
Abstract
The influence of sampling strategy on estimates of effective population size (N e ) from single-sample genetic methods has not been rigorously examined, though these methods are increasingly used. For headwater salmonids, spatially close kin association among age-0 individuals suggests that sampling strategy (number of individuals and location from which they are collected) will influence estimates of N e through family representation effects. We collected age-0 brook trout by completely sampling three headwater habitat patches, and used microsatellite data and empirically parameterized simulations to test the effects of different combinations of sample size (S = 25, 50, 75, 100, 150, or 200) and number of equally-spaced sample starting locations (SL = 1, 2, 3, 4, or random) on estimates of mean family size and effective number of breeders (N b ). Both S and SL had a strong influence on estimates of mean family size and \( \hat{N}_{b} , \) however the strength of the effects varied among habitat patches that varied in family spatial distributions. The sampling strategy that resulted in an optimal balance between precise estimates of N b and sampling effort regardless of family structure occurred with S = 75 and SL = 3. This strategy limited bias by ensuring samples contained individuals from a high proportion of available families while providing a large enough sample size for precise estimates. Because this sampling effort performed well for populations that vary in family structure, it should provide a generally applicable approach for genetic monitoring of iteroparous headwater stream fishes that have overlapping generations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Landscape changes (deforestation, dams, road systems, impassable culverts, invasive species) have greatly reduced patch size and connectivity among populations of headwater stream fishes (Dunham et al. 1997; Morita and Yamamoto 2002; Letcher et al. 2007). Future climate change predicts increased population isolation and further reductions in patch size (Hudy et al. 2008; Isaak et al. 2010; Wenger et al. 2011). An essential management goal for stream fish species is to identify existing populations that are likely to be resilient to environmental change and populations that are at greatest risk. One important determinant of likely population persistence in highly fragmented landscapes is effective population size (N e ), defined as the size of an ideal population that has the same rate of change of allele frequencies or heterozygosity as the observed population (Wright 1931). N e is of central importance for conservation genetics and evolutionary biology (Waples 2005; Hare et al. 2011). It strongly influences the rate of loss of genetic variation due to genetic drift, the rate of inbreeding, and the efficacy of natural selection and migration (Crow and Kimura 1970). Collection of unbiased estimates of effective population size (N e ) can be useful for monitoring past landscape fragmentation and future restoration efforts.
Methods for estimating N e fall into two broad categories—single-sample (Pudovkin et al. 1996; Tallmon et al. 2008; Waples and Do 2008; Wang 2009) and repeated sample (Waples 1989; Wang and Whitlock 2003) techniques. For management situations involving a large number of small populations at a landscape scale, single-sample techniques have a major advantage in terms of cost and effort. The most widely used and evaluated single-sample estimator is based on the magnitude of linkage disequilibrium (LD) in a population sample (Hill 1981; Waples and Do 2008). This LD-N e method, including the implementation of a recently derived bias correction (Waples 2006), provides an estimate of contemporary N e that applies to the past one-to-few generations (Luikart et al. 2010). This estimator appears to provide high precision and low bias over a range of effective sizes (up to approximately 500), sample sizes, number of loci and number of alleles relevant to many conservation applications (Tallmon et al. 2010; Waples and Do 2010). However this method is based on the assumption that that the source of LD is from small N e (Luikart et al. 2010). LD arising from other factors can lead to biased N e estimates. Factors that can cause LD include nonrandom mating (but see Waples 2006 for a treatment of likely effects of assumed monogamy), immigration, population substructure, overlapping generations, linked markers that are not selectively neutral, and nonrandom sampling of individuals from the population of interest.
Two of these assumptions are highly relevant to stream fishes such as the brook trout (Salvelinus fontinalis). First, brook trout populations almost always have overlapping generations (Curry et al. 2010). N e estimates obtained from mixed-cohort samples that include members of multiple age classes might often be biased low (Waples 2010). This potential problem can be overcome by restricting analyses to a single cohort or age-class. For stream-dwelling brook trout the likely target age-class would be age-0 individuals as they are usually readily distinguishable based on body size (Hudy et al. 2000). Use of a sufficient number of age-0 individuals yields an unbiased estimate of N b , the effective number of parents or breeding adults of the age-0 cohort (Waples 2010). Unbiased estimates of N b can then be the focus of genetic monitoring efforts for species with overlapping generations. Second, because offspring emerge from discrete nests (redds) and show limited dispersal (Hunt and Brynildson 1964; Miller 1970; Hudy et al. 2010) there is a high probability of non-random sampling of close kin, particularly for small samples (relatively few individuals) collected over limited areas (Hansen and Jensen 2005). Further, typical sampling protocols for headwater salmonids involve starting in one location and working upstream until the desired sample size is achieved, which is likely to increase the probability of family over-representation. Family over-representation is likely to cause downward bias for estimates from the LD-N e approach (Luikart et al. 2010). However, to date, no study has systematically tested the effects of violation of the assumption that individuals are collected at random.
In this paper, we examine the effect of sampling effort on the performance of the single-sample LD-N e method for estimating N b of headwater brook trout populations. We obtained large samples (total N = 1,440) of young-of-the-year (age-0) brook trout from each of three separate and currently isolated headwater habitat patches (the entire available habitat area within each creek) in Virginia, USA. Previous work in one of the habitat patches demonstrated close kin associations within four months of emergence (Hudy et al. 2010) and therefore there is a strong possibility that sampling strategy will influence N b estimates through its effect on family representation. Our goal was to identify the appropriate sampling effort for obtaining precise N b estimates across habitat patches that vary in family structure. We used sibship reconstruction to assign age-0 individuals to full-sibling families based on microsatellite genotypes. Simulations based on the empirical data were then used to define “true” N b and to test the accuracy of sibship reconstruction. We subjected the empirical and simulated data sets to combinations of sampling effort to assess effects on bias and precision of estimates of family size and N b . Sampling effort differed in two primary aspects that are most relevant to headwater brook trout populations: number of sampled individuals (S) and the number of sample starting locations (SL) used to obtain S.
Methods
Brook trout sampling
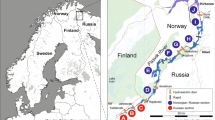
Complete surveys were conducted on three brook trout populations located in Rockingham County, Virginia, USA (Table 1; Fig. 1). The sampling protocol consisted of single-pass electrofishing surveys of the entire watershed during July 2004 (Fridley Gap, FG) and August 2010 (Above Switzer Dam, hereafter Above Switzer, AS; and Little River, LR). Sampling during late summer allowed age-0 brook trout to become large enough to be captured efficiently while still enabling year-class differentiation based upon length (Hudy et al. 2000). Additionally, habitat area was reduced at this time, which allowed larger watersheds to be surveyed. We conducted mark-capture population estimates for the entire habitat patch on both young-of-the-year (YOY) and adults. Mean detection probabilities were 65% for adult brook trout (>100 mm) and 35% for young of the year (<100 mm). Only results for YOY are presented here. Upon capture, individual length (nearest mm, total length (TL)) and location (nearest upstream meter) were recorded, and a tissue sample (anal fin clip) was taken as a source of genetic material.

Map of North-Central Virginia, USA showing the three watersheds that contained the brook trout populations used for this study
Genotyping
Individual genotypes of age-0 brook trout were used to estimate the effective number of breeders (N b ) that produced the cohort and to reconstruct full-sibling families. All populations were genotyped at eight microsatellite loci (SfoC-113, SfoD-75, SfoC-88, SfoD-100, SfoC-115, SfoC-129, SfoC-24 (King et al. 2003), and SsaD-237 (King et al. 2005) following protocols for DNA extraction and amplification detailed in King et al. (2005). Loci were electrophoresed on either an ABI Prism 3100-Avant or an ABI Prism 3130xl genetic analyzer (Applied Biosystems Inc., Foster City, California), and alleles were hand-scored using GENEMAPPER version 3.2 and PEAK SCANNER version 1.0 software (Applied Biosystems Inc.).
Population simulation
Populations were simulated (1) to enable calculation of “true” N b values for comparison with estimates of N b derived from sampling strategy results and (2) to estimate the accuracy of sibship reconstruction in the empirical datasets. We conducted simulations with PEDAGOG version 1.24 (Coombs et al. 2010a). Simulated populations had life history characteristics similar to brook trout (Supplemental Information). We initialized simulations for each population based on observed allele frequencies in each of the three study sites. To allow direct comparison between the matched empirical and simulated data sets, we performed an additional post hoc step on each simulated population to equalize number of families and family sizes. Simulated families were rank-ordered and individuals within families were randomly trimmed to match the empirical results. We then directly assigned stream location information for simulated individuals from the location of their empirical counterparts (Supplemental Information).
Population sub-sampling
To evaluate the effects of S and SL on estimates of N b , we varied these factors for both the empirical and simulated datasets. We evaluated S = 25, 50, 75, 100, 150, and 200. We evaluated SL = 1, 2, 3, 4, or Random. SL refers to 1, 2, 3, or 4 spatially discrete starting locations that corresponded to either a single start to sampling (SL = 1) or to the division of the stream into halves (SL = 2), thirds (SL = 3), or quarters (SL = 4). SL = Random represented a spatial control where individuals were selected at random from throughout the habitat patch. Individuals were sorted by stream location within each habitat patch. For SL = 1, a random number between 1 and N–S was selected, where N was the total sample size for a patch. This number represented the initial individual to be sampled (therefore the single initial starting location), with the remainder of the sub-sample composed of the next S-1 consecutively sampled fish in the upstream direction. SL = 2 was implemented in a similar manner, with the exception that each stream was divided in equal halves based on its length and sampling proceeded consecutively from two starting points, until S was reached. Half of the total sub-sample was collected from each half of the stream. Within each stream half, a random number between 1 and X-(S/2) was selected, where X was the number of age-0 brook trout inhabiting that half of the stream. SL = 3 and 4 were collected in an identical manner, but used three and four stream divisions instead of two. Due to spatially uneven fish distribution in LR, we were unable to perform SL = 4 in this habitat patch. The random strategy selected a random number between 1 and N without replacement until the target S was reached. Twenty replicates were performed for each combination of S and SL.
Genetic analyses
All N b estimates were generated using the single-sample linkage disequilibrium method within the program LDNe version 1.31 (Waples and Do 2008). A monogamous mating model was assumed based on a report that 80% of parents contributed to only a single family in two headwater stream brook trout populations (Coombs 2010). N b estimates were derived using a minimum allele frequency cutoff (P crit) of 0.02 for each value of S. This choice of P crit allowed us to exclude bias-inducing singletons across all values of S while keeping P crit constant. P crit = 0.02 has been shown to provide an adequate balance between precision and bias across values of S (Waples and Do 2008). 95% confidence intervals were generated using the jackknife approach. For both the empirical and simulated data sets, N b estimates were obtained for all twenty replicates within each sample size/starting location combination, and for all age-0 fish captured in a population. The later estimates, based on all sampled fish, provided a best estimate to assess bias. We calculated a “true” N b for the simulated data sets. True N b values were calculated for all age-0 fish with equation 6 in Waples and Waples (2011)
where P equals the total number of parents that produced the target cohort, and k i equals the number of captured brook trout contributed by parent i. Parental reproductive success was computed using complete pedigree output produced during simulations by PEDAGOG.
Mean heterozygosity and number of alleles were calculated using GDA version 1.0 (Lewis and Zaykin 2001). We tested for departures from Hardy–Weinberg (HW) proportions with exact tests implemented in GENEPOP version 4.0.10 (Rousset 2008). We tested for a deficit of heterozygotes due to possible population substructure (Wahlund effect) and because we suspected that a null allele was present at one locus in FG. We corrected for multiple tests for deviation from HW proportions with the sequential Bonferroni procedure (Rice 1989). Null allele frequencies were estimated with ML-RELATE version 090408 (Kalinowski et al. 2006). All input files for all genetic analysis programs were generated using CREATE version 1.33 (Coombs et al. 2008). For all populations, sibship reconstruction was performed using COLONY version 1.2 (Wang 2004). We estimated the mean number of individuals per full-sibling family by fitting a Poisson distribution to a frequency distribution of full-sibling family size. To estimate family spatial ranges, we calculated 95% confidence intervals for the spatial location of full-sibs within each family, assuming a normal distribution. We then calculated the ratio of the mean of the 95% CIs within a habitat patch to the inhabited stream length of each patch. Accuracy of reconstructed sibships for simulated datasets was calculated using PEDAGREE version 1.05 (Coombs et al. 2010b). For each simulated population, a single replicate was used to address the effects of sampling strategy on N b estimation, while ten replicates were used to estimate sibship reconstruction accuracy.
Statistical analyses
We fitted a general linear model to examine the relative effect of individual variables (number of sample locations, 5-level factor; sample size, 6-level factor; and habitat patch, 3-level factor) on estimates of N b with the stats package in R version 2.12.0 (R Development Core Team 2006). To standardize among rivers, we used relative bias in estimates of N b as the dependent variable instead of \( \hat{N}_{b} . \) Relative bias was calculated as the difference between \( \hat{N}_{b} \) for each combination of SL, S, and habitat patch (HP) and the \( \hat{N}_{b} \) obtained from all individuals examined in each patch. We also fitted a linear model with number of individuals per family (family size) as the dependent variable and the same predictor variables (SL, S, and HP). Models were fitted to the empirical data only.
Results
Mean sample size for the three study sites was 480 individuals (range 299–838; Table 1). Age-0 (YOY) abundance estimates based on mark-recapture ranged from 463 to 4,904 (Table 1). Inhabited stream length ranged from 518 to 4,875 m and watershed area ranged from 590 to 4,121 ha (Table 1). The mean number of alleles per locus (A) ranged from 9.5 to 10.8 (Table 2). Observed heterozygosity (H O) ranged from 0.74 to 0.81 (Table 2). Genotypic proportions at SSaD-237 in FG deviated significantly from Hardy–Weinberg (HW) proportions (Table 2). The deficit of heterozygotes at this locus was consistent with a null allele (estimated frequency = 0.288). We did not detect evidence of a null allele at this locus in the other two populations. We incorporated a null allele at this frequency for the FG simulations. Tests for deviation from HW proportions across populations for the other loci or across loci within populations were not significant following sequential Bonferroni correction (Table 2) and therefore there was no evidence for a Wahlund effect due to population substructure.
There were a large number of full-sib families in each site and the distribution of full-sibling family sizes was similar among the three study locations (Fig. 2). In FG, sibship reconstruction revealed a total of 180 full-sibling families with a mean family size (\( \hat{\lambda } \)) of 4.65 (Fig. 2a). In LR, there were 90 full-sibling families with a mean family size (\( \hat{\lambda } \)) of 3.32 (Fig. 2b). In AS, the total number of full-sib families was 82 and the mean family size (\( \hat{\lambda } \)) was 3.70 (Fig. 2c). Family representation was strongly skewed in all three sites. The proportion of families that accounted for 50% of the offspring was 16% (FG), 13% (LR), and 15% (AS). Based on the simulations, sibship reconstruction accuracies ranged from 87.4 to 93.2% (Table 1).

Distribution of full-sibling family sizes for each of the three study sites (FG panel a; LR panel b; and AS panel c). Sample size (N), number of estimated families (\( \hat{N}_{\text{fam}} \)) and mean of a fitted Poisson distribution (\( \hat{\lambda } \)) are shown for each site
Family spatial distributions relative to inhabited stream length differed among the three habitat patches (Fig. 3). Inhabited stream lengths were 518 m (AS), 1,750 m (FG), and 4,875 m (LR; Table 1). In FG, families occurred throughout the 1,750 m habitat patch but tended to be highly clumped spatially (Fig. 3a). In LR, families tended to occur in either the lower portion of the patch (0–3,000 m) or the upper portion (>5,000 m)(Fig. 3b). Full-siblings from only one of the 20 largest families (family #2; Fig. 3b) occurred in both portions of the habitat patch. AS had the smallest inhabited stream length and families tended to occur throughout the patch (Fig. 3c). Mean family spatial range was lowest in FG (137.5 m), intermediate in AS (200.1 m), and greatest in LR (314.2 m). The ratio of family spatial range to inhabited stream length was low in FG (0.08) and LR (0.06) and much greater in AS (0.39).

Stream locations of individuals within 20 largest full-sibling families for each study site (FG panel a; LR panel b; and AS panel c). Families were rank-ordered by size. Individuals within full-sib families were jittered along only the x-axis for heuristic purposes. Horizontal lines represent the upstream-most location of fish in each habitat patch
Estimates of family size increased with increasing S and decreasing SL (Fig. 4). S had the greatest relative effect on estimated family size in the linear model (Table 3) and the relationship was positive and of similar magnitude in all three habitat patches (Fig. 4). SL had a smaller relative effect than S on estimated family size but there was a strong SL × HP interaction (Table 3). The negative relationship between SL and estimated family size was most pronounced in FG, intermediate in LR, and hardly apparent in AS (Fig. 4). A greater number of individuals per family translates directly to detection of fewer families for a given S (Fig. S1). Thus, we consistently detected fewer full-sibling families, each with more members, with SL = 1 and 2. The simulated data revealed patterns for estimates of family size that were highly similar to the empirical data (Fig. 4).

Box plots of family size (number of individuals per full-sibling family) of brook trout from the three study sites (FG panels a and d; LR panels b and e; and AS panels c and f). Family size for each site was estimated for a combination of six sample sizes (S) and five sample starting locations (SL). Empirical data are shown in panels a–c, simulated data are in panels d–f. The box represents 50% of all values, whiskers represent the first quartile −1.5 × IQR (interquartile range) and the third quartile +1.5 × IQR. The line within each box is the median. Outliers are shown as circles
SL further removed from random sampling (fewer sample starting locations) caused greater bias in empirically-based \( \hat{N}_{b} \) (Fig. 5). The best estimate of N b obtained for all of the empirical data for FG was 111.6 (95% CI 94.2–131.3), for LR was 46.0 (95% CI 39.6–53.2), and for AS was 54.8 (95% CI 48.1–62.3). Overall, bias relative to these best estimates of N b was reduced in AS relative to FG and LR. This was reflected by the greatest relative effect of habitat patch in the linear model (Table 3). SL also had a strong relative effect and there was a strong SL × HP interaction (Table 3). That is, fewer starting locations led to substantial downward relative bias in \( \hat{N}_{b} \) in FG and LR but not in AS. Averaged across S and HP, median relative bias was below 10% for the empirical data for SL = 3, 4, and Random (Fig. 6). Relative bias across S was lowest for SL = 4 for FG (Table S1; Fig. 5). For LR, relative bias was below 10% for SL = 3 and was lowest for SL = Random (Table S1; Fig. 5). For AS, relative bias was below 10% for all SL (Table S1; Fig. 5). S had a relatively weak effect on relative bias in \( \hat{N}_{b} \) (Table 3; Fig. 5) but larger S generally led to more precise estimates of N b (reduced coefficients of variation; Fig. 7).

Box plots of estimates of N b (\( \hat{N}_{b} \)) of brook trout from the three study sites (FG panels a and d; LR panels b and e; and AS panels c and f). N b for each site was estimated for a combination of six sample sizes (S) and five sample starting locations (SL). Empirical data are shown in panels a–c, simulated data are in panels d–f. Point estimates of N b based on all of the empirical data (solid horizontal lines) were 111.6, 46.0, and 54.8 for FG, LR, and AS, respectively. Point estimates of N b based on all of the simulated data (solid horizontal lines) were 109.6, 64.5, and 58.4 for FG, LR, and AS, respectively. Point estimates of true N b based on the simulated data and Eq. 1 (dashed lines) were 108.4, 56.7, and 52.9 for FG, LR, and AS, respectively. Note log scale on the y-axis. See Fig. 4 for details on box plots

Bias in \( \hat{N}_{b} \) as a function of number of sample starting locations (SL). Box plots show estimates for each sampling strategy with data collapsed over sample size (S) and habitat patches. Bias was estimated as \( \hat{N}_{b} \) for each combination of S and SL relative to \( \hat{N}_{b} \) obtained from all individuals from each site (relative bias; empirical data, panel a) or relative to true N b (true bias; simulated data, panel b). True N b was calculated with Eq. 1. See Fig. 4 for details on box plots

Coefficient of variation for \( \hat{N}_{b} \). Box plots show estimates for each sample size (S) with data collapsed over number of sample starting locations (SL) and habitat patches. Empirical data are shown in panel a, simulated data in panel b. See Fig. 4 for details on box plots
Observed effects of SL, HP, and S on true N b (calculated based on Eq. 1) were similar for the simulated data sets (Fig. 5). For the simulated data, best estimates of N b based on all of the simulated individuals in a patch were similar in value to true N b (Fig. 5). The best estimate of N b based on all of the simulated data for FG was 109.6 (95% CI 95.9–124.6), for LR was 64.5 (95% CI 52.8–78.4), and for AS was 58.4 (95% CI 48.8–69.4). True N b for FG was 108.4, for LR was 56.7, and for AS was 52.9. Similarly to the empirical data, bias for the simulated data was lowest in AS relative to FG and LR. SL had a greater effect on bias in \( \hat{N}_{b} \) at fewer sample start locations, especially in FG and LR. Mean bias (averaged across S and HP) was lowest for SL = 3 (Table S1; Fig. 6). Finally, increasing S led to more precise (lower coefficients of variation) estimates of N b (Fig. 7).
Discussion
Our analyses revealed that obtaining samples of at least 75 individuals using multiple starting locations along habitat patches allowed robust estimates of N b for headwater brook trout populations. While the strength of these effects varied somewhat across streams, smaller sample sizes, or samples obtained from only a single starting location were likely to produce biased estimates. These results provide some broad rules of thumb for designing management sampling protocols, or when determining whether existing sampling methods are likely to be amenable for estimating N b for stream fishes. Further, our quantitative approach allowed us to incorporate system-specific information on allelic diversity and spatial population (family) structure to allow system-specific estimates of potential bias associated with sample size and sampling design. Our recommended sampling strategy performed well across varying family structures and therefore provides a powerful tool for a potentially wide range of species and conditions.
Our goal was to define a logistically feasible sampling strategy for minimizing both effort and bias for estimating N b of headwater brook trout populations. Bias in estimates of N b was lowest for SL = Random for the empirical data and below 10% for SL = 3 or 4 (Fig. 6). For the simulated data, SL = 3 had the least bias relative to true N b (Fig. 6). SL = 3 performed substantially better (in terms of bias) than SL = 1 or 2 in FG. Performance between SL = 3 and 4 was similar in FG. Of these two strategies, the one that involves less sampling effort (SL = 3) emerges as the best option. SL = 3 also outperformed SL = 1 and 2 in LR. In AS, SL = 3 performed well but the difference among sampling strategies was less pronounced. For streams with underlying family structures like AS (high degree of overlap in family spatial distributions), use of multiple starting locations for sampling is not critical. However, since family structure cannot be known prior to sampling, SL = 3 remains the best alternative for minimizing bias for all rivers that we considered, whether family structure was pronounced or not. Importantly, SL = 1 was clearly not an effective strategy at any sample size for either FG or LR. \( \hat{N}_{b} \) from SL = 1 for these two habitat patches were relatively precise but consistently biased low. The use of this sampling strategy, which is arguably the most commonly employed strategy currently, is likely to underestimate N b .
Increasing S led to increased precision in \( \hat{N}_{b} \). S = 75 provided a 15% decrease in the coefficient of variation over S = 50 for the empirical data and a 37% decrease in the coefficient of variation for the simulated data. Waples and Do (2008) demonstrated a tradeoff between precision and bias in estimates of N e as function of the critical allele frequency cutoff (P crit). The use of a lower P crit value in our study would likely have strengthened the observed pattern of increased precision at higher S but would have come at expense of increased bias across values of S (especially at smaller S). Based on the results of Waples and Do (2008) the use of a lower P crit value would not have changed our recommendations, which is an S of at least 75. This sample size is consistent with recommendations from simulations based on a Wright-Fisher model (Tallmon et al. 2010). S = 75 of age-0 individuals should be obtainable under most circumstances. However, if 75 age-0 fish are not present in a stream at the time of sampling, we recommend sampling fewer fish from multiple starting locations, or possibly sampling all of the age-0 fish present in a habitat patch. It should also be noted that estimates of N b in the habitat patches examined here were relatively small (range 46–112). Populations with substantially larger effective numbers of breeders (approx. >500) will likely require larger S (Tallmon et al. 2010).
Family spatial structure is the primary reason why sampling design (number of locations) and sample size had such an important influence on N b estimates. In our study, environmental variation was likely responsible for observed variation in family spatial distributions. The LR and AS samples were collected in 2010, an extremely low flow year (M.H. unpublished data). Stream habitat was reduced to pools with little intervening habitat and low dispersal opportunity following mid-summer. This likely led to the high degree of overlap of family spatial distributions in AS and the discrete upstream and downstream patches of habitat in LR. The FG sample was collected in 2004, a year with relatively favorable environmental conditions. Families were the most clumped spatially in FG but occurred throughout the available habitat. For the empirical data as well as simulations based on these three very different site parameterizations, sampling from a single location and obtaining small sample sizes increased the probability that some families were over-represented in the sample, biasing N b estimates downward.
Use of a sampling strategy that avoids family over-representation effects is of general importance for genetic monitoring populations of stream fishes. Early (soon after hatching) non-random kin associations have been demonstrated in other stream fishes, including Atlantic salmon (Salmo salar)(Olsen et al. 2004; Einum and Nislow 2005), brown trout (Salmo trutta)(Hansen et al. 1997; Hansen and Jensen 2005; Carlsson 2007; Sanz et al. 2011) and high predation populations of guppies (Poecilia reticulata) (Piyapong et al. 2011) and are likely to be demonstrated in future studies of additional species. Population-specific spatial variation in kin associations is likely to vary with environmental conditions and age of the individuals under consideration. A robust procedure for estimating effective population size that performs well regardless of the family structure encountered is needed. Our recommended sampling strategy (S = 75, SL = 3) provided unbiased and precise N b estimates for brook trout populations with clumped or dispersed family spatial distributions. A distinct advantage of this conservative approach is that it can be used without prior knowledge of family structure.
Researchers interested in monitoring effective population size are faced with the option to obtain mixed- or single-cohort population samples. Iteroparous species with overlapping generations violate the discrete generation assumption made by most single sample N e estimators (Waples 2005). Use of the LD-N e approach with mixed-cohort samples provides an estimate of N b that produced the cohort(s) from which the sample was taken, not generational N e (Waples and Do 2010). Degree of iteroparity and lifetime variance in reproductive success can contribute to uncertainty and bias in estimates based on mixed-cohort samples that are presumed to represent generational N e (Waples 2010). However, if all or most of the cohorts within a generation are represented in a sample, Waples and Do (2010) speculate that estimates from the LD-N e approach should roughly correspond to generational N e . This speculation remains untested (Waples 2010; Waples and Do 2010). A thorough evaluation, including the S necessary for each cohort, is needed. At least one empirical study to date suggests that mixed-cohort based estimates of N e might be biased. \( \hat{N}_{e} \) based on the LD-N e approach and obtained from combining individuals from successive cohorts did not correspond well with the \( \hat{N}_{e} \) obtained from the Jorde and Ryman (1995) modified temporal approach for sandbar sharks, Carcharhinus plumbeus (Portnoy et al. 2009).
We have taken the approach of using single-cohort samples to obtain LD-N e -based estimates of N b for an iteroparous organism. We have demonstrated that single-cohort based estimates of N b will be precise and unbiased for a wide range of family structures and realistic sample effort. A problem with this approach is that it does not provide an estimate of generational N e , which is the parameter needed for inference regarding rate of loss of genetic variation and adaptive potential (Hare et al. 2011). Unless it can be shown that reasonably sized mixed-cohort samples can provide unbiased single-sample estimates of N e over a wide range of life-history parameter space, we recommend focusing on N b from single-cohort samples because it is the parameter that can be estimated with minimal bias and precision as long as appropriate sampling effort is applied. \( \hat{N}_{b} \) obtained in this manner can be compared over time for iteroparous species to monitor for population trend, and single-cohort based estimates of N b separated by a number of generations (at least 3–5 or more; Waples and Yokota 2007) can be used to obtain temporal estimates of generational N e (Waples and Yokota 2007). We suggest that this approach is preferable to obtaining estimates of N e from mixed-cohorts that are more difficult to interpret. It is also not clear if mixed-cohort based N e estimates taken at one point in time can be compared to estimates obtained from either a second mixed-cohort or a single-cohort sample taken from the same population at a later time to examine population trend.
Future efforts to clarify the relationship between generational N e and N b in age-structured populations may allow estimates of N b obtained from our approach to be translated to estimates of generational N e . N e and N b are approximately related as N e ≈ generation length * N b when iteroparity is low (Hare et al. 2011). However, rates of iteroparity for most species with overlapping generations are rarely known and would need to be resolved for a species prior to using this equation to translate N b into an estimate of generational N e .
Conclusions
Concrete recommendations that are robust to varying population attributes emerge from our analysis. We recommend the sampling strategy that involves three equally spaced starting locations (SL = 3) and samples sizes (S) of at least 75 individuals from the young-of-the-year cohort. This combination of sampling strategy and sample size will minimize bias and provide precise estimates of N b across conditions realistically encountered in headwater brook trout populations. We also recommend estimates of N b from single cohorts as the most interpretable and straightforward focal point for genetic monitoring efforts for iteroparous species with overlapping generations. Our recommended sampling strategy was applied across three habitat patches with varying family structure and therefore should be widely applicable to headwater brook trout populations. Our recommendations should also apply to additional stream fishes, based on the similarity of family spatial structure across species living in headwater streams. Our approach is specifically designed for organisms that inhabit linear stream networks, however, any effective size genetic monitoring effort should guard against the bias associated with family over-representation effects observed in this study.
References
Carlsson J (2007) The effect of family structure on the likelihood for kin-biased distribution: an empirical study of brown trout populations. J Fish Biol 71:98–110
Coombs JA (2010) Reproduction in the wild: the effect of individual life history strategies on population dynamics and persistence. University of Massachusetts Amherst, Dissertation AAI3427513
Coombs JA, Letcher BH, Nislow KH (2008) CREATE: a software to create input files from diploid genotypic data for 52 genetic software programs. Mol Ecol Resour 8:578–580
Coombs JA, Letcher BH, Nislow KH (2010a) PEDAGOG: Software for simulating eco-evolutionary population dynamics. Mol Ecol Resour 10:558–563
Coombs JA, Letcher BH, Nislow KH (2010b) PedAgree: software to quantify error and assess accuracy and congruence for genetically reconstructed pedigree relationships. Conserv Genet Resour 2:147–150
Crow JF, Kimura M (1970) An Introduction to Population Genetics Theory. Burgess Publishing Company, Minneapolis
Curry RA, Bernatchez L, Whoriskey F, Audet C (2010) The origins and persistence of anadromy in brook charr. Rev Fish Biol Fish 20:557–570
Dunham JB, Vinyard GL, Rieman BE (1997) Habitat fragmentation and extinction risk of Lahontan cutthroat trout. N Am J Fish Manag 17:1126–1133
Einum S, Nislow KH (2005) Local-scale density dependent survival of mobile organisms in continuous habitats: an experimental test using Atlantic salmon. Oecologia 143:203–210
Hansen MM, Jensen LF (2005) Sibship within samples of brown trout (Salmo trutta) and implications for supportive breeding. Conserv Genet 6:297–305
Hansen MM, Nielsen EE, Mensberg KLD (1997) The problem of sampling families rather than populations: relatedness among individuals in samples of juvenile brown trout Salmo trutta L. Mol Ecol 6:469–474
Hare MP, Nunney L, Schwartz MK, Ruzzante DE, Burford M, Waples RS, Ruegg K, Palstra F (2011) Understanding and estimating effective population size for practical application in marine species management. Conserv Biol 25:438–449
Hill WG (1981) Estimation of effective population-size from data on linkage disequilibrium. Genet Res 38:209–216
Hudy M, Downey DM, Bowman DW (2000) Succesful restoration of an acidified native brook trout stream through mitigation with limestone sand. N Am J Fish Manag 20:453–466
Hudy M, Theiling TM, Gillespie N, Smith EP (2008) Distribution, status, and land use characteristics of subwatersheds within the native range of brook trout in the eastern United States. N Am J Fish Manag 28:1069–1085
Hudy M, Coombs JA, Nislow KH, Letcher BH (2010) Dispersal and within-stream spatial population structure of brook trout revealed by pedigree reconstruction analysis. Trans Am Fish Soc 139:1276–1287
Hunt RL, Brynildson OM (1964) A five-year study of a headwaters trout refuge. Trans Am Fish Soc 93:194–197
Isaak DJ, Luce CH, Rieman BE, Nagel DE, Peterson EE, Horan DL, Parkes S, Chandler GL (2010) Effects of climate change and wildfire on stream temperatures and salmonid thermal habitat in a mountain river network. Ecol Appl 20:1350–1371
Jorde PE, Ryman N (1995) Temporal allele frequency change and estimation of effective size in populations with overlapping generations. Genetics 139:1077–1090
Kalinowski ST, Wagner AP, Taper ML (2006) ML-RELATE: a computer program for maximum likelihood estimation of relatedness and relationship. Mol Ecol Notes 6:576–579
King TL, Julian SE, Coleman RL, Burnham-Curtis MK (2003) Isolation and characterization of novel tri- and tetranucleotide microsatellite DNA markers for brook trout Salvelinus fontinalis: GenBank submission numbers AY168187, AY168192, AY168193, AY168194, AY168195, AY168197, AY168199. Available: ncbi:nlm.nih.gov/nucleotide/
King TL, Eackles MS, Letcher BH (2005) Microsatellite DNA markers for the study of Atlantic salmon (Salmo salar): kinship, population structure, and mixed-fishery analyses. Mol Ecol Notes 5:130–132
Letcher BH, Nislow KH, Coombs JA, O’Donnell MJ, Dubreuil TL (2007) Population response to habitat fragmentation in a stream-dwelling brook trout population. PLoS ONE 2:e1139. doi:1110.1371/journal.pone.0001139
Lewis PO, Zaykin D (2001) Genetic data analysis: computer program for the analysis of allelic data (version 1.1). Available at http://www.eeb.uconn.edu/people/plewis/software.php
Luikart G, Ryman N, Tallmon DA, Schwartz MK, Allendorf FW (2010) Estimation of census and effective population sizes: the increasing usefulness of DNA-based approaches. Conserv Genet 11:355–373
Miller JM (1970) An analysis of the distribution of young-of-the-year brook trout, Salvelinus fontinalis (Mitchill), in Lawerence Creek, Wisconsin. University of Wisconsin, Madison
Morita K, Yamamoto S (2002) Effects of habitat fragmentation by damming on the persistence of stream-dwelling charr populations. Conserv Biol 16:1318–1323
Olsen KH, Petersson E, Ragnarsson B, Lundqvist H, Jarvi T (2004) Downstream migration in Atlantic salmon (Salmo salar) smolt sibling groups. Can J Fish Aquat Sci 61:328–331
Piyapong C, Butlin RK, Faria JJ, Scruton KJ, Wang J, Krause J (2011) Kin assortment in juvenile shoals in wild guppy populations. Heredity 106:749–756
Portnoy DS, McDowell JR, McCandless CT, Musick JA, Graves JE (2009) Effective size closely approximates the census size in the heavily exploited western Atlantic population of the sandbar shark, Carcharhinus plumbeus. Conserv Genet 10:1697–1705
Pudovkin AI, Zaykin DV, Hedgecock D (1996) On the potential for estimating the effective number of breeders from heterozygote-excess in progeny. Genetics 144:383–387
R Development Core Team (2006) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Rousset F (2008) GENEPOP ‘ 007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106
Sanz N, Fernandez-Cebrian R, Casals F, Araguas RM, Garcia-Marin JL (2011) Dispersal and demography of brown trout, Salmo trutta, inferred from population and family structure in unstable Mediterranean streams. Hydrobiologia 671:105–119
Tallmon DA, Koyuk A, Luikart G, Beaumont M (2008) ONeSAMP: a program to estimate effective population size using approximate Bayesian computation. Mol Ecol Notes 8:299–301
Tallmon DA, Gregovich D, Waples RS, Baker CS, Jackson J, Taylor BL, Archer E, Martien KK, Allendorf FW, Schwartz MK (2010) When are genetic methods useful for estimating contemporary abundance and detecting population trends? Mol Ecol Resour 10:684–692
Wang J (2009) A new method for estimating effective population sizes from a single sample of multilocus genotypes. Mol Ecol 18:2148–2164
Wang J, Whitlock MC (2003) Estimating effective population size and migration rates form genetic samples over space and time. Genetics 163:429–446
Waples RS (1989) Temporal variation in allele frequencies: testing the right hypothesis. Evolution 43:1236–1251
Waples RS (2005) Genetic estimates of contemporary effective population size: to what time periods do the estimates apply? Mol Ecol 14:3335–3352
Waples RS (2006) A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conserv Genet 7:167–184
Waples RS (2010) Spatial-temporal stratifications in natural populations and how they affect understanding and estimation of effective population size. Mol Ecol Resour 10:785–796
Waples RS, Do C (2008) LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resour 8:753–756
Waples RS, Do C (2010) Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evol Appl 3:244–262
Waples RS, Waples RK (2011) Inbreeding effective population size and parentage analysis without parents. Mol Ecol Resour 11:162–171
Waples RS, Yokota M (2007) Temporal estimates of effective population size in species with overlapping generations. Genetics 175:219–233
Wenger SJ, Isaak DJ, Luce CH, Neville HM, Fausch KD, Dunham JB, Dauwalter DC, Young MK, Elsner MM, Rieman BE, Hamlet AF, Williams JE (2011) Flow regime, temperature, and biotic interactions drive differential declines of trout species under climate change. Proc Natl Acad Sci USA 108:14175–14180
Wright S (1931) Evolution in mendelian populations. Genetics 16:97–159
Acknowledgments
M. Burak and M. Page helped with genetic analyses. The following organizations provided financial assistance or volunteer support: James Madison University, George Washington and Jefferson National Forest; Virginia Department of Game and Inland Fisheries; U.S. Forest Service, Northern Research Station; University of Massachusetts Amherst; U.S. Geological Survey, Leetown Science Center; and Conte Anadromous Fish Research Laboratory.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Whiteley, A.R., Coombs, J.A., Hudy, M. et al. Sampling strategies for estimating brook trout effective population size. Conserv Genet 13, 625–637 (2012). https://doi.org/10.1007/s10592-011-0313-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-011-0313-y