
Abstract
β-defensins are important components of the vertebrate innate immune system responsible for encoding a variety of anti-microbial peptides. Pathogen-mediated selection is thought to act on immune genes and potentially maintain allelic variation in the face of genetic drift. The Seychelles warbler, Acrocephalus sechellensis, is an endemic passerine that underwent a recent bottleneck in its last remaining population, resulting in a considerable reduction in genome-wide variation. We genotyped avian β-defensin (AvBD) genes in contemporary (2000–2008) and museum samples (1876–1940) of the Seychelles warbler to investigate whether immunogenetic variation was lost through this bottleneck, and examined AvBD variation across four other Acrocephalus species with varying demographic histories. No variation was detected at four of the six AvBD loci screened in the post-bottleneck population of Seychelles warbler, but two silent nucleotide polymorphisms were identified at AvBD8 and one potentially functional amino-acid variation was observed at AvBD11. Variation in the Seychelles warbler was significantly lower than in the mainland migratory congeneric species investigated, but it similar to that found in other bottlenecked species. In addition, screening AvBD7 in 15 museum specimens of Seychelles warblers sampled prior to the bottleneck (1877–1905) revealed that this locus possessed two alleles previously, compared to the single allele in the contemporary population. Overall, the results show that little AvBD variation remains in the Seychelles warbler, probably as a result of having low AvBD diversity historically rather than the loss of variation due to drift associated with past demographic history. Given the limited pathogen fauna, this lack of variation at the AvBD loci may currently not pose a problem for this isolate population of Seychelles warblers, but it may be detrimental to the species’ long-term survival if new pathogens reach the population in the future.
Similar content being viewed by others


Introduction
Drift is the predominant evolutionary force shaping genetic variation in small populations (Hedrick et al. 2001; Miller and Lambert 2004; Jensen et al. 2013), and its effects on genetic variation often outweigh the influence of selection (Miller and Lambert 2004; Alcaide 2010; Grueber et al. 2013). Nevertheless, various studies have shown that within small natural populations, variation at specific key loci can be elevated above that of the genome-wide average, and be maintained across bottleneck events as a result of balancing selection (Aguilar et al. 2004; Tompkins 2007; van Oosterhout et al. 2006). Given that a loss of genetic variation within a population impacts on both inbreeding depression and adaptive potential (for review, see Garrigan and Hedrick 2003), the maintenance of polymorphisms at key loci will be important to a populations’ long-term viability (Meyers and Bull 2002; Ellegren and Sheldon 2008; Zhu et al. 2013). However, not all immune genes are under balancing selection (Mukherjee et al. 2009). By performing a temporal analysis comparing variation before and after a bottleneck event it is possible to discern the effects of balancing selection. Furthermore, taking a candidate-gene approach and focusing on those loci most likely to be under selection in natural populations (Fitzpatrick et al. 2005), avoids inflating the possibility of type 1 errors.
Genes that contribute to immune function are ideal candidates with which to assess the roles of drift and selection in maintaining functional diversity within natural populations (for review, see Acevedo-Whitehouse and Cunningham 2006). Many such studies have focused on the highly polymorphic genes of the major histocompatibility complex (MHC), which play a central role in the acquired immune system (Doherty and Zinkernagel 1975; Klein 1986; Piertney and Oliver 2006). However, there are complex interacting evolutionary forces acting upon the MHC, including the effects of epistasis and selection against the so-called ‘sheltered load’ (van Oosterhout 2009). Additionally, frequent gene duplication (Eimes et al. 2011) and recombination-like processes i.e., gene conversion (Ohta 1995; Spurgin et al. 2011) confound the interpretation of the population genetic mechanisms maintaining variation at these genes. In contrast, studies of variation within natural populations in genes that play a role in the innate immune system are relatively scarce (Sutton et al. 2011), despite the fact that these genes are often simpler in form and function than the MHC (for review, see Kaiser 2007). Variation in such genes may be crucial, given that the innate immune response is the first line of defence against pathogens. Moreover, a number of innate immune gene families, including the toll-like receptors (TLRs) and cytokines, have been shown to be targets of balancing selection (for examples, see Schlenke and Begun 2003; Ferrer-admetlla et al. 2008; Mukherjee et al. 2009).
Anti-microbial peptides (AMPs) are effector molecules involved in the innate immune system. AMPs directly kill invading pathogens via the disruption of membranes through cationic attack mechanisms (Hancock and Sahl 2006). All defensin molecules have six cysteine residues but are sorted into three classes based on their physical structure (Yang et al. 2002). Both α-defensins and β-defensins form beta-sheet dimers but they have different lengths and pairing of cysteine linkages, whereas γ-defensins have a cyclic structure (Sugiarto and Yu 2004). Different taxonomic groups have different classes and numbers of defensins in their immune repertoire (Selsted and Ouellette 2005). For example, birds have only β-defensins, of which 14 different loci have been identified in the domestic chicken, Gallus gallus domesticus (Lynn et al. 2004; Xiao et al. 2004), whereas mammals have both α and β-defensins (Yang et al. 2002). The number of β-defensins in a species has been shown to highly relevant to the ever-changing microbial challenges of the environment in which that species’ inhabits (Tu et al. 2015). It has been well-shown that different defensin alleles have different antimicrobial activities in vitro in a range of vertebrate hosts (Meredith et al. 2008; Mukherjee et al. 2009; Hellgren et al. 2010; Chow et al. 2012). These studies suggest that the greater the variety of AMPs encoded, the greater the ability to combat a range of bacteria. These studies therefore suggest that there could be an advantage to individuals (and populations) which are heterozygous at these loci.
Birds provide excellent systems in which to study the causes and consequences of innate immune gene variation under natural conditions. Functional variation at defensin genes has been shown to exist within and among species (for review, see van Dijk et al. 2008) and locus-specific protocols have been developed to screen for avian β-defensins (AvBDs) in passerines (Hellgren and Sheldon 2011). Importantly, variation within these loci has been shown to influence anti-microbial properties in vitro (Hellgren and Ekblom 2010; Hellgren et al. 2010). Specific defensin alleles have also been shown in vitro to be associated with avian pathogens (Higgs et al. 2007; Ma et al. 2012; Ramasamy et al. 2012), although whether individual heterozygosity is advantageous has yet to be shown.
The Seychelles warbler, Acrocephalus sechellensis, is an ideal species in which to study the influence of different evolutionary forces on AvBD genes. As a result of anthropogenic factors- this population experienced a bottleneck during the last century when it was on the verge of extinction with ca 26 individuals remaining on a single island (Collar and Stuart 1985). As a result, considerable variation has been lost across the warblers genome (Spurgin et al. 2014), although diversity appears to have been maintained at MHC class I loci (Richardson and Westerdahl 2003; Hansson and Richardson 2005) due to a combination of natural and sexual selection (Richardson et al. 2005; Brouwer et al. 2010). Given these patterns, we hypothesise that genetic variation could also have been maintained at other immune loci. If we can identify loci at which variation has been maintained then we can carry out association analysis between this immunogenetic variation and individual fitness parameters using data collected over the last two decades.
Here, we screened six AvBD loci in the contemporary bottlenecked population of the Seychelles warbler. For one AvBD locus, AvBD7, that was identified to be polymorphic in most other passerine species for which AvBD genes have been characterised already (Hellgren and Ekblom 2010), we used museum samples of the Seychelles warbler dating from 1877 to 1940 to assess variation that existed at this locus prior to the population bottleneck. This enables us to compare the variation in pre- and post-bottleneck populations at this locus. We also screened AvBD variation in a small sample of individuals from four other Acrocephalus species to provide a comparison for the levels of AvBD variation observed in the Seychelles warbler, and to test for signatures of selection within the sequences across the genus.
Materials and methods
Study species and sampling
The Seychelles warbler is a small (ca 12–15 g) insectivorous passerine endemic to the Seychelles archipelago (Safford and Hawkins 2013). As a result of anthropogenic factors, the species’ global population was dramatically reduced to an estimated low of 26 individuals on the single small island of Cousin in the 1960s (Collar and Stuart 1985). This reduced the species effective population size from 2600 to 9700 in the early 1800s to <50 in the contemporary population (Spurgin et al. 2014). After conservation intervention, the population on Cousin recovered and reached saturation by 1982 (Komdeur 1992) remaining relatively stable at ca 320 adults ever since (Brouwer et al. 2009; Wright et al. 2014a, b). Four translocations have been undertaken from the original population on Cousin as part of a conservation programme. A total of 29 birds were translocated to both Aride in 1988 and to Cousine island in 1990 (Komdeur 1994). A further 58 birds were translocated to Denis in 2004 (Richardson et al. 2006) and 59 to Frégate in 2011 (Wright et al. 2014a, b). This species has been intensively studied as a model system for evolutionary, ecological and conservation questions (Komdeur 1992; Richardson et al. 2003; van de Crommenacker et al. 2011; Barrett et al. 2013). Since 1997, >96 % of the Cousin population has been caught, blood-sampled and marked with a unique combination of colour rings and a metal British Trust for Ornithology (BTO) ring (Richardson et al. 2002).
The great reed warbler, A. arundinaceus, and Eurasian reed warbler, A. scirpaceus, are two mainland migratory species classified as ‘under least concern’ with estimated populations (Nc) in Europe of 950,000 and 3.1 million respectively (after Hagemeijer and Blair 1997; IUCN 2015). In contrast, the Cape Verde warbler, A. brevipennis, and Henderson’s Island warbler, A. taiti, are two island species with restricted but stable populations of an Nc estimated at 1000–1500 (Schulze-Hagen and Leisler 2011) and ca 7000 individuals (Brooke and Hartley 1995; IUCN 2015) respectively. The Cape Verde warbler is endemic to the Cape Verde islands and until recently, was thought to be confined to just Santiago island until small populations were discovered in São Nicolau and Fogo in 1998 and 2004, respectively (IUCN 2015). All samples used in this study are from the Santiago population. The population of Henderson’s Island warbler appears to have remained stable despite the observed severe population bottlenecks in other endemic species on the island during the human colonisation of Henderson Island in the early 1900s (Brooke 2010).
Estimates of effective population sizes (Ne) are available for the great reed warbler at ca 20,000 (Bensch and Hasselquist 1999). However, for the other warbler species with only a census population size (Nc) known, we can only estimate that the Ne will be ca 10 % or less of the population size (Frankham 1995). Samples were taken from all Seychelles warbler museum specimens known to exist (n = 26) (Spurgin et al. 2014) including 19 from Cousin Island and seven from Marianne Island, all collected between 1876 and 1940 (Table S1). A small (ca 1.5 × 1.5 × 3.0 mm) piece of skin was cut from the ventral surface of the foot and stored at room temperature in a sterile microfuge tube. All other Acrocephalus samples were from unrelated adults (>1 year old) from single populations with details as follows: 23 individuals were sampled for the Seychelles warbler between 2000 and 2008 from the Cousin Island population (ca 320 adults, 0.3 km2, Wright et al. 2014a, b). The Cape Verde warbler samples (n = 5) were sourced from the Santiago Island population in 2011 (ca 500 adults, 991 km2, Batahla unpublished) and the Henderson’s Island warbler were from Henderson Island (n = 5) and randomly chosen from an extant population in the 2000s (ca 7200 adults, 41 km2, IUCN 2015) (Brooke and Hartley 1995). The two migratory Acrocephalus species A. scirpaceus (n = 5) and A. arundinaceus (n = 6), were both sampled from breeding areas in central Sweden and Belgium, respectively, and randomly chosen from the same cohort used and outlined in previous studies (Richardson et al. 2000; Hansson and Richardson 2005; Hansson et al. 2006).
Molecular methods
Genomic DNA was extracted from the Seychelles warbler blood samples using a salt extraction method (Richardson et al. 2001). The same procedure had been used for the Cape Verde warbler blood samples (provided by Juan-Carlos Illera) and the Eurasian reed warbler and the great reed warbler DNA samples (provided by Andrew Dixon and Bengt Hansson, respectively). The Henderson’s Island warbler DNA samples were provided by Mike Brooke and extracted by Ian Hartley using a phenol–chloroform protocol (Brooke and Hartley 1995). We extracted DNA from Seychelles warbler museum samples (Table S1) using a Qiagen DNeasy tissue kit (Qiagen, Crawley, UK) under the manufacturer’s instructions with the following changes: (i) each sample was finely chopped in a small volume of ATL buffer prior to digestion with proteinase K, (ii) 20 µl 1 M DTT (Dithiothreitol, Sigma-Aldrich, UK) was added at incubation; and (iii) 1 µl (Qiagen, final concentration = 20 pg/ml) was added during the precipitation phase. All extractions and PCRs based on historical DNA were carried out in a laminar flow cabinet in a ‘clean room’ isolated from the main laboratory with no record of passerine DNA use in that facility with sample controls (see Spurgin et al. 2014, for further details).
Locus-specific primers (Hellgren and Sheldon 2011) were used to screen six AvBD genes: AvBD4, AvBD7, AvBD8, AvBD9, AvBD11 and AvBD13. These loci were chosen based on their successful amplification in congeneric species (Table S2) (Hellgren and Sheldon 2011). All AvBD loci and the available primer sets used produced amplicon lengths short enough for amplification in the degraded DNA we obtained from the museum samples, as they are all >200 base-pairs (bp) (Spurgin et al. 2014). However, we have very limited volumes of DNA from these samples and thus could only choose one candidate AvBD locus to characterise. AvBD7 was chosen because it was polymorphic in most bird species examined (Hellgren et al. 2010) and was a short enough fragment to amplify in the degraded museum sample DNA.
For each locus, PCRs were carried out in volumes of 10 µl with genomic DNA at a concentration of 5–10 ng/µl. Taq PCR Master Mix was used (Qiagen, UK), which included: Taq-DNA Polymerase, QIAGEN PCR Buffer, 1.5 Mm MgCl2, and 200 µM of ultrapure dNTPs. PCRs were carried out using the following conditions: 30 s at 94 °C, 30 s at the locus-specific annealing temperature of 55 °C (AvBD4, AvBD7, AvBD8, AvBD9) and 60 °C (AvBD11, AvBD13), 45 s at 72 °C, run for a total of 40 cycles. All PCRs started with an incubation step of 3 min at 94 °C and finished with an incubation step of 10 min at 72 °C. PCR products were electrophoresed on a 2 % agarose gel containing ethidium bromide to confirm successful amplification of the expected size fragment. Positive samples were submitted to the Genome Analysis Centre, Norwich, for Sanger-sequencing. All sequence polymorphisms were confirmed by sequencing in both the forward and reverse direction.
All sequences were aligned against target sequences of the given loci from other passerine species available using the basic local alignment search tool (BLAST) from the nucleotide database (NCBI) using BioEdit (Hall 1999) via ClustalW codon alignment. Each chromatogram was examined by eye to identify single-nucleotide polymorphisms (SNPs) and haplotypes were constructed using Phase v 2.1 (Stephens et al. 2001; Stephens and Donnelly 2003) in DnaSP (Librado and Rozas 2009). Given that Phase has an estimated rate of ca 5 % (Marchini et al. 2006), all reconstructed haplotypes were also checked by eye. Amino acid sequences were translated in BioEdit (Hall 1999).
Phylogenetic trees were constructed in Mega v6 (Tamura et al. 2007) using the maximum-likelihood method based on the models best suited to that clustal-sequence alignment, as determined by Mega. The trees were used to infer evolutionary history both within and between AvBD loci across the Acrocephalus genus. The trees with the highest log likelihood are presented, based on nucleotide variation given the short sequence sizes of <150 bp. All Acrocephalus sequences used originate from this study. Outgroup non-Acrocephalus passerine species sequences were obtained using the NCBI BLAST database and included: Eurasian blackcap, Sylvia atricapilla, house sparrow, Passer domesticus, icterine warbler, Hippolais icterina, lesser redpoll, Carduelis cabaret and zebra finch, Taeniopygia guttata (Table S3). Model details are as follows, AvBD4: Jukes-Cantor model (Jukes and Cantor 1969) all gaps and missing data eliminated and total of 42 positions analysed in the final dataset. AvBD7: Kimura 2-parameter model (Kimura 1980), all gaps and missing data eliminated and total of 102 positions analysed in the final dataset. AvBD8: Tamura 3-parameter model (Tamura 1992), all sites are considered due to an entire codon insertion in some sequences and total of 93 positions analysed in the final dataset. AvBD9: Jukes-Cantor model, all gaps and missing data eliminated and total of 66 positions considered in final dataset. AvBD11: Kimura 2-parameter model, all gaps and missing data eliminated and total of 113 positions analysed in final dataset. AvBD13: Kimura 2-parameter model, all gaps and missing data eliminated and total of 68 positions analysed in final data set. These trees were constructed to examine allelic richness at each locus for the Seychelles warbler, and provide further insight into AvBD loci evolution across the Acrocephalus genus. An overall tree was constructed to encompass all AvBD loci with the single most common allele at each locus used for each Acrocephalus species. The evolutionary history was inferred using the Neighbour-Joining method (Saitou and Nei 1987). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree (Felsenstein 1985). The evolutionary distances were computed using the number of differences method (Nei and Kumar 2000) and are in the units of the number of base differences per sequence. All positions containing gaps and missing data were eliminated. Only bootstrap values above 50 % are presented.
Analyses
Tests for linkage disequilibrium and deviation from the Hardy–Weinberg equilibrium (HWE) were carried out using GenePop (Raymond and Rousset 1995) and tests were based on (i) heterozygote excess and (ii) heterozygote deficiency. Polymorphism statistics and tests for neutrality were carried out in the Seychelles warbler, including: Tajima’s D statistic (Tajima 1989), Fu and Li’s D (Fu and Li 1993) and Fu and Li’s F statistics (Fu 1996) in the program DnaSP (Librado and Rozas 2009).
Site-specific dN/dS tests were then carried out using two different models (i) MEME and (ii) FUBAR to identify any individual codons under putative selection. MEME is a mixed effects model of evolution where the significance level of 0.1 is used to classify a site as positively or negatively selected as this method tends to be more conservative than empirical Bayesian approaches (Murrell et al. 2012). FUBAR is a fast unconstrained Bayesian approximation model using a Markov chain Monte Carlo routine which has a Bayes Factor/posterior probability set at 0.9 as a minimum value for inclusion in the inferred Bayesian graph (Murrell et al. 2013). Both models come highly recommended as part of the HyPhy package available for detecting individual sites under episodic diversifying selection using the DataMonkey web application (Delport et al. 2010).
Measures of variation were compared between mainland migratory Acrocephalus species and island endemic Acrocephalus species using Welch’s t tests of unequal variances. Population size for each species was obtained from IUCN (2015) and cross-referenced in Schulze-Hagen and Leisler’s publication (2011). The relationship between population size and AvBD haplotype diversity was analysed (and then the diversity of haplotypes which only resulted in amino acid variation) by log-transforming population size before using a simple linear regression analysis in Sigmaplot from Systat Software Inc., San Jose California USA. Haplotype diversity is a measure of the uniqueness of a given haplotype in a given population of individuals and includes a measure of the relative haplotype frequency (xi) in the sample of individuals as well as any difference in sample size (N) (Nei 1987).
Results
Four out of six AvBD loci were found to be monomorphic in the contemporary Seychelles warbler population (Table 1). In the two that were variable we identified two synonymous single-nucleotide polymorphisms (SNPs) within AvBD8 and one non-synonymous SNP within AvBD11 (from 20 screened individuals) (Fig S1). Of the 26 museum DNA samples screened, only 15 successfully amplified the AvBD7 locus. From these, two alleles were identified, but one allele was found in just one individual (Table S1). This novel allele, just one non-synonymous nucleotide different from the common allele, was confirmed by independent PCRs. Given the low levels of variation identified, no meaningful statistical analysis of the difference in AvBD7 variation between the pre- and post-bottleneck populations, or the intra-specific variation at AvBD8 and AvBD11, were possible. There was no evidence of selection at AvBD8 or AvBD11 based on the tests of neutrality or results from the Z-tests of selection based on dN/dS (Table S4). There was no evidence found of linkage disequilibrium between all pairwise combinations of polymorphic loci. Furthermore, there was no evidence of significant deviation from Hardy–Weinberg equilibrium based on the observed allele frequencies (P > 0.1) although it must be noted that these test would have low power and only test for deviations within that single generation (Fig. 1).

Mean AvBD haplotype diversity (Hd) verses population size (Nc) (log-transformed) in five Acrocephalus species; a All nucleotide substitutions (raw data presented on the left, means on the right), b only putatively functional substitutions resulting in different amino-acids (raw data presented on the left, means on the right). Mean standard errors are shown. The dashed lines represent the regression and adjusted R-squared values are given based on the regression analyses carried out on the raw data
Across the Acrocephalus genus, five out of six loci screened were polymorphic (Table 1; Fig S1) and only AvBD9 was monomorphic across all five Acrocephalus species. However, one of these polymorphic loci AvBD4, only had one SNP (and additional allele) in the Eurasian reed warbler and there was no other variation across the other species. In the Seychelles warbler, there was no evidence for selection within any of the six AvBD loci using Tajima’s D, Fu and Li’s F and D statistical tests (P > 0.1). However, AvBD8, the most polymorphic locus observed showed evidence for negative (purifying) selection in the Z-test of selection looking across the Acrocephalus genus (Z = 1.72, df = 10, P = 0.04) (Table S4). Site-specific dN/dS based tests were carried out on AvBD7, AvBD8 and AvBD11 as a minimum of three unique haplotype sequences are needed. The MEME model failed to detect any sites under episodic diversifying selection across the Acrocephalus genus, but the FUBAR model which focuses on putative selection detected one site under diversifying selection at the AvBD8 locus (posterior probability dN > dS = 0.90, dN−dS = 1.19). It also detected two sites under purifying selection at the same locus (posterior probability dN < dS = 0.90 and 0.91, dN−dS = −2.89 and −0.86 respectively), in addition to one site each at AvBD7 (posterior probability dN < dS = 0.98, dN−dS = −4.04) and AvBD11 (posterior probability dN < dS = 0.98, dN−dS = −4.18).
Welch’s t tests showed that mainland migratory species A. arundinaceus and A. scirpaceus had significantly more nucleotide variation observed across the AvBD loci in comparison to the island endemic species, A. taiti, A. brevipennis and A. sechellensis (mean vs mean respectively, t = 2.427, df = 27, P = 0.022). Even when only considering amino acid variation (only dN substitutions) the difference between mainland and island species was still significant (t = 2.844, df = 27, P = 0.008). This is further supported by the significant difference in overall number of alleles observed within these two categories of species (t = 2.732, df = 27, P = 0.011).
When exploring the association between census population size and mean AvBD haplotype diversity, there was a significant difference between different population sizes and AvBD variation—whether focusing on all variation (F = 12.32, df = 27, P = 0.002) or just amino acid variation (F = 6.96, df = 27, P = 0.014). There was a positive linear relationship between population size (log-transformed) and AvBD variation both, for nucleotide variation (t = 3.51, df = 27, P = 0.002) and amino acid variation (t = 2.64, df = 27, P = 0.014).
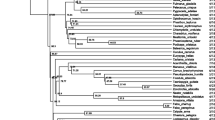
The maximum-likelihood trees show the levels of polymorphism that occur within and between the Acrocephalus species for each locus (Fig. 2). Outgroup passerine species consistently cluster separately from the Acrocephalus species for each AvBD locus. The tree for all AvBD loci combined using the single most common haplotype for each Acrocephalus species and the reference sequences for outgroup species, shows definite segregation by locus and confirm independent locus-specific evolution of these immune genes (Fig. 3).


Trees inferring intra-locus evolutionary history of AvBD genes across five Acrocephalus species, inferred by using the maximum likelihood method based on different models and parameters, optimised dependent upon the locus. Non-Acrocephalus passerine species are included as outgroups (see “Materials and methods” section). Trees are drawn to scale with haplotype number given in brackets and branch lengths measured in the number of substitutions per site. Only bootstrap values above 50 % are presented

Phylogenetic tree for all AvBD loci across avian lineages. The evolutionary history was inferred using the Neighbour-Joining method. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the number of differences method and are in the units of the number of base differences per sequence. All positions containing gaps and missing data were eliminated. Only bootstrap values above 50 % are presented. There were a total of 35 positions in the final dataset
Discussion
We characterised variation within the AvBD gene group in the Seychelles warbler. Four out of the six AvBD loci examined were monomorphic in the contemporary post-bottleneck population, while two loci had low levels of polymorphism with only a single nucleotide polymorphism causing a change in the protein translated at one locus (AvBD11). In the historical samples, we detected only two alleles, diverging by a single nucleotide substitution, in the usually highly polymorphic AvBD7 locus (Hellgren and Ekblom 2010). These low levels of polymorphism meant we were unable to perform meaningful tests of selection using traditional population genetic tests. In order to increase power, we characterised variation within the AvBD gene group in a small number (3–5) individuals from four other Acrocephalus species’ populations and looked at the same loci across the genus. One locus, AvBD8, was inferred to be under purifying selection given its high ratio of synonymous substitutions compared to non-synonymous substitution across the haplotype. Looking at specific sites within the haplotype sequence, we identified one site to be under putative diversifying selection when all other loci failed to identify any sites under episodic or putative positive selection. However, the reliability of results from tree-based models can be controversial (Anisimova et al. 2003; Wong 2004). We found that when changing the tree-build from neighbour-joining to maximum-likelihood methods, this single site was no longer identified as being under diversifying selection. Overall, the lack of variation at these loci in the Seychelles warbler (and other island species) suggests that balancing selection has not maintained AvBD variation in this bottlenecked population.
Significantly more variation at AvBD loci was observed in the two outbred migratory species, the great reed warbler and Eurasian reed warbler, in contrast to the three island species, the Seychelles warbler, Cape Verde warbler and Henderson’s Island warbler, where there was little or no variation. This was found for nucleotide variation overall and when only considering amino-acid variation i.e., non-synonymous polymorphisms. Interestingly, the recently bottlenecked Seychelles warbler has more variation observed across all AvBD loci than the Henderson’s Island warbler, despite the fact that the former species now exists within a smaller population than the latter. Henderson’s Island is, however, an uplifted coral atoll at the end of a chain of small volcanic islands which are very isolated in the middle of the Pacific Ocean. Consequently it is highly likely that Henderson’s island warbler has undergone multiple sequential bottleneck events in colonising this island, resulting in the low levels of genetic variation observed in our study of AvBDs, and in studies looking at neutral genetic markers (Brooke and Hartley 1995). In contrast, until recently the Seychelles warbler existed in a larger population across multiple islands (Spurgin et al. 2014) and only lost ca 25 % of its genomic variation in the recent bottleneck. Unfortunately, as far as we know there are no other studies on AvBDs in bottlenecked wild populations to compare our findings with.
Our results show that almost no functional variation exists at the AvBD loci in the Seychelles warbler and this refutes our a priori hypothesis that pathogen-mediated selection would maintain variation at these immunologically-important loci. Similar losses in diversity in immune defence genes associated with bottleneck events have been reported in other endangered vertebrates (Eimes et al. 2011; Jamieson 2011; Basu et al. 2012; Zhu et al. 2013). The majority of heterozygous Seychelles warbler individuals have the rare variants observed, which suggests there may be a selective advantage with heterozygosity. However, a lack of any deviation from Hardy–Weinberg proportions, suggest that this is not the case. Therefore, it is likely that the alternate variants are merely in the heterozygous form because they are rare (it is unlikely that both parents possess the same rare variant to pass onto offspring). Our results do not, therefore, mirror those from an outbred population of the blue tit, Parus major, where all but one of 40 individuals screened showed functional heterozygosity within the exon coding for the mature defensin peptide of AvBD2, 4, 7, 9, 10 and 12, thus supporting a heterozygote advantage (Hellgren 2015). Furthermore, when comparing patterns of variation at AvBD loci in the Seychelles warbler with variation at neutral markers in the same population, AvBD variation appeared to be lower than expected. Hansson and Richardson (2005) found seven out of ten neutral microsatellite markers (used to avoid ascertainment bias) were polymorphic (70 %), whereas only two out of six AvBD markers were polymorphic (33 %) in our study: AvBD8 and AvBD11.
Given the near-absence of variation found in both pre- and post-bottleneck populations of this species, it is impossible to statistically assess the roles that drift and selection may have played in shaping AvBD variation through this particular bottleneck. The AvBD7 locus shows considerable intra-specific variation in other species with many nucleotide substitutions among the Acrocephalus genus and entire codon insertions between different families in the Passeriformes (Hellgren et al. 2010). At this locus in the Seychelles warbler, we only detected two alleles in the population prior to the bottleneck and one thereafter. Given the low frequency of the additional allele in the historical sample (1/15 individuals) a large sample would need to be screened to confirm its absence in the contemporary population. Here we screened 20 individuals, so if the allele is present it is probably at a frequency <0.05. Therefore, we have no evidence for higher diversity in the ancient samples, which supports the idea that AvBDs had low diversity historically, rather than lose variation due to bottlenecks and stochastic processes.
Pathogen-mediated selection (PMS) has been shown to be an important force in maintaining variation at immune genes such as the MHC and innate immune components like cytokines (Potts and Slev 1995; Jeffery and Bangham 2000; Spurgin and Richardson 2010; Turner et al. 2012). However, while a number of studies on β-defensins have been carried out on laboratory populations and in humans (Hollox and Armour 2008; Lazzaro 2008; Ardia et al. 2011), to our knowledge there is as yet no information on PMS acting on β-defensins in wild populations. Furthermore, remote isolated populations often have fewer pathogens, as shown recently in a study of haematozoans, bacteria and viruses in avian populations (Vögeli et al. 2011). Indeed, the diversity of pathogens in the Seychelles warbler population is very low; despite extensive screening efforts, no gastro-intestinal parasites or signs of virus infection have been detected, and only one strain of avian malaria (GRW1) has ever been observed (Hutchings 2009). This shows that processes which prevail in small island populations cannot only erode immunogenetic variation (i.e., due to drift), but can reduce pathogen biodiversity (Vögeli et al. 2011). The combination of increased drift and reduced pathogen-mediated selection may therefore explain why variation at the AvBD genes is lost in bottlenecked island populations, such as the Seychelles warbler. In addition, if the parasite biodiversity is reduced such that only one (or a few) parasite strains are retained, the effects of pathogen-mediated selection on immunogenetic variation might be reversed and become purifying (Mukherjee et al. 2009). For example, the AvBD alleles observed at each locus may have become fixed in the Seychelles warbler because they provided adequate defence against the limited pathogens remaining in the environment. In such a situation, positive selection may have acted in concert with neutral effects to eliminate variation. Several studies have found that immunogenetic variation eroded faster than (neutral) microsatellite variation in small isolated populations (Bollmer et al. 2011; Eimes et al. 2011; Sutton et al. 2011).
Investigating variation at a combination of both neutral and critical markers, as we have done within this study, can help us to understand the genetic vulnerabilities of any wild population and species (Grueber and Carolyn 2015). For example, patterns of neutral variation across individuals have been compared to that observed at MHC markers in the Seychelles warbler. There is evidence that MHC class I genes have historically been under balancing selection (Richardson and Westerdahl 2003) and that the recent bottleneck resulted in ca 25 % loss of variation across the entire genome in this species (Spurgin et al. 2014). However, when comparing the rates of loss between the two markers, the rate is slower at MHC loci compared to neutral microsatellite loci, and this is more apparent when also looking at patterns of variation in other congeneric species with different demographic histories (Hansson and Richardson 2005).
Pathogens are being increasingly cited as major threats in conservation (for review, see Tompkins and Poulin 2006). When developing conservation plans and management for species and populations, the importance of pathogens within the system is not often considered, despite their roles in maintaining overall biodiversity (Hall 1999). Pathogens can have severe consequences in naïve populations, but endemic pathogens may play an important role in maintaining genetic diversity. If PMS generates balancing selection that can maintain diversity at immune genes, then a paucity of pathogens could have important consequences for the long-term genetic viability of a host population. This is exacerbated in translocated or populations which undergo a series of bottleneck events and already suffer from reduced genetic variability (Frankham 1995). If further variation is lost at their immune loci, they will be more vulnerable to infectious diseases in the long-term (O’Brien and Evermann 1988). Ironically, this may suggest that it would be unwise to deliberately remove or exclude pathogens from a host system, unless the host was on the brink of extinction and those actions were necessary for an immediate recovery. By analysing immunogenetic variation, the direct implications of a depauperate parasite biodiversity can be assessed and monitored (for examples, see Van Oosterhout et al. 2007; Knowles et al. 2011; Radwan et al. 2012; Sutton et al. 2013). Immune genes that are under balancing selection can be identified using population genetic analyses, and this knowledge can be used in both in situ and ex situ (captive) breeding. For example, population viability can be increased by genetic supplementation, and increased individual fitness has been directly attributable to outbreeding carried out in a natural setting in genetic rescue (for examples, see Vila et al. 2003; Pimm et al. 2006), including fewer studies where fitness benefits are directly related to infectious diseases (Hogg et al. 2006; Van Oosterhout et al. 2007). In particular, studies on natural populations are informative to elucidate the effects of advantageous or deleterious genetic variants, because the fitness effects of genetic variation is often condition-dependent (for review, see Sommer 2005). By assessing the current status of immunogenetic variation across different populations under the same pathogen-selection regimes will increase our knowledge on the importance of adaptive genetic variability with respect to the role of candidate immune genes in evolutionary ecology and conservation biology.
In conclusion, our results show that the low levels of AvBD variation observed in the Seychelles warbler are in line with the low levels observed in other small island populations of Acrocephalus, and contrast to the higher levels found in mainland migratory congeneric populations. This suggests that drift may be the main force driving the patterns of variation seen these bottlenecked species. Nevertheless, it does not totally rule out the possibility that balancing selection may have attenuated the loss of variation caused by a reduction in population size. However, in the Seychelles warbler the effect must be very limited as we only found one functional variant at just one of the five AvBD loci and little evidence of this gene group having more diversity before the bottleneck occurred. It is important to report observations of invariant genes within natural populations, such as observed here in this bottlenecked species. Firstly, it prevents a publication-bias towards studies that outline where and when genes are polymorphic, potentially leading to erroneous conclusions. Secondly, studies that show depleted genetic variation at loci that are typically polymorphic can be of conservation interest as they may identify populations that are particularly vulnerable to future challenges such as pathogen infections and have effective conservation applications (Frankel 1974; Hedrick 2001; Pertoldi et al. 2007).
References
Acevedo-Whitehouse K, Cunningham AA (2006) Is MHC enough for understanding wildlife immunogenetics? Trends Ecol Evol 21:433–438
Aguilar A, Roemer G, Debenham S et al (2004) High MHC diversity maintained by balancing selection in an otherwise genetically monomorphic mammal. Proc Natl Acad Sci U S A 101:3490–3494
Alcaide M (2010) On the relative roles of selection and genetic drift in shaping MHC variation. Mol Ecol 19:3842–3844
Anisimova M, Nielsen R, Yang Z (2003) Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics 164:1229–1236
Ardia DR, Parmentier HK, Vogel LA (2011) The role of constraints and limitation in driving individual variation in immune response. Funct Ecol 25:61–73
Barrett ELB, Burke TA, Hammers M et al (2013) Telomere length and dynamics predict mortality in a wild longitudinal study. Mol Ecol 22:249–259
Basu M, Das T, Ghosh A et al (2012) Gene-gene interaction and functional impact of polymorphisms on innate immune genes in controlling Plasmodium falciparum blood infection level. PLoS One 7:e46441
Bensch S, Hasselquist D (1999) Phylogeographic population structure of great reed warblers: an analysis of mtDNA control region sequences. Biol J Linn Soc 66:171–185
Bollmer JL, Ruder EA, Johnson JA et al (2011) Drift and selection influence geographic variation at immune loci of prairie-chickens. Mol Ecol 20:4695–4706
Brooke M (2010) Henderson Island. Br Birds 103:428–444
Brooke MDL, Hartley IR (1995) Nesting Henderson reed-warblers (Acrocephalus vaughani taiti) studied by DNA fingerprinting : unrelated coalitions in a stable habitat? Auk 112:77–86
Brouwer L, Tinbergen JM, Both C et al (2009) Experimental evidence for density-dependent reproduction in a cooperatively breeding passerine. Ecology 90:729–741
Brouwer L, Barr I, van de Pol M et al (2010) MHC-dependent survival in a wild population: evidence for hidden genetic benefits gained through extra-pair fertilizations. Mol Ecol 19:3444–3455
Chow BT, Soto M, Lo BL et al (2012) Antibacterial activity of four human beta-defensins: HBD-19, HBD-23, HBD-27, and HBD-29. Polymers (Basel) 4:747–758
Collar NJ, Stuart SN (1985) Threatened birds of Africa and related islands. International Council for Bird Preservation, Cambridge
Delport W, Poon AFY, Frost SDW, Kosakovsky Pond SL (2010) Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26:2455–2457
Doherty PC, Zinkernagel RM (1975) Enhanced immunological surveillance in mice heterozygous at the H-2 gene complex. Nature 256:50–52
Eimes JA, Bollmer JL, Whittingham LA et al (2011) Rapid loss of MHC class II variation in a bottlenecked population is explained by drift and loss of copy number variation. J Evol Biol 24:1847–1856
Ellegren H, Sheldon BC (2008) Genetic basis of fitness differences in natural populations. Nature 452:169–175
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Ferrer-admetlla A, Bosch E, Sikora M et al (2008) Balancing selection is the main force shaping the evolution of innate immunity. J Immunol 181:1315–1322
Fitzpatrick MJ, Ben-Shahar Y, Smid HM et al (2005) Candidate genes for behavioural ecology. Trends Ecol Evol 20:96–104
Frankel OH (1974) Genetic conservation: our evolutionary responsibility. Genetics 78:53–65
Frankham R (1995) Conservation genetics. Annu Rev Genet 29:305–327
Fu Y-X (1996) New statistical tests of neutrality for DNA samples from a population. Genetics 143:557–570
Fu Y-X, Li W-H (1993) Statistical tests of neutrality of mutations. Genetics 133:693–709
Garrigan D, Hedrick PW (2003) Perspective: detecting adaptive molecular polymorphism: lessons from the MHC. Evolution 57:1707–1722
Grueber CE, Carolyn J (2015) Impacts of early viability selection on management of inbreeding and genetic diversity in conservation. Mol Ecol 24:1645–1653
Grueber CE, Wallis GP, Jamieson IG (2013) Genetic drift outweighs natural selection at toll-like receptor (TLR) immunity loci in a re-introduced population of a threatened species. Mol Ecol 22:4470–4482
Hagemeijer WJM, Blair MJ (1997) The EBCC Atlas of European Breeding Birds. T & AD Poyser, London
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp 41:95–98
Hancock REW, Sahl H-G (2006) Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol 24:1551–1557
Hansson B, Richardson DS (2005) Genetic variation in two endangered Acrocephalus species compared to a widespread congener: estimates based on functional and random loci. Anim Conserv 8:83–90
Hansson B, Jack L, Christians JK et al (2006) No evidence for inbreeding avoidance in a great reed warbler population. Behav Ecol 18:157–164
Hedrick PW (2001) Conservation genetics: where are we now? Trends Ecol Evol 16:629–636
Hedrick PW, Gutierrez-Espeleta GA, Lee RN (2001) Founder effect in an island population of bighorn sheep. Mol Ecol 10:851–857
Hellgren O (2015) Allelic variation at innate immune genes (avian β-defensins), within a natural population of great tits. J Avian Biol 46:113–118
Hellgren O, Ekblom R (2010) Evolution of a cluster of innate immune genes (beta-defensins) along the ancestral lines of chicken and zebra finch. Immunome Res 6:3
Hellgren O, Sheldon BC (2011) Locus-specific protocol for nine different innate immune genes (antimicrobial peptides: beta-defensins) across passerine bird species reveals within-species coding variation and a case of trans-species polymorphisms. Mol Ecol Resour 11:686–692
Hellgren O, Sheldon BC, Buckling A (2010) In vitro tests of natural allelic variation of innate immune genes (avian β-defensins) reveal functional differences in microbial inhibition. J Evol Biol 23:2726–2730
Higgs R, Lynn DJ, Cahalane S et al (2007) Modification of chicken avian beta-defensin-8 at positively selected amino acid sites enhances specific antimicrobial activity. Immunogenetics 59:573–580
Hogg JT, Forbes SH, Steele BM, Luikart G (2006) Genetic rescue of an insular population of large mammals. Proc Biol Sci 273:1491–1499
Hollox EJ, Armour JA (2008) Directional and balancing selection in human beta-defensins. BMC Evol Biol 8:113
Hutchings K (2009) Parasite-mediated selection in an island endemic, the Seychelles warbler (Acrocephalus sechellensis). University of East Anglia
IUCN (2015) The IUCN Red List of Threatened Species. Version 2015-4. http://www.iucnredlist.org
Jamieson IG (2011) Founder effects, inbreeding, and loss of genetic diversity in four avian reintroduction programs. Conserv Biol 25:115–123
Jeffery KJM, Bangham CRM (2000) Do infectious diseases drive MHC diversity? Microbes Infect 2:1335–1341
Jensen H, Moe R, Hagen IJ et al (2013) Genetic variation and structure of house sparrow populations: is there an island effect? Mol Ecol 22:1792–1805
Jukes TH, Cantor CR (1969) Evolution of protein molecules. In: Munro HN (ed) Mammalian protein metabolism. Academic Press, New York
Kaiser P (2007) The avian immune genome–a glass half-full or half-empty? Cytogenet Genome Res 117:221–230
Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Klein J (1986) Natural history of the major histocompatibility complex. Wiley, New York
Knowles SC, Wood MJ, Alves R et al (2011) Molecular epidemiology of malaria prevalence and parasitaemia in a wild bird population. Mol Ecol 20:1062–1076
Komdeur J (1992) Importance of habitat saturation and territory quality for evolution of cooperative breeding in the Seychelles warbler. Nature 358:493–495
Komdeur J (1994) Conserving the Seychelles warbler Acrocephalus sechellensis by translocation from Cousin island to the islands of Aride and Cousine. Biol Conserv 67:143–152
Lazzaro BP (2008) Natural selection on the Drosophila antimicrobial immune system. Curr Opin Microbiol 11:284–289
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Lynn DJ, Higgs R, Gaines S et al (2004) Bioinformatic discovery and initial characterisation of nine novel antimicrobial peptide genes in the chicken. Immunogenetics 56:170–177
Ma D, Zhou C, Zhang M et al (2012) Functional analysis and induction of four novel goose (Anser cygnoides) avian beta-defensins in response to salmonella enteritidis infection. Comp Immunol Microbiol Infect Dis 35:197–207
Marchini J, Cutler D, Patterson N et al (2006) A comparison of phasing algorithms for trios and unrelated individuals. Am J Hum Genet 78:437–450
Meredith JM, Hurd H, Lehane MJ, Eggleston P (2008) The malaria vector mosquito Anopheles gambiae expresses a suite of larval-specific defensin genes. Insect Mol Biol 17:103–112
Meyers LA, Bull JJ (2002) Fighting change with change: adaptive variation in an uncertain world. Trends Ecol Evol 17:551–557
Miller HC, Lambert DM (2004) Genetic drift outweighs balancing selection in shaping post-bottleneck major histocompatibility complex variation in New Zealand robins (Petroicidae). Mol Ecol 13:3709–3721
Mukherjee S, Sarkar-roy N, Wagener DK, Majumder PP (2009) Signatures of natural selection are not uniform across genes of innate immune system, but purifying. PNAS 106:7073–7078
Murrell B, Wertheim JO, Moola S et al (2012) Detecting individual sites subject to episodic diversifying selection. PLoS Genet 8:e1002764
Murrell B, Moola S, Mabona A et al (2013) FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Mol Biol Evol Evol 30:1196–1205
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Nei M, Kumar S (2000) Molecular evolution and phylogenetics. Oxford University Press, New York
O’Brien SJ, Evermann J (1988) FInteractive influence of infectious disease and genetic diversity in natural populations. Trends Ecol Evol 3:254–259
Ohta T (1995) Gene conversion vs point mutation in generating variability at the antigen recognition site of major histocompatibility complex loci. J Mol Evol 41:115–119
Pertoldi C, Bijlsma R, Loeschcke V (2007) Conservation genetics in a globally changing environment: present problems, paradoxes and future challenges. Biodivers Conserv 16:4147–4163
Piertney SB, Oliver MK (2006) The evolutionary ecology of the major histocompatibility complex. Heredity 96:7–21
Pimm SL, Dollar L, Bass OL (2006) The genetic rescue of the Florida panther. Anim Conserv 9:115–122
Potts WK, Slev PR (1995) Pathogen-based models favoring MHC genetic diversity. Immunol Rev 143:181–197
Radwan J, Zagalska-Neubauer M, Cichon M et al (2012) MHC diversity, malaria and lifetime reproductive success in collared flycatchers. Mol Ecol 21:2469–2479
Ramasamy KT, Verma P, Reddy MR (2012) Differential gene expression of antimicrobial peptides beta defensins in the gastrointestinal tract of Salmonella serovar Pullorum infected broiler chickens. Vet Res Commun 36:57–62
Raymond M, Rousset F (1995) GENEPOP (Version 1.2): population genetics software for exact tests and ecumenicism. Heredity 86:248–249
Richardson DS, Westerdahl H (2003) MHC diversity in two Acrocephalus species: the outbred Great reed warbler and the inbred Seychelles warbler. Mol Evol 12:3523–3529
Richardson DL, Jury FL, Dawson DA et al (2000) Fifty Seychelles warbler (Acrocephalus sechellensis) microsatellite loci polymorphic in Sylviidae species and their cross-species amplification in other passerine birds. Mol Ecol 9:2155–2157
Richardson DS, Jury FL, Blaakmeer K et al (2001) Parentage assignment and extra-group paternity in a cooperative breeder: the Seychelles warbler (Acrocephalus sechellensis). Mol Ecol 10:2263–2273
Richardson DS, Burke T, Komdeur J (2002) Direct benefits and the evolution of female-biased cooperative breeding in Seychelles warblers. Evolution 56:2313–2321
Richardson DS, Komdeur J, Burke T, Lusk C (2003) Altruism and infidelity among warblers. Nature 422:580–581
Richardson DS, Komdeur J, Burke T, von Schantz T (2005) MHC-based patterns of social and extra-pair mate choice in the Seychelles warbler. Proc R Soc B 272:759–767
Richardson DS, Bristol R, Shah NJ (2006) Translocation of the Seychelles warbler Acrocephalus sechellensis to establish a new population on Denis Island, Seychelles. Conserv Evid 3:54–57
Safford R, Hawkins F (2013) The birds of Africa, vol 8. The Malagasy Region, London
Saitou N, Nei M (1987) The neighbour-joining method: a new method for reconstructing phylogentic trees. Mol Biol Evol 4:406–425
Schlenke TA, Begun DJ (2003) Natural selection drives Drosophila immune system evolution. Genetics 164:1471–1480
Schulze-Hagen K, Leisler B (2011) Reed warblers: diversity in a uniform bird family, 1st edn. KNNV Uitgeverij, Zeist
Selsted ME, Ouellette AJ (2005) Mammalian defensins in the antimicrobial immune response. Nat Immunol 6:551–557
Sommer S (2005) The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front Zool 2:16
Spurgin LG, Richardson DS (2010) How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc R Soc B 277:979–988
Spurgin LG, van Oosterhout C, Illera JC et al (2011) Gene conversion rapidly generates major histocompatibility complex diversity in recently founded bird populations. Mol Ecol 20:5213–5225
Spurgin LG, Wright DJ, van der Velde M et al (2014) Museum DNA reveals the demographic history of the endangered Seychelles warbler. Evol Appl 7:1134–1143
Stephens M, Donnelly P (2003) A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 73:1162–1169
Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989
Sugiarto H, Yu PL (2004) Avian antimicrobial peptides: the defense role of beta-defensins. Biochem Biophys Res Commun 323:721–727
Sutton JT, Nakagawa S, Robertson BC, Jamieson IG (2011) Disentangling the roles of natural selection and genetic drift in shaping variation at MHC immunity genes. Mol Ecol 20:4408–4420
Sutton JT, Robertson BC, Grueber CE et al (2013) Characterization of MHC class II B polymorphism in bottlenecked New Zealand saddlebacks reveals low levels of genetic diversity. Immunogenetics 65:619–633
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K (1992) Estimation of the number of nucleotide substitutions When there are strong transition-transversion and G+C-content biases. Mol Biol Evol 9:678–687
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Tompkins DM (2007) Population bottlenecks and avian immunity: implications for conservation. Anim Conserv 10:11–13
Tompkins DM, Poulin R (2006) Parasites and biological invasions. Biol Invasions New Zeal Ecol Stud 186:67–84
Tu J, Li D, Li Q et al (2015) Molecular evolutionary analysis of β-defensin peptides in vertebrates. Evol Bioinform 11:105–114
Turner AK, Begon M, Jackson JA, Paterson S (2012) Evidence for selection at cytokine loci in a natural population of field voles (Microtus agrestis). Mol Ecol 21:1632–1646
van de Crommenacker J, Komdeur J, Burke T, Richardson DS (2011) Spatio-temporal variation in territory quality and oxidative status: a natural experiment in the Seychelles warbler (Acrocephalus sechellensis). J Anim Ecol 80:668–680
van Dijk A, Veldhuizen EJ, Haagsman HP (2008) Avian defensins. Vet Immunol Immunopathol 124:1–18
van Oosterhout C (2009) A new theory of MHC evolution: beyond selection on the immune genes. Proc Biol Sci 276:657–665
van Oosterhout C, Joyce DA, Cummings SA et al (2006) Balancing selection, random genetic drift, and genetic variation at the major histocompatibility complex in two wild populations of guppies (Poecilia reticulata). Evolution 60:2562–2574
Van Oosterhout C, Smith AM, Hänfling B et al (2007) The guppy as a conservation model: implications of parasitism and inbreeding for reintroduction success. Conserv Biol 21:1573–1583
Vila C, Sundqvist A-K, Flagstad O et al (2003) Rescue of a severely bottlenecked wolf (Canis lupus) population by a single immigrant. Proc R Soc B Biol Sci 270:91–97
Vögeli M, Lemus JA, Serrano D et al (2011) An island paradigm on the mainland: host population fragmentation impairs the community of avian pathogens. Proc R Soc B 278:2668–2676
Wong WSW (2004) Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics 168:1041–1051
Wright DJ, Shah NJ, Richardson DS (2014a) Translocation of the Seychelles warbler Acrocephalus sechellensis to establish a new population on Frégate Island, Seychelles. Conserv Evid 11:20–24
Wright DJ, Spurgin LG, Collar NJ et al (2014b) The impact of translocations on neutral and functional genetic diversity within and among populations of the Seychelles warbler. Mol Ecol 23:2165–2177
Xiao Y, Hughes AL, Ando J et al (2004) A genome-wide screen identifies a single beta-defensin gene cluster in the chicken: implications for the origin and evolution of mammalian defensins. BMC Genom 5:56
Yang D, Biragyn A, Kwak LW, Oppenheim JJ (2002) Mammalian defensins in immunity: more than just microbicidal. Trends Immunol 23:291–296
Zhu L, Hu Y, Qi D et al (2013) Genetic consequences of historical anthropogenic and ecological events on giant pandas. Ecology 94:2346–2357
Acknowledgments
Nature Seychelles kindly facilitate and support our long-term Seychelles warbler study on Cousin Island. The Seychelles Bureau of Standards and the Department of Environment gave permission for sampling and fieldwork. We thank Prof Terry Burke for the use of the NERC Biomolecular Analysis Facility at the University of Sheffield, and also would like to thank a number of collaborators for providing Acrocephalus DNA samples: Dr Deborah Dawson, Dr Juan Carlos Illera, Andrew Dixon, Dr Bengt Hansson, Dr Michael Brooke and Dr Ian Hartley. This work was funded by a VH-C Dean’s PhD Studentship at the University of East Anglia awarded to DLG, by a Natural Environmental Research Council grant to DSR (NE/F02083X/1) and by an additional contribution provided by JK from the University of Groningen. CvO is funded by the Earth and Life Systems Alliance (ELSA).
Author contributions
DSR, CVO and DLG designed the study. DSR obtained the Seychelles warbler samples; DLG undertook the molecular lab work and associated analysis. DLG drafted the manuscript all authors revised and agreed the final manuscript.
Data Accession Statement
GenBank do not accept sequences which are <200 bp, therefore, we have provided all sequences originating from this study in the supplementary material (Table S5) for easy and full access.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gilroy, D., van Oosterhout, C., Komdeur, J. et al. Avian β-defensin variation in bottlenecked populations: the Seychelles warbler and other congeners. Conserv Genet 17, 661–674 (2016). https://doi.org/10.1007/s10592-016-0813-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-016-0813-x