suitability model for bighorn sheep. Restor Ecol 8:38–46
Acknowledgements
This translocation project was the idea of Bern W. Tryon (late Director of Animal Collections/Herpetology) at Zoo Knoxville. We thank the private landowners and the Nature Conservancy for living accommodations and access to sites, our volunteers and/or field technicians, Annlynn Eastin, Doug Rice, Stephen Nelson, Brad Moxley, Tessa Bickhart, and Ann Somers for their assistance collecting tissue samples. A special thanks to Todd Pierson for his invaluable assistance in the lab and the Project Bog Turtle organization for their support. Financial support was provided by the Association of Zoos and Aquariums (AZA) Conservation Grants Fund (CGF) [14-1245] from the Disney Worldwide Conservation Fund (DWCF), the Ecology and Evolutionary Biology Department at the University of Tennessee, and Zoo Knoxville. Approved by IACUC at the University of Tennessee [2436-0316].
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dresser, C.M., Ogle, R.M. & Fitzpatrick, B.M. Genome scale assessment of a species translocation program. Conserv Genet 18, 1191–1199 (2017). https://doi.org/10.1007/s10592-017-0970-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-017-0970-6