
Abstract
Captive bred individuals are often released into natural environments to supplement resident populations. Captive bred salmonid fishes often exhibit lower survival rates than their wild brethren and stocking measures may have a negative influence on the overall fitness of natural populations. Stocked fish often stem from a different evolutionary lineage than the resident population and thus may be maladapted for life in the wild, but this phenomenon has also been linked to genetic changes that occur in captivity. In addition to overall loss of genetic diversity via captive breeding, adaptation to captivity has become a major concern. Altered selection pressure in captivity may favour alleles at adaptive loci like the Major Histocompatibility Complex (MHC) that are maladaptive in natural environments. We investigated neutral and MHC-linked genetic variation in three autochthonous and three hatchery populations of Austrian brown trout (Salmo trutta). We confirm a positive selection pressure acting on the MHC II β locus, whereby the signal for positive selection was stronger in hatchery versus wild populations. Additionally, diversity at the MHC II β locus was higher, and more uniform among hatchery samples compared to wild populations, despite equal levels of diversity at neutral loci. We postulate that this stems from a combination of stronger genetic drift and a weakening of positive selection at this locus in wild populations that already have well adapted alleles for their specific environments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Animals have been kept (and bred) under human care since the Neolithic. The environmental conditions in captivity often differ drastically from natural surroundings and include altered diet, increased or reduced populations sizes, different pathogen stress and the release from ecological forces such as competition or predation. These changes manifest themselves in behavioural (Price 1999; Reinhardt 2001), morphological (O’Regan and Kitchener 2005; Stringwell et al. 2014) and physiological changes (Quispe et al. 2014; Roznere et al. 2014).
Rare or endangered species are often bred in captivity to support reintroduction programs, such as those for the Przewalski horse (Equus ferus przewalskii; Roznere et al. 2014), the black-footed ferret (Mustela nigripes; Miller et al. 1996) and Mexican and Red wolves (Canis lupus baileyi and Canis lupus rufus; Hedrick and Fredrickson 2008). However, only about 13% of captive breeding and reintroduction efforts are successful, whereas reintroductions not involving captive breeding (termed relocations) have a higher success rate (31%; Fischer and Lindenmayer 2000; Williams and Hoffman 2009). The causes of reintroduction failures have been linked to ecological factors or mismanagement, but genetic factors have also been suggested (Fischer and Lindenmayer 2000; Jiménez et al. 1994).
Interest has grown concerning the genetic changes that arise during captivity, especially those that may be disadvantageous in natural environments (Kitada et al. 2009). Frankham et al. (2002) summarise these changes to include: (1) loss of genetic diversity, (2) inbreeding depression, (3) accumulation of mildly deleterious mutations, and (4) genetic adaptation to captivity. While the first three changes result from small population sizes, genetic adaptation to captivity is strongest at large effective population sizes. Most traits selected for in captivity are disadvantageous in natural environments (Frankham 2008). While measures to reduce both loss of genetic diversity and artificial selection (i.e. adaptation to captivity) in breeding programs are often considered (Ballou and Foose 1996; Fraser 2008), these measures can be quite contradictory (Frankham 2008). Therefore, to optimize management of a captive population and to maximize its viability in the wild, it is of crucial importance to understand the alterations in evolutionary forces (e.g. genetic drift or selection) and genetic characteristics of specific captive population relative to their wild counterparts.
The rearing and transport of salmonid fishes in Europe can be traced back to the Middle Ages (Pechlaner 1984) while artificial propagation of trout in Germany began around the eighteenth century (Borgstrom 2012). Genetic studies on Austrian brown trout have shown that stocked hatchery fish are mainly of the Atlantic mtDNA lineage whereas limited numbers of native populations were found to consist mainly of Danubian mtDNA lineage fish (Bernatchez 2001; Lerceteau-Köhler et al. 2013).
Aside from a broad contact zone between the upper Danubian watershed and north-flowing drainage systems (e.g. Rhine or Elbe), the Danubian and Atlantic lineages existed largely in allopatry before human stocking activities began (Lerceteau-Köhler et al. 2013). The natural range of the Danubian lineage encompasses the drainages of the Black, Caspian, Azov and Aral seas, reaching from Bavaria, Germany (upper Danube) to eastern tributaries of the Aral Sea in Afghanistan and Pakistan. While most Atlantic lineage brood stocks used in Austria stem from central Europe (e.g. Czech Republic) or southern Scandinavia (i.e. Denmark), the Atlantic lineage naturally ranges from North Africa as well as some Mediterranean islands, up the entire Atlantic coast of Europe and into Central Europe and Scandinavia (Bernatchez 2001). Thus, both of these so-called lineages encompass enormous regions of highly heterogeneous habitats with thousands of isolated populations in small to large rivers, lakes, and estuary habitats.
Evidence for lower survival of hatchery-reared salmonids in natural environments is overwhelming, has been documented since the 1960s (Flick and Webster 1967; McGinnity 1997; Hansen et al. 2000; McGinnity et al. 2003; Fraser 2008), and includes a study on Austrian brown trout (Weiss and Schmutz 1999). Even if captive-reared fish stem from the local population they are being stocked into, the maladaptive effect of captivity can be quite drastic. Araki et al. (2007) demonstrated a reduction in the reproduction of hatchery-reared steelhead (i.e. anadromous rainbow trout Oncorhynchus mykiss) in the wild within three generations of captivity. This genetic effect of domestication, stemmed solely from differing environments as the captive-reared and wild fish had the same source population. Christie et al. (2016) demonstrated large-scale gene expression differences (i.e. across 723 genes) in steelhead attributed to a single generation in captivity, with the involved genes associated with wound healing, immunity and metabolism.
The Major Histocompatibility Complex (MHC) comprises a well-studied functional group of genes with a key role in the immune system. Two main subfamilies of the MHC, namely MHC class I, and MHC class II, encode for glycoproteins that present peptides to specialist immune cells and are therefore crucial for pathogen recognition and immune response. Some MHC genes have revealed astoundingly high levels of variation in many species (Klein 1986; Hughes and Yeager 1998). Empirical data suggests that positive, parasite-driven selection underlies this high variation whereby the exact mechanism behind the positive selection is still debated (Paterson et al. 2004; Sommer 2005; Piertney and Oliver 2006; Zhang et al. 2015). Nonetheless, the high variability and adaptive significance of MHC genes make them ideal candidates to study selection pressure in non-model species (e.g. Shafer et al. 2012; Chen et al. 2015).
For salmonids, the genetic variability of MHC loci has been linked to kin discrimination (Olsén et al. 1998), mate choice (Landry et al. 2001; Forsberg et al. 2007), embryo viability (Jacob et al. 2010) and pathogen infections (Consuegra and Garcia de Leaniz 2008; Lamaze et al. 2014), and also play an important role in the viability of invasive species or non-native strains (O’Farrell et al. 2013; Monzón-Argüello et al. 2014). The negative effects that stocking can have on immunogenetic traits (including MHC variability and expression) have also been documented in salmonids (Currens et al. 1997; Lamaze et al. 2014).
We genotyped 17 persumably neutral microsatellite loci and three MHC-linked microsatellite loci and cloned and sequenced a specific MHC class II locus (MHC II β, called Satr-DAB in S. trutta) in three wild Austrian and three hatchery populations of brown trout to assess both neutral and adaptive genetic variation in captive versus wild populations. The results reveal insights into the relative roles of genetic drift and selection pressure in captive and wild populations and may help steer captive breeding strategies toward more sustainable and ecologically beneficial management strategies.
Materials and methods
Sample populations
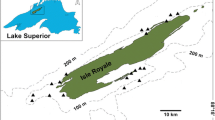
Six brown trout populations (three wild populations and three hatcheries) were used in the analysis (Table 1; Fig. 1). Whereas some tissue samples are identical to those used in Lerceteau-Köhler et al. (2013), sample sizes were increased in 2012 and 2013 using non-invasive tissue sampling during standard electrofishing monitoring, approved by the local responsible authorities. The three wild populations (Etrachbach-ETR, Lohnbach-LOH and Wolfsgrabenbach-WOL) represent the native Danubian lineage with no sign of introgression from foreign sources (Lerceteau-Köhler et al. 2013; Schenekar et al. 2014). Etrachbach and Wolfsgrabenbach are in the southern provinces of Austria (Styria and Carinthia), whereas Lohnbach lies in Lower Austria, a province northeast of the Alps. Hatchery 1 (Hat1) represents a local hatchery near Graz, Styria, hatchery 2 (Hat2) a commercial hatchery in Lower Austria, and hatchery 3 (Hat3) a large commercial hatchery in Denmark. These three hatcheries represent widely distributed strains used for stocking in Austria and consist primarily of Atlantic lineage fish (Lerceteau-Köhler et al. 2013). Based on previous analyses these hatchery populations are all relatively homogeneous and represent the same major sub-lineage of the Atlantic basin (Lerceteau-Köhler et al. 2013; Schenekar et al. 2014). All finclips were preserved in 96% ethanol, total DNA was extracted using a high-salt (ammonium acetate) protocol (Sambrook et al. 1989).

a Locations of the three wild populations analysed in this study: ETR Etrachbach, LOH Lohnbach and WOL Wolfsgrabenbach, b relative location of Austria in Europe in relation to the major rivers
Microsatellite (SSR) typing
A total of 17 neutral microsatellite loci (neutral SSRs) were amplified in three multiplex reactions, as in Schenekar et al. (2014). The data for the neutral SSRs for the hatchery populations and LOH stem from Schenekar et al. (2014), whereas all other data were newly generated. Three additional microsatellites linked to MHC loci (adaptive SSRs), and first described in Atlantic salmon (Salmo salar; Grimholt 1997; Grimholt et al. 2002; Gharbi et al. 2009) were amplified in a 3-plex reaction: (1) Satr-UBA is a tightly linked microsatellite in the 3′-untranslated region of the MHC I locus Satr-UBA and has been applied in brown trout (Coughlan et al. 2006; O’Farrell et al. 2012). (2) Satr-TAP2b (nomenclature of Lukacs et al. 2007, corresponding to TAP2A of; Grimholt et al. 2002) is located in intron 5 of the TAP2A gene. The TAP protein is involved in transporting peptides into the endoplasmic reticulum, where they bind to MHC class I molecules. The TAP locus is on a different linkage group than UBA and was successfully employed in S. trutta (Hansen et al. 2007). (3) Ssa60NVH is linked to the MHC II DAA locus and has also been used in S. trutta (Gharbi et al. 2006; Keller et al. 2011). These markers were chosen to serve as proxies for additional adaptive markers, assuming that they should retain some signal of selection acting on the linked adaptive genes.
Each PCR reaction contained 0.5 µl primer mix (containing 2 µM of each primer), 2.5 µl Type-it Multiplex PCR Master Mix (Qiagen) and 20–40 ng template DNA. The final reaction volume was 6 µl. The forward primers were fluorescently labelled and reverse primers carried a “pig-tail”-sequence (5′-GTTTCTT-3′) at their 5′-end to increase genotyping accuracy. The primer sequences used were as follows: (1) Satr-UBA-fwd 5′-GGAGAGCTGCCCAGATGACTT-3′ and Satr-UBA-rev-pt 5′-GTTTCTTCAATTACCACAAGCCCGCTC-3′, (2) Satr-TAP2-fwd 5′-GCGGGACACCGTCAGGGCAGT-3′ and Satr-TAP2-rev-pt 5′-GTTTCTTGTCCTGATGTTGGCTCCCAGG-3′, and (3) Ssa60NVH-fwd 5′-TTGTGGAGTATTTAGCAATC-3′ and Ssa60NVH-rev-pt 5′-GTTTCTTATTGGCAGACATGCACTC-3′. PCR cycling conditions were as follows: An initial denaturation at 95 °C for 5 min, followed by 25 cycles of denaturation at 95 °C for 30 s, annealing for 1:30 min at 54 °C and extension at 72 °C for 30 s with a final extension step at 60 °C for 30 min. PCR products were mixed with 0.25 µl size standard (GeneScan 500 ROX Size Standard, life technologies) and 10 µl formamide and run on an ABI 3130xL Genetic Analyzer. Allele calling and microsatellite typing was performed in GeneMapper Software v3.7 (Applied Biosystems). Raw SSR data was tested for potential null-alleles with the program Micro-Checker 2.2.3 (Van Oosterhout et al. 2006) and the software COLONY 2.0 (Jones and Wang 2010) was used to screen for identical multi-locus genotypes in order to identify potential recaptured individuals.
MHC II β sequencing
We amplified, cloned and sequenced a 254–257 bp fragment of exon 2 of the MHC II β (Satr-DAB) locus, which exists as a single copy in salmonids, including brown trout (Hordvik et al. 1993; Jacob et al. 2010). The MHC II β locus is called Satr-DAB in S. trutta, but in the interest of uniformity with other cross-taxa studies, we will refer to it as MHC II β. We used primers CL007 5′-GATCTGTATTATGTTTTCCTTCCAG-3′ and AL1002 5′-CACCTGTCTTGTCCAGTATG-3# (Olsén et al. 1998). Each PCR reaction contained 5 µl TopTaq Master Mix (Qiagen), 0.2 μM of each primer and 40–80 ng template DNA, brought up with water to a final reaction volume of 10 μl. PCR cycling conditions were as follows: an initial denaturation at 94 °C for 3 min, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing for 30 s at 62 °C, and extension at 72 °C for 1 min, with a final extension step at 72 °C for 10 min. PCR products were cloned in E. coli DH5α cells into a pGEM®-5Zf(+) Vector (Promga) that we modified to add T-overhangs using the protocol of Marchuk et al. (1991). A total of 6–8 positive clones (using blue-white selection) were amplified with primers T7 5′-TAATACGACTCACTATAGGG-3′ and SP6 5′-ATTTAGGTGACACTATAG-3′ using a High-Fidelity Polymerase (KAPA-HiFi; Peqlab). Positive clones were sequenced in both directions using primers CL007 and AL1002. Sequencing products were run on an ABI 3130xl Genetic Analyzer.
Genetic diversity and differentiation—microsatellites (SSR)
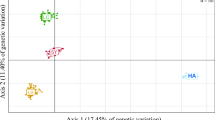
Estimations of the number of alleles (N A), allelic richness (A R), and testing for Hardy–Weinberg-Equilibrium (based on F IS- values using 1000 permutations) were carried out in FSTAT 2.9.3 (Goudet 1995). Observed and expected heterozygosity (H O and H E, respectively) as well as pairwise F ST-values were calculated in Arlequin 3.5 (Excoffier and Lischer 2010). To aid in comparing markers with differing (or very high) mutation rates, we additionally calculated Hedrick’s standardized G ST-values (G′ ST; Hedrick 2005) using the software SMOGD (Crawford 2010). Mantel tests were used to test for a correlation between values (of both, F ST and G′ ST) of neutral and adaptive SSRs, but also between neutral SSRs and MHC II β sequences. Linkage disequilibrium between adaptive SSRs and MHC II β haplotypes was also tested in Arlequin. Three-dimensional Factorial Correspondence Analyses (FCAs) were used to depict general genetic relationships among populations for the 17 neutral SSRs, and the three adaptive SSRs in GENETIX 4.05.2 (Belkhir et al. 1996). Effective population sizes (N es) were estimated with the program LDNe (Waples and Do 2008). We calculated a “relative adaptive allelic richness” for each population, by subtracting the mean A R of the 17 neutral SSRs from the A R of the MHC II β sequences or the MHC-linked SSRs. Wilcoxon signed rank-tests were used to test whether relative adaptive variability differed between hatcheries and wild populations.
Genetic diversity and differentiation—MHC II β
Sequences were edited and aligned in MEGA 5.2 (Tamura et al. 2011). To retain an allele it had to be, (1) sequenced in at least 3 bacterial clones, and (2), found in clones from at least two independent PCRs or had to be a 100% match to a previously described allele in GenBank. Individuals were retained if there were (1), at least five cloned sequences after filtering out potential false alleles, and (2), less than three alleles after allele filtering. After filtering, all haplotypes were BLASTed to the NCBI GenBank database and new alleles were named following Klein et al. (1990) and the rules of the IPD-MHC Database for fish, continuing the allele numbering described in Shum et al. (2001) and Monzón-Argüello et al. (2014). Number and frequency of alleles, diversity indices (allelic richness—A R, expected and observed heterozygosity—H E & H O, etc), F IS values and further analyses concerning genetic diversity and differentiation were calculated similarly as described for SSRs. Gene diversity and Tajima’s D were calculated in DnaSP 5.10.01 (Librado and Rozas 2009). To test for selection signals in sequence diversity, Tajima’s D test and Fu & Li’s tests were carried out in DNaSP and Slatkins implementation of the Ewens–Watterson neutrality test was carried out in PyPop 0.7.0 (Lancester et al. 2003).
Selection and recombination on the MHC II β locus
Selection at the protein-coding level of the MHC II β locus was evaluated using the ratio of the nonsynonymous substitutions per nonsynonymous site (dN) to the synonymous substitutions per synonymous site (dS) (i.e. the dN/dS ratio), calculated in DnaSP using a Jukes-Cantor correction for multiple hits. A codon-based Z-test implemented in MEGA was carried out to test for positive selection using 1000 bootstrap replicates. Comparisons of dN/dS ratios between hatcheries and wild populations were done with a Mann Whitney-U-test and a Wilcoxon signed rank-test.
Selection on individual codons was evaluated using codon-specific dN/dS ratios using the program CODEML of the PAML 4.7 software package (Yang 2007). Likelihood ratio tests (LRTs) were used to test for varying dN/dS ratios along the sequence and for positive selection comparing four nested model-pairs (Wong et al. 2004). A Bayes empirical Bayes (BEB) estimation was used to identify sites under positive selection (Yang et al. 2005). Each model was run for all six populations separately and for all haplotypes found in the whole dataset. Tree topologies were calculated by the Maximum Likelihood algorithm implemented in MEGA 5.2 whereby the best fitting substitution model was evaluated with jModeltest 2.1.4 (Darriba et al. 2012) for each dataset separately. A Wilcoxon signed-rank test was used to test for differing dN/dS ratios between antigen-binding sites (ABS) and non-ABS, as defined by the human model (Brown et al. 1993) and applied to salmonid fishes (Aguilar and Garza 2007; Gómez et al. 2010).
Since the MHC is highly recombinogenic in many species, including brown trout (Shum et al. 2001; O’Farrell et al. 2013), an additional Bayesian approach accounting for recombination was implemented in the software omegaMap 0.5 (Wilson and McVean 2006). The dN/dS ratios, posterior probabilities of selection and population recombination rate (ρ) were calculated for each codon separately (independent models of ρ & ω; details see Online Resource 1).
To test for recombination, we ran a permutation test implemented in the program Permute of the omegaMap software package. The program uses three LD statistics: (1), r 2 = the square of the correlation coefficient (2), D′ = the standardized LD coefficient (Lewontin 1964) and (3), G4 = the statistic of the four-gamete test (Meunier and Eyre-Walker 2001). Permute was run for each population separately and over the whole dataset with 10,000 permutations for each. The minimum number of recombination events was calculated by the method of Hudson and Kaplan (1985) implemented in DnaSP. The program LDhat v2.2 was used to estimate the population-scaled recombination rate ρ = 4N e r, where N e is the effective population size and r is the recombination rate per gene per generation (McVean et al. 2002).
Results
Genetic diversity and differentiation: adaptive and neutral SSRs and MHC II β
Patterns of genetic diversity across groups of markers and populations were heterogeneous, but generally highest for the MHC II β locus in hatchery populations, and lowest for both MHC-linked SSRs and the MHC II β in wild populations (Table 2). Both, the pairwise FST (Fig. 2) and the FCA of the 17 neutral SSRs showed clearly support distinction among all wild populations as well as between wild and hatchery populations, however, the three hatchery populations themselves could not be clearly distinguished. The clear distinctions among wild, or between wild and hatchery populations were less evident based on the three adaptive SSRs. All three marker types gave significant F ST- and G′ ST-values between all pairs of populations (p < 0.0001; Online Resource 2). Mantel tests revealed a significant correlation between the neutral versus adaptive SSRs, F ST- and G′ ST, but no significant correlation between the neutral SSRs and MHC II β (Fig. 2). For comparisons among wild populations, both, F ST- and G′ ST-values of MHC II β were clearly higher than the respective F ST- and G′ ST-values of neutral SSRs, however, due to the lack of power, there was no significant difference among those values (at α = 0.05). Estimates of effective population sizes (N es) ranged from 20 to 162 individuals (Table 3) and were slightly higher (on average) for hatchery than for wild populations (104 versus 78 individuals) but the difference was not significant (Wilcoxon signed rank-test, W = 0.667, p = 0.41).

Correlation between (a) F ST–values of 17 neutral microsatellites (SSRs) and F ST–values of the 3 adaptive SSRs, b F ST–values of 17 neutral microsatellites (SSRs) and F ST–values of the MHC II β locus, c G′ ST–values of 17 neutral microsatellites (SSRs) and G′ ST–values of the 3 adaptive SSRs, and d G′ ST–values of 17 neutral microsatellites (SSRs) and F ST–values of the MHC II β locus. Data points are labelled according to population types (hatchery or wild) of the population pair for the respective F ST–value. The dashed line indicates a 1:1 correlation; r correlation coefficient of the Mantel test, p p value of the Mantel test
The relative adaptive A R of MHC II β of hatchery populations was significantly higher than for wild populations (Median Test; p = 0.014) using an asymptotic estimation but not using an exact estimation (p = 0.100). For the three adaptive SSRs, no significant differences between hatcheries and wild populations could be observed, even though the MHC II β-linked SSR (Ssa60NVH), showed a similar pattern than the MHC II β locus itself.
MHC II β sequence diversity, selection and recombination
The final 257 bp MHC II β alignment (N = 153 individuals) revealed 37 haplotypes, of which 19 were novel (Online Resource 3). Eight additional haplotypes were previously described but using sequences that were a few base pairs shorter in length than ours. These eight alleles received the identical allele number as the previously published sequence but a suffix (−a) was added to denote the difference in length. Our global alignment contained 72 polymorphic sites including a 3 bp—indel polymorphism, which has been previously reported (e.g. Shum et al. 2001; Campos et al. 2006; Forsberg et al. 2007).
Significant positive selection across the data set was supported by Fu and Li’s D* and F* as well as the Codon-based Z-test, in addition to several significant tests for individual populations (Table 3). The Tajima’s D test was only significant for ETR but with a negative value, indicating directional rather than positive selection.
The null hypothesis of no recombination was rejected in all three Permute LD tests concerning the wild populations and the whole dataset (Online Resource 4). In hatchery populations, only the permutation test for r 2 was significant. However, the minimum number of recombination events R m and the population recombination rate ρ were on average higher in hatchery compared to wild populations, but the difference was only significant for ρ (Wilcoxon signed rank-test, W = 6.0 and p = 0.014).
Selection on the MHC II β locus based on dN/dS ratios
The average pairwise dN/dS ratio indicated strong positive selection (mean dN/dS ratio overall: 3.33), which was higher in hatcheries versus wild populations (means of 3.67 versus 2.78 respectively; Mann Whitney-U and Wilcoxon signed rank-test, U = 12.496 and R = 29.4, both p < 0.001; Fig. 3; Table 3).

Codon-specific dN/dS ratios of MHC II β from PAML analyses. The dN/dS ratios are based on model M8 for the all-haplotypes dataset. Bars represent mean dN/dS ratios and error bars indicate standard deviations. Asterisks above a bar indicate a position suggested to be under positive selection with a posterior probability of ** >0.99, or * >0.95. Arrows above the panel indicate antigen binding sites (ABSs) derived from the human HLA-DR1 locus (Brown et al. 1993). Codon positions correspond to GenBank entry DQ257393
Likelihood ratio tests based on the PAML analysis revealed varying dN/dS ratios along the sequence over all haplotypes (M0 versus M3), as well as strong positive selection (all three nested-model pair tests were significant). At least one of these three tests of positive selection was highly significant in each population, with the exception of WOL. This population was moreover the only population that did not show significant signals of a varying dN/dS ratio along the sequence.
A total of 18 sites were suggested to be under positive selection with a posterior probability >0.99, whereby 13 of these were at antigen binding sites (ABS). ABSs had a significantly higher dN/dS ratio than non-ABS (Wilcoxon signed rank-test, W = 11.3, p < 0.001). In total, four codon positions to be under positive selection were shared among all hatcheries, whereas none were shared among wild populations (Online Resource 5). In general, fewer codon positions were suggested to be under positive selection in wild (mean = 3) compared to hatchery (mean = 12) populations.
The Bayesian analysis revealed a similar pattern of dN/dS ratios. Of the 20 sites revealing support for positive selection >0.99, 14 are suggested to be ABSs. Point estimates for the population recombination rate (ρ) ranged from 0.07 to 10.16.
OmegaMap analyses for individual populations, revealed sites suggested to be under positive selection in all populations. Twelve of these sites were shared among all three hatchery populations, whereas only four were shared among all three wild populations. However, only two sites were unique to a single hatchery population, whereas nine sites were unique to single wild populations. There was a significant difference in the frequency distribution of sites under positive selection in the three categories (1), unique to a population (2), shared by two populations, and (3), shared by all three population between the hatcheries and wild populations (χ2 = 9.07, p = 0.011).
Discussion
We found significantly more variation at adaptive markers (MHC II β and adaptive SSRs) in hatchery versus wild populations. Results varied from locus to locus but differences between hatchery and wild populations were most pronounced at MHC II β, underscored by the stronger signals of positive selection in hatchery versus wild populations. The increased genetic diversity at this locus could not be explained by higher effective population sizes (N es), as there was no significant differences in mean N e for hatcheries versus wild populations, and there was additionally no correlation between overall diversity of neutral versus adaptive markers.
Furthermore, we attempted to correct for potential effects of differing N e by applying an index reflecting relative adaptive allelic richness. This index was also significantly higher in hatcheries compared to wild populations for MHC II β, further supporting our inference of stronger, positive selection pressure in hatcheries. The linked adaptive microsatellite Ssa60NVH shows a quite similar, but not so pronounced pattern. Both, Satr-UBA and TAP2b show either no signs of selection or negative selection pressure in both hatchery and wild populations.
Further insights into the active mechanism responsible for these differences can be elucidated through closer inspection of the differentiation estimates. F ST-values among wild populations were much higher than among hatchery populations and neutral SSRs could clearly separate wild populations but not hatchery populations from each other. Clearly, genetic drift is acting stronger on these wild populations compared to hatchery populations. The smaller separation at the three MHC linked SSRs may be due to the lower resolution of three versus 17 loci, but could also be a sign of balancing selection (Schierup et al. 2000). However, the MHC II β F ST and G′ ST values among wild populations were all higher than F ST and G′ ST values of neutral SSRs (similarly for adaptive SSRs, with one exception), a pattern better explained by directional rather than balancing selection (Holsinger and Weir 2009).
Selection pressure on MHC IIβ
While selection tests based on sequence diversity (Tajima’s D, Fu and Li’s and Ewens-Watterson neutrality test) were non-significant, tests at the protein-coding level showed strong signals of positive selection, seen by an overall dN/dS ratio >3 and significant likelihood ratio tests in the PAML analyses. These signals were also more pronounced in hatchery than in wild populations (dN/dS ratios of 3.67 versus 2.78). Both, PAML and omegaMap analyses showed similar patterns of varying dN/dS ratio along exon 2 of MHC II β, with particularly high dN/dS ratios at antigen binding sites (ABSs). This pattern has been repeatedly found in various taxa and has been interpreted to result from pathogen-driven selection leading to extremely high intra-population genetic diversity, (e.g. Hughes and Nei 1989; Hedrick and Kim 1998; Kamath and Getz 2011; Chen et al. 2015).
Our omegaMap analyses also supported a higher number of codon positions under selection in hatchery versus wild populations. In contrast, more sites unique to single populations were suggested to be under positive selection in wild compared to hatchery populations. PAML analyses did not detect any codon positions under positive selection that were shared between all three wild populations but PAML may not be as reliable as omegaMap due to recombination in our dataset.
There was no obvious correlation between the dN/dS ratios and ρ along the sequence and no recombination hotspot could be observed. Therefore, it is more likely that selection is a much stronger force than recombination in shaping the genetic diversity of MHC II β, similar to what was observed at a MHC I locus, outside an important recombination hotspot (O’Farrell et al. 2013).
The differing selection patterns of MHC II β between hatcheries and wild populations may result from one or both of two scenarios:
Wild brown trout populations in our study are highly fragmented and found in isolated river stretches with wholly different environmental characteristics (e.g. with respect to hydrology, geology and overall fauna). Thus they are also less likely challenged by migrants from distant populations, more likely to carry new pathogens. In comparison, our hatcheries use similar infrastructure, as well as periodic supplementation leading to considerably more homogeneity in environmental conditions and pathogen exposure. Thus, this selection landscape markedly differs from that experienced by wild populations. Altered selection pressure is a logical consequence of life in an artificial environment and has long been a major concern in conservation (Lynch and Hely 2001). Our wild brown trout populations may be more strongly influenced by genetic drift than by selection (already suggested by Jorde and Ryman 1996). Campos et al. (2006) also observed a strong effect of genetic drift that might have eroded the effect of balancing selection in wild brown trout populations in Spain. In hatcheries, selection appears to outweigh drift, albeit selection for an artificial environment. Similarly, Atlantic salmon also revealed a relatively higher genetic diversity at MHC loci in Australian hatchery populations compared to their ancestral Canadian natural populations suggesting a strong impact of selection in the hatchery-environment (Wynne et al. 2007).
Individual wild brown trout populations may exhibit specialized, well-adapted alleles at the MHC II β locus that have the highest fitness advantage in this specific environment and pathogen milieu. Additionally, based on lower individual- and pathogen density in wild populations, the pathogen-driven balancing selection is slowed and/or weakened. This may shift the selection regime on the MHC towards “directional-selection”, leading to weaker signals of positive selection. This has also been suggested by Keller et al. (2011), investigating wild brown trout populations along an altitudinal gradient.
Implications for stocking measures in Austria
Our results suggest that stocking of non-autochthonous brown trout in Austria is not only non-advisable because of the intermixture of (neutral) genetic variability and loss of differentiation among populations throughout their natural range, but also because of the introduction of disadvantageous alleles of adaptive loci like the MHC genes. The introduction of hatchery fish that stem from a different genetic lineage can result in introgression of MHC alleles that have a fitness disadvantage for that specific environment thus leading to a lower population viability (Bert et al. 2007 and citations therein). Therefore, the introduction of maladaptive MHC alleles may be among the causative factors in of these population declines or/fitness reductions in heavily stocked populations.
Additionally, the introduction of fish reared in high-density hatcheries is very likely to introduce non-native pathogens into the wild populations. These pathogens may not be pathogenic to the hatchery population but indeed so in wild populations. This could lead to the frequently observed population declines in stocked populations. An example of the negative impact of stocked fish on wild populations by parasite spread is the pink salmon (Oncorhynchus gorbuscha), being infected with the salmon lice (Lepeophtheirus salmonis; Krkosek et al. 2007).
Our results support that stocking measures in autochthonous populations should be avoided, especially with non-native fish. If stocking measures are inevitable in natural habitats, ideally, locally established brood-stocks with local genetic material should be used. Adaptation to captivity should be minimized, e.g. by the continuous supplementation of new “natural” genetic material in order to keep the genetic composition of the captive population as close to its source population as possible. Nonetheless, genetic or epigenetic changes can begin in the first generation of captivity (Christie et al. 2016) and thus it appears to be extremely difficult or impossible to use hatchery operations in any capacity without risking deleterious effects to the wild population. While stocking in Austria will continue to be used to support various forms of sport fishing, we strongly advocate that the few remaining purely native populations be spared from such potentially damaging activity.
References
Aguilar A, Garza JC (2007) Patterns of historical balancing selection on the salmonid major histocompatibility complex class II beta gene. J Mol Evol 65:34–43. doi:10.1007/s00239-006-0222-8
Araki H, Cooper B, Blouin MS (2007) Genetic effects of captive breeding cause a rapid, cumulative fitness decline in the wild. Science 318:100–103. doi:10.1126/science.1145621
Ballou JD, Foose T (1996) Demographic and genetic management of captive populations. In: Kleiman DG, Allen ME, Thompson KV, Lumpkin S (eds) Wild animals in captivity; principles and techniques. Univ. Chicago Press, Chicago, pp 263–283
Belkhir K, Borsa P, Chikhi L et al (1996) GENETIX 4.05, logiciel sous Windows TM pour la genetique des populations
Bernatchez L (2001) The evolutionary history of brown trout (Salmo trutta L.) inferred from phylogeographic, nested clade, and mismatch analyses of mitochondrial DNA variation. Evolution (N Y) 55:351–379. doi:10.1111/j.0014-3820.2001.tb01300.x
Bert TM, Crawford CR, Tringali MD et al (2007) Genetic management of hatchery-based stock enhancement. Ecol Genet Implic Aquac Act 6:123–174
Borgstrom G (2012) Fish as food V1: production, biochemistry, and microbiology. Elsevier, Elsevier
Brown JH, Jardetzky TS, Gorga JC et al (1993) Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364:33–39
Campos JL, Posada D, Morán P (2006) Genetic variation at MHC, mitochondrial and microsatellite loci in isolated populations of Brown trout (Salmo trutta). Conserv Genet 7:515–530. doi:10.1007/s10592-005-9063-z
Chen W, Bei Y, Li H (2015) Genetic variation of the major histocompatibility complex (MHC class II B gene) in the threatened Hume pheasant, Syrmaticus humiae. PLoS ONE 10:e0116499. doi:10.1371/journal.pone.0116499
Christie MR, Marine ML, Fox SE et al (2016) A single generation of domestication heritably alters the expression of hundreds of genes. Nat Commun 7:10676. doi:10.1038/ncomms10676
Consuegra S, Garcia de Leaniz C (2008) MHC-mediated mate choice increases parasite resistance in salmon. Proc R Soc B 275:1397–1403. doi:10.1098/rspb.2008.0066
Coughlan J, McGinnity P, O’Farrel B et al (2006) Temporal variation in an immune response gene (MHC I) in anadromous Salmo trutta in an Irish river before and during aquaculture activities. ICES J Mar Sci 63:1248–1255. doi:10.1016/j.icesjms.2006.03.025
Crawford NG (2010) Smogd: software for the measurement of genetic diversity. Mol Ecol Resour 10:556–557. doi:10.1111/j.1755-0998.2009.02801.x
Currens KP, Hemmingsen AR, French RA et al (1997) Introgression and susceptibility to disease in a wild production of rainbow trout. North Am J Fish Manag 17:1065–1078
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772–772
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. doi:10.1111/j.1755-0998.2010.02847.x
Fischer J, Lindenmayer DB (2000) An assessment of the published results of animal relocations. Biol Conserv 96:1–11
Flick WA, Webster DA (1967) Comparative first year survival and production in wild and domestic strains of brook trout, Salvelinus fontinalis. Trans Am Fish Soc 93:58–69
Forsberg LA, Dannewitz J, Petersson E, Grahn M (2007) Influence of genetic dissimilarity in the reproductive success and mate choice of brown trout-females fishing for optimal MHC dissimilarity. J Evol Biol 20:1859–1869. doi:10.1111/j.1420-9101.2007.01380.x
Frankham R (2008) Genetic adaptation to captivity in species conservation programs. Mol Ecol 17:325–333. doi:10.1111/j.1365-294X.2007.03399.x
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to Conservation Genetics. Cambridge University Press, New York
Fraser DJ (2008) How well can captive breeding programs conserve biodiversity? A review of salmonids. Evol Appl 1:535–586. doi:10.1111/j.1752-4571.2008.00036.x
Gharbi K, Gautier A, Danzmann RG et al (2006) A linkage map for brown trout (Salmo trutta): chromosome homeologies and comparative genome organization with other salmonid fish. Genetics 172:2405–2419. doi:10.1534/genetics.105.048330
Gharbi K, Glover KA, Stone LC, MacDonald ES, Matthews L, Grimholt U, Stear MJ (2009) Genetic dissection of MHC-associated susceptibility to Lepeophtheirus salmonis in Atlantic salmon. BMC Genet 10:20. doi:10.1186/1471-2156-10-20
Gómez D, Conejeros P, Marshall SH, Consuegra S (2010) MHC evolution in three salmonid species: a comparison between class II alpha and beta genes. Immunogenetics 62:531–542. doi:10.1007/s00251-010-0456-x
Goudet J (1995) FSTAT (Version 1.2): A Computer program to calculate F-statistics. J Hered 86:485–486
Grimholt U (1997) Transport-associated proteins in Atlantic salmon (Salmo salar). Immunogenetics 46:213–221. doi:10.1007/s002510050264
Grimholt U, Drabløs F, Jørgensen SM et al (2002) The major histocompatibility class I locus in Atlantic salmon (Salmo salar L.): polymorphism, linkage analysis and protein modelling. Immunogenetics 54:570–581. doi:10.1007/s00251-002-0499-8
Hansen MM, Ruzzante DE, Nielsen EE, Mensberg KD (2000) Microsatellite and mitochondrial DNA polymorphism reveals life-history dependent interbreeding between hatchery and wild brown trout (Salmo trutta L.). Mol Ecol 9:583–594. doi:10.1046/j.1365-294x.2000.00898.x
Hansen MM, Skaala O, Jensen LF et al (2007) Gene flow, effective population size and selection at major histocompatibility complex genes: brown trout in the Hardanger Fjord, Norway. Mol Ecol 16:1413–1425. doi:10.1111/j.1365-294X.2007.03255.x
Hedrick PW (2005) A standardized genetic differentiation measure. Evolution (N Y) 59:1633–1638
Hedrick PW, Fredrickson RJ (2008) Captive breeding and the reintroduction of Mexican and red wolves. Mol Ecol 17:344–350. doi:10.1111/j.1365-294X.2007.03400.x
Hedrick P, Kim T (1998) Genetics of complex polymorphisms: parasites and maintenance of MHC variation. In: Sing R, Krimbas C (eds) Genetics, evolution, and society. Harvard University Press, Cambridge, pp 205–233
Holsinger KE, Weir BS (2009) Genetics in geographically structured populations: defining, estimating and interpreting F(ST). Nat Rev Genet 10:639–650. doi:10.1038/nrg2611
Hordvik V, Grimholt U, Fosse V et al (1993) Cloning and sequence analysis of cDNAs encoding the MHC class II beta chain in Atlantic salmon (Salmo salar). Immunogenetics 37:437–441
Hudson R, Kaplan N (1985) Statistical properties of the number of recombination events in the history of a sample of DNA-sequences. Genetics 111:147–164
Hughes AL, Nei M (1989) Nucleotide substitution at major histocompatibility complex class II loci: evidence for overdominant selection. Proc Natl Acad Sci 86:958–962. doi:10.1073/pnas.86.3.958
Hughes AL, Yeager M (1998) Natural selection at major histocompatibility complex loci of vertebrates. Annu Rev Genet 32:415–435. doi:10.1146/annurev.genet.32.1.415
Jacob A, Evanno G, Von Siebenthal BA et al (2010) Effects of different mating scenarios on embryo viability in brown trout. Mol Ecol 19:5296–5307. doi:10.1111/j.1365-294X.2010.04884.x
Jiménez JA, Hughes KA, Alaks G, Graham L, Lacy RC (1994) An experimental study of inbreeding depression in a natural habitat. Science 265:271–273. doi:10.1126/science.7939661
Jones OR, Wang J (2010) COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol Ecol Resour 10:551–555. doi:10.1111/j.1755-0998.2009.02787.x
Jorde P, Ryman N (1996) Demographic genetics of brown trout (Salmo trutta) and estimation of effective population size from temporal change of allele frequencies. Genetics 143:1369–1381
Kamath PL, Getz WM (2011) Adaptive molecular evolution of the major histocompatibility complex genes, DRA and DQA, in the genus Equus. BMC Evol Biol 11:128. doi:10.1186/1471-2148-11-128
Keller I, Taverna A, Seehausen O (2011) Evidence of neutral and adaptive genetic divergence between European trout populations sampled along altitudinal gradients. Mol Ecol 20:1888–1904. doi:10.1111/j.1365-294X.2011.05067.x
Kitada S, Shishidou H, Sugaya T et al (2009) Genetic effects of long-term stock enhancement programs. Aquaculture 290:69–79. doi:10.1016/j.aquaculture.2009.02.011
Klein J (1986) The natural history of the major histocompatibility complex. Wiley, New York
Klein J, Bontrop R, Dawkins R et al (1990) Nomenclature for the major histocompatibility complexes of different species: a proposal. Immunogenetics 31:217–219
Krkosek M, Ford JS, Morton A et al (2007) Declining wild salmon populations in relation to parasites from farm salmon. Science 318:1772–1775. doi:10.1126/science.1148744
Lamaze FC, Pavey SA, Normandeau E et al (2014) Neutral and selective processes shape MHC gene diversity and expression in stocked brook charr populations (Salvelinus fontinalis). Mol Ecol 23:1730–1748. doi:10.1111/mec.12684
Lancester A, Nelson MP, Meyer D et al (2003) PyPop: a software framework for population genomics: analyzing large-scale multi-locus genotype data. Pac Symp Biocomput 8:514–525
Landry C, Garant D, Duchesne P, Bernatchez L (2001) “Good genes as heterozygosity”: the major histocompatibility complex and mate choice in Atlantic salmon (Salmo salar). Proc R Soc B 268:1279–1285. doi:10.1098/rspb.2001.1659
Lerceteau-Köhler E, Schliewen U, Kopun T, Weiss S (2013) Genetic variation in brown trout Salmo trutta across the Danube, Rhine, and Elbe headwaters: a failure of the phylogeographic paradigm? BMC Evol Biol 13:176. doi:10.1186/1471-2148-13-176
Lewontin R (1964) The interaction of selection and linkage. I. General considerations; heterotic models. Genetics 49:49–67
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. doi:10.1093/bioinformatics/btp187
Lukacs MF, Harstad H, Grimholt U et al (2007) Genomic organization of duplicated major histocompatibility complex class I regions in Atlantic salmon (Salmo salar). BMC Genomics 8:251. doi:10.1186/1471-2164-8-251
Lynch M, Hely MO (2001) Captive breeding and the genetic fitness of natural populations. Conserv Genet 2:363–378
Marchuk D, Drumm M, Saulino A, Collins FS (1991) Construction of T-vectors, a rapid and general system for direct cloning of unmodified PCR products. Nucleic Acids Res 19:1154–1154. doi:10.1093/nar/19.5.1154
McGinnity P (1997) Genetic impact of escaped farmed Atlantic salmon (Salmo salar L.) on native populations: use of DNA profiling to assess freshwater performance of wild, farmed, and hybrid progeny in a natural river environment. ICES J Mar Sci 54:998–1008. doi:10.1016/S1054-3139(97)80004-5
McGinnity P, Prodöhl P, Ferguson A et al (2003) Fitness reduction and potential extinction of wild populations of Atlantic salmon, Salmo salar, as a result of interactions with escaped farm salmon. Proc R Soc B 270:2443–2450. doi:10.1098/rspb.2003.2520
McVean G, Awadalla P, Fearnhead P (2002) A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160:1231–1241
Meunier J, Eyre-Walker A (2001) The correlation between linkage disequilibrium and distance: implications for recombination in hominid mitochondria. Mol Biol Evol 18:2132–2135
Miller B, Reading R, Forrest S (1996) Prairie night: black-footed ferrets and the recovery of endangered species. Smithsonian Institution Press, Washington, DC
Monzón-Argüello C, Garcia de Leaniz C, Gajardo G, Consuegra S (2014) Eco-immunology of fish invasions: the role of MHC variation. Immunogenetics 66:393–402. doi:10.1007/s00251-014-0771-8
O’Farrell B, Dennis C, Benzie JAH et al (2012) Balancing selection on MHC class I in wild brown trout Salmo trutta. J Fish Biol 81:1357–1374. doi:10.1111/j.1095-8649.2012.03421.x
O’Farrell B, Benzie JAH, McGinnity P et al (2013) Selection and phylogenetics of salmonid MHC class I: wild brown trout (Salmo trutta) differ from a non-native introduced strain. PLoS ONE 8:e63035. doi:10.1371/journal.pone.0063035
O’Regan H, Kitchener AC (2005) The effects of captivity on the morphology of captive, domesticated and feral mammals. Mamm Rev 35:215–230. doi:10.1111/j.1365-2907.2005.00070.x
Olsén K, Grahn M, Lohm J, Langefors à (1998) MHC and kin discrimination in juvenile Arctic charr, Salvelinus alpinus (L.). Anim Behav 56:319–327. doi:10.1006/anbe.1998.0837
Paterson S, Piertney SB, Knox D et al (2004) Characterization and PCR multiplexing of novel highly variable tetranucleotide Atlantic salmon (Salmo salar L.) microsatellites. Mol Ecol Notes 4:160–162. doi:10.1111/j.1471-8286.2004.00598.x
Pechlaner R (1984) Historical evidence for the introduction of Arctic charr into high-moutain lakes in the Alps by man. In: Johnson L, Burns L (eds) Biology of the Arctic charr. University of Manitoba Press, Winnipeg, pp 549–557
Piertney SB, Oliver MK (2006) The evolutionary ecology of the major histocompatibility complex. Heredity (Edinb) 96:7–21. doi:10.1038/sj.hdy.6800724
Price EO (1999) Behavioral development in animals undergoing domestication. Appl Anim Behav Sci 65:245–271. doi:10.1016/S0168-1591(99)00087-8
Quispe R, Villavicencio CP, Addis E et al (2014) Seasonal variations of basal cortisol and high stress response to captivity in Octodon degus, a mammalian model species. Gen Comp Endocrinol 197:65–72. doi:10.1016/j.ygcen.2013.12.007
Reinhardt U (2001) Selection for surface feeding in farmed and sea-ranched masu salmon juveniles. Trans Am Fish Soc 130:155–158. doi:10.1577/1548-8659(2001)1302.0.CO;2
Roznere I, Watters GT, Wolfe BA, Daly M (2014) Nontargeted metabolomics reveals biochemical pathways altered in response to captivity and food limitation in the freshwater mussel Amblema plicata. Comp Biochem Physiol Part D Genomics Proteomics 12:53–60. doi:10.1016/j.cbd.2014.09.004
Sambrook J, Fritsch E, Maniatis T (1989) Molecular cloning. A laboratory manual. Cold Spring Harbor Laboratory Press, New York
Schenekar T, Lerceteau-Köhler E, Weiss S (2014) Fine-scale phylogeographic contact zone in Austrian brown trout Salmo trutta reveals multiple waves of post-glacial colonization and a pre-dominance of natural versus anthropogenic admixture. Conserv Genet 15:561–572. doi:10.1007/s10592-013-0561-0
Schierup MH, Vekemans X, Charlesworth D (2000) The effect of subdivision on variation at multi-allelic loci under balancing selection. Genet Res 76:51–62. doi:10.1017/S0016672300004535
Shafer ABA, Fan CW, Côté SD, Coltman DW (2012) (Lack of) genetic diversity in immune genes predates glacial isolation in the North American mountain goat (Oreamnos americanus). J Hered 103:371–379. doi:10.1093/jhered/esr138
Shum BP, Guethlein L, Flodin LR et al (2001) Modes of salmonid MHC class I and II evolution differ from the primate paradigm. J Hered 166:3297–3308
Sommer S (2005) The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front Zool 2:16
Stringwell R, Lock A, Stutchbury CJ et al (2014) Maladaptation and phenotypic mismatch in hatchery-reared Atlantic salmon Salmo salar released in the wild. J Fish Biol 85:1927–1945. doi:10.1111/jfb.12543
Tamura K, Peterson D, Peterson N et al (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi:10.1093/molbev/msr121
Van Oosterhout C, Weetman D, Hutchinson WF (2006) Estimation and adjustment of microsatellite null alleles in nonequilibrium populations. Mol Ecol Notes 6:255–256. doi:10.1111/j.1471-8286.2005.01082.x
Waples RS, Do C (2008) ldne: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resour 8:753–756. doi:10.1111/j.1755-0998.2007.02061.x
Weiss S, Schmutz S (1999) Performance of hatchery-reared brown trout and their effects on wild fish in two small Austrian streams. Trans Am Fish Soc 128:302–316. doi:10.1577/1548-8659(1999)1282.0.CO;2
Williams SE, Hoffman EA (2009) Minimizing genetic adaptation in captive breeding programs: a review. Biol Conserv 142:2388–2400. doi:10.1016/j.biocon.2009.05.034
Wilson DJ, McVean G (2006) Estimating diversifying selection and functional constraint in the presence of recombination. Genetics 172:1411–1425. doi:10.1534/genetics.105.044917
Wong WSW, Yang Z, Goldman N, Nielsen R (2004) Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics 168:1041–1051. doi:10.1534/genetics.104.031153
Wynne JW, Cook MT, Holmes BH, Elliott NG (2007) Allelic and haplotypic diversity at the major histocompatibility class II within domesticated Australian Atlantic salmon (Salmo salar L.). J Fish Biol 70:45–59. doi:10.1111/j.1095-8649.2007.01364.x
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591. doi:10.1093/molbev/msm088
Yang Z, Wong WSW, Nielsen R (2005) Bayes empirical bayes inference of amino acid sites under positive selection. Mol Biol Evol 22:1107–1118. doi:10.1093/molbev/msi097
Zhang L, Wu Q, Hu Y et al (2015) Major histocompatibility complex alleles associated with parasite susceptibility in wild giant pandas. Heredity (Edinb) 114:85–93. doi:10.1038/hdy.2014.73
Acknowledgements
Open access funding provided by University of Graz. The authors would like to thank Günter Unfer of the Institute of Hydrobiology and Aquatic Ecosystem Management of the University of Natural Resources and Life Sciences, Vienna and Klaus Kugi of the Nature Protection Agency of Carinthia for their helpful assistance during sample collection. Also, special thanks go to Matthias Romauch and Achim Lass of the Institute of Molecular Biosciences at the University of Graz for their continuous assistance during the development of the cloning protocol. Tamara Schenekar is a recipient of a DOC-fFORTE-fellowship (23406) of the Austrian Academy of Sciences at the Institute of Zoology, University of Graz.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Schenekar, T., Weiss, S. Selection and genetic drift in captive versus wild populations: an assessment of neutral and adaptive (MHC-linked) genetic variation in wild and hatchery brown trout (Salmo trutta) populations. Conserv Genet 18, 1011–1022 (2017). https://doi.org/10.1007/s10592-017-0949-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-017-0949-3