
Abstract
Maintaining genetic diversity and population viability in endangered and threatened species is a primary concern of conservation biology. Genetic diversity depends on population connectivity and effective population size (Ne), both of which are often compromised in endangered taxa. While the importance of population connectivity and gene flow has been well studied, investigating effective population sizes in natural systems has received far less attention. However, Ne plays a prominent role in the maintenance of genetic diversity, the prevention of inbreeding depression, and in determining the probability of population persistence. In this study, we examined the relationship between breeding pond characteristics and Ne in the endangered California tiger salamander, Ambystoma californiense. We sampled 203 individuals from 10 breeding ponds on a local landscape, and used 11 polymorphic microsatellite loci to quantify genetic structure, gene flow, and effective population sizes. We also measured the areas of each pond using satellite imagery and classified ponds as either hydrologically-modified perennial ponds or naturally occurring vernal pools, the latter of which constitute the natural breeding habitat for A. californiense. We found no correlation between pond area and heterozygosity or allelic diversity, but we identified a strong positive relationship between breeding pond area and Ne, particularly for vernal pools. Our results provide some of the first empirical evidence that variation in breeding habitat can be associated with differences in Ne and suggest that a more complete understanding of the environmental features that influence Ne is an important component of conservation genetics and management.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Quantifying the effects of landscape features on the effective population sizes of natural populations remains an important challenge in conservation genetics (Andersen et al. 2004; Rowe and Beebee 2004; Beebee 2005; Wang 2009a). Effective population sizes (Ne) in wild animal systems vary widely (Scribner et al. 1997; Crawford 2003; Hoffman et al. 2004; Palstra and Rruzzante 2008) and determining Ne for threatened and endangered species is a key component of their conservation and management (Andersen et al. 2004; Rowe and Beebee 2004; Beebee 2005; Schwartz et al. 2007). Reduced effective population sizes can lead to deleterious effects, including inbreeding depression and loss of genetic diversity, which can diminish the ability of a species or population to adapt to environmental change and to respond to risks from diseases and pathogens (Crnokrak and Roff 1999; Nieminen et al. 2001; Keller et al. 2002; Frankham 2005). In addition, if small effective populations tend to have small census populations, then measures of Ne will also be informative regarding the demographic consequences of small populations. Thus, understanding the effects of environmental variables on the effective sizes of natural populations can contribute substantially to scientifically-informed conservation practices (Palstra and Rruzzante 2008; Wang 2009a).
Determining how environmental variation affects Ne is also fundamental to a more complete understanding of variation in population dynamics and patterns of local adaptation. Population genetics theory emphasizes the central role of Ne on the relative efficacy of selection and drift, levels of genetic variability within populations, and rates of molecular evolution (Wright 1938; Kimura 1968). In populations with smaller Ne, drift plays a larger role in changes in allele frequencies, genetic variation tends to decrease over time, and molecular evolution occurs at a slower rate due to decreased mutational opportunities (Hartl and Clark 1997; Gillespie 2004; Charlesworth and Willis 2009).
There are many factors that can potentially influence Ne, including demographic history, the carrying capacity of habitat patches, and population connectivity (Waples and Gaggiotti 2006; Mills 2007). While new methods in landscape genetics are increasingly sophisticated in their ability to assess the effects that the landscapes separating populations have on dispersal probability and the resulting population genetic structure (Manel et al. 2003; Storfer et al. 2007; Spear et al. 2005; Giordano et al. 2007; Wang 2010), few conservation and landscape genetics studies consider the effects that environmental variation has on intra-population variables such as demographic history and effective population size (Funk et al. 1999; Driscoll 1999; Wang 2009a). For instance, in pond-breeding amphibians, substantial progress has been made in understanding the role of upland habitat in genetic structure and dispersal among populations (Trenham et al. 2001; Wang et al. 2009), but the effects of pond characteristics on population demography are relatively unknown.
Several species of amphibians provide excellent systems for quantifying the roles of residential and breeding habitats in population demography and Ne. The aquatic life history stage of many amphibians means that pond and stream, as well as terrestrial habitat characteristics, may contribute to variation in Ne among populations. Pond-breeding amphibians provide especially good study systems for examining the effects of aquatic breeding habitats on population parameters, because breeding ponds have quantifiable features and often demarcate discrete yearly cohorts of breeding populations. Additionally, amphibians are facing local and global declines (Fisher and Shaffer 1996; Houlahan et al. 2000; Collins and Storfer 2003; Stuart et al. 2004) and are often sensitive to anthropogenic habitat alteration (Guerry and Hunter 2002; Zamudio and Wieczorek 2007). Thus, studies linking breeding habitat and population persistence may play a critical role in conservation and management strategies (Gibbs 1998; Guerry and Hunter 2002; Funk et al. 2005; Rittenhouse and Semlitsch 2006).
In this study, we examined population structure and demography in the California tiger salamander, Ambystoma californiense, in a network of breeding ponds on a relatively undisturbed landscape in Merced County, CA. This region has little topographic relief (National Elevation Dataset; http://ned.usgs.gov) and ponds are separated only by grassland, a preferred terrestrial habitat for the species (Shaffer and Trenham 2005; Wang et al. 2009). The homogeneity of upland habitat in this region, compared to other study sites (e.g. Trenham et al. 2001; Wang et al. 2009), allowed us to isolate the effects of pond characteristics on population processes. Specifically, we used eleven microsatellite markers and population genetic analyses to test for associations between pond size and effective population size, allelic diversity, and heterogeneity in A. californiense. Our results suggest that pond size may play an important role in Ne (and therefore population persistence) and have important implications for the conservation of this species and amphibian conservation genetics more broadly.
Materials and methods
Sampling and study system
The California tiger salamander, A. californiense, is a federally protected, pond-breeding amphibian endemic to central California (Loredo et al. 1996; Shaffer and Trenham 2005; Fitzpatrick et al. 2009). A. californiense breeds in seasonal and permanent ponds that are free of fish and other introduced predators (Fisher and Shaffer 1996; Shaffer and Trenham 2005; Ryan et al. 2009). Aquatic larvae grow and develop in these pools for 3–6 months, at which time they metamorphose and disperse onto the surrounding terrestrial landscape. Except for a few weeks of breeding activity, they are thereafter primarily fossorial, residing in small mammal (primarily California ground squirrel, Spermophilus beecheyi and Botta’s pocket gopher, Thomomys bottae) burrows, which provide protection against predation and dessication (Loredo et al. 1996; Shaffer and Trenham 2005; Trenham and Shaffer 2005; Searcy and Shaffer 2008). The importance of adequate terrestrial (residential) habitat to maintain viable populations of A. californiense is now well-established (Trenham et al. 2001; Trenham and Shaffer 2005; Searcy and Shaffer 2008), although the relationship between upland terrestrial and aquatic breeding habitat remains relatively unknown.
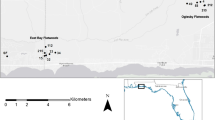
We collected tissue samples via tail-clip from a total of 203 larvae from the 10 breeding ponds in which A. californiense occurs in our study region (Fig. 1). All tissues were collected on March 30 and 31, 2006. Tissues were stored in 95% ethanol and maintained at −20°C. Pond areas were calculated using tools provided in Google Earth Pro v5.2. We traced the perimeter of the pond basin (as opposed to the water level) based on apparent transitions in vegetation cover using satellite imagery captured on December 30, 2005. The breeding period for A. californiense typically occurs between late November and early March, depending on the initiation of winter rains and pond filling. Ponds were categorized as either vernal pools (VP) or artificial perennial ponds (PP) based on expected differences in pond hydroperiods (Table 1); vernal pools are natural, shallow depressions that typically dry each year, whereas perennial ponds are artificial, deeper ponds that frequently retain water throughout the year.

Shaded relief map indicating the locations of the 10 sampled breeding ponds from Merced County, California, used in our study: Flying M Conservario Pond (FMC), One Eyed Vernal Pool (OEV), Virginia Smith Trust Pond 1 (VS1), Virginia Smith Trust Pond 2 (VS2), Virginia Smith Trust Pond 3 (VS3), Virginia Smith Trust Pond 4 (VS4), Chance Ranch Corral Pond (CCR), Chance Ranch Salamander Pond (SCR), Virginia Smith Trust La Paloma Pond (LPR), and Cyril Smith Trust Bull Pond (BCS). Underlined acronyms represent vernal pools
Genotyping
Tissues were digested in lysis buffer with Proteinase K, and genomic DNA was purified using a standard ethanol precipitation. Extracted samples were diluted to 10 ng/μl and used as template in PCR reactions for eleven tetra-nucleotide microsatellite loci (Savage 2008). Forward primers for each PCR were labeled with a 5′ fluorescent tag (6-FAM, NED, VIC, or PET) for visualization. We amplified loci individually and ran PCR products on an ABI 3730 Genetic Analyzer (Applied Biosystems, Costa Mesa, CA). Fragments were sized with LIZ-500 size standard and collected with GeneScan v3.1 (Applied Biosystems). Scoring was performed with STRand v.2.9.34 (University of California, Davis, CA). We used Micro-Checker v2.2.3 (van Oosterhout et al. 2004) to check for potential scoring errors, the presence of null alleles, linkage disequilibrium, and departures from Hardy–Weinberg equilibrium (HWE).
Genetic variation and population structure
We investigated the distribution of genetic variation by performing an analysis of molecular variance (AMOVA) and calculating allelic diversity, heterozygosity, and pairwise values of F ST among breeding ponds using GenAlEx v2.6 (Peakall and Smouse 2006). We also examined the genetic clustering of samples using BAPS v4.14 (Corander et al. 2004), Structure v2.3.2 (Pritchard et al. 2000), and by performing an analysis of molecular variation (AMOVA). BAPS (Corander et al. 2004) uses Bayesian mixture modeling to describe variation within subpopulations using a separate joint probability distribution over multiple loci. The mixture model parameters include the assignment of individuals to clusters to optimize genetic similarity within clusters. We performed the ‘clustering of individuals’ analysis, using 1–11 as maximum numbers of clusters and 10,000 iterations to estimate the admixture coefficient for each sample. Structure implements a genotypic clustering method that assigns samples to groups in order to maximize within-cluster conformity to HWE and minimize linkage disequilibrium. Structure calculates the likelihood for an assumed number of clusters, K, and so allows comparisons between different values of K. We performed 20 independent replicates for each value of K (K = 1–11) with 10,000 iterations of the MCMC following a 10,000 iteration burn-in. For these runs, we assumed independence among loci, allowed admixture in the clustering model, and used the sampling localities as prior information. A drawback of Structure is that the number of genetic clusters can often be overestimated, and, in many cases, the inferred number and membership of populations do not appear biologically reasonable. To correct for this, we took into account the shape of the log-likelihood curve and variance among multiple runs as recommended by Evanno et al. (2005) and calculated ΔK to find the optimal estimate for number of genetic clusters.
Effective population size estimation
To estimate effective population sizes of A. californiense in each of the ten breeding ponds, we performed the sibship assignment method implemented in Colony v2.0 (Wang 2009b). The sibship assignment method first determines the probabilities of all pairs of samples from a population being full-sibs and half-sibs. These assignment probabilities are then used to determine Ne based on a predictive equation that relates the probability of drawing these assignments from a randomly sampled, single cohort to the number of effective breeding adults. Extensive simulation studies, as well as some empirical data, have shown that the sibship assignment method is much more accurate than other common methods for estimating Ne, such as the heterozygote excess method, the linkage disequilibrium method, and the temporal method (Wang 2009b).
Bottleneck detection
We used two methods to detect molecular signatures of historical bottlenecks. We used Bottleneck v1.2.02 (Cornuet et al. 1999; Piry et al. 1999) to investigate deviations from expected heterozygote excess relative to allelic diversity. During population bottlenecks, rare alleles are lost due to drift at a faster rate than the overall loss of heterozygosity, and Bottleneck utilizes this disparity to detect past bottlenecks (Spencer et al. 2000; Pearse and Crandall 2004; Williamson-Natesan 2005). We performed the analysis under all three microsatellite mutational models available: the infinite alleles model (IAM), stepwise mutational model (SMM), and two phase mutational model (TPM) with 80% single-step mutations and 20% multiple-step mutations, based on empirical estimates of microsatellite mutation rates (Goldstein and Schlotterer 1999). We also calculated the statistical significance of the M statistic in each population compared to that from a simulated population in M-Ratio (Garza and Williamson 2001). In this test, M is the ratio between the number of alleles at a locus and the total range in allele sizes. Unless all rare alleles are at the ends of the allele size distribution, bottlenecks will tend to eliminate rare alleles but leave the allele size distribution intact, and M will be reduced in populations that have experienced a significant population size reduction (Pearse and Crandall 2004; Williamson-Natesan 2005). We parameterized the TPM used in M-Ratio by assigning an 80% rate of single-step mutations and a mean of 2.8 repeats to the size change of multiple-step mutations, based upon empirical estimates of microsatellite evolution (Garza and Williamson 2001), and used a mutation rate of 5 × 10−4 (Goldstein and Schlotterer 1999). We performed the analysis by choosing a range of pre-bottleneck effective population sizes of 100, 200, and 1000 individuals, resulting in θ = 0.05, 0.10, and 0.50. Because some recent studies have shown that gene flow among populations can sometimes produce false signals of population bottlenecks (Chikhi et al. 2010; Peter et al. 2010), we corrected for this possible artifact by combining samples from several populations (Chikhi et al. 2010). Specifically, we performed both analyses again using all 10 populations and using all combinations of 9 populations. For these analyses, we used proportionally adjusted pre-bottleneck effective population sizes of 1000, 2000, and 10000 individuals (for 10 populations) and 900, 1800, and 9000 individuals (for 9 populations).
Gene flow estimation
We estimated ongoing gene flow between populations using a genetic assignment method implemented in BayesAss+ v1.3 (Wilson and Rannala 2003). BayesAss+ uses a fully Bayesian MCMC resampling method to estimate asymmetrical rates of recent gene flow between populations and also calculates a confidence interval for results that would be returned from uninformative data, typically those that do not contain sufficient variation to estimate dispersal with high confidence (Wilson and Rannala 2003; Pearse and Crandall 2004). We performed our BayesAss+ run with 5 million generations, discarded 2 million as burn-in, sampled the chain every 2,000 generations, and used default parameter settings.
Regression analysis
Pond area measurements were log-transformed and simple linear regressions were performed using the ‘lm’ function of the ‘stats’ package in R (R Development Core Team 2008). We evaluated the relationship between pond area and each of four response variables: effective population size (Ne), observed heterozygosity (HO), average pairwise F ST, and allelic richness (Â).
Results
Genotyping
The microsatellite loci we used were highly polymorphic, ranging from 6 to 14 alleles per locus, with a mean of 11.2 ± 2.4 alleles; these values are nearly identical to those found in A. californiense in a different part of their range (Wang et al. 2009). Micro-Checker did not indicate the presence of null alleles, scoring errors, or linkage disequilibrium. We found deviations from HWE for four loci in one or two populations each, but no consistent evidence that these loci did not conform to HWE expectations, and thus we did not exclude any loci from our analysis. We were unable to unambiguously score 9.3% of genotypes and coded these as missing data.
Genetic variation and population structure
We found slight variations in allelic diversity and heterozygosity among populations (Table 1). Estimates of pairwise F ST ranged from 0.037 to 0.182 (Table 2), with an overall mean F ST = 0.087 ± 0.032. The AMOVA indicated that 81% of molecular variation occurs within populations and 19% between populations. We found no evidence of isolation by distance, based on a Mantel test for correlation between pairwise F ST and geographic distance (r = −0.117; P = 0.635).
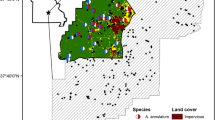
The results of our Structure runs indicated an optimal log likelihood for K = 8. However, after applying the correction described by Evanno et al. (2005), we found that the optimal number of clusters was K = 6 (Fig. 2a), corresponding to FMC + OEV + BCS, VS1 + LPR1, VS2 + VS3, VS4, CCR, and SCR. BAPS identified 10 genetic clusters, which corresponded to our 10 sampled breeding ponds (Fig. 2b).

Results of genetic clustering in Structure (a) and BAPS (b) based on 11 microsatellite loci. Each vertical bar represents an individual sample, and the height of each colored segment within each bar represents the probability of that sample being assigned to each cluster. The results depicted in a were derived for the optimal inferred value of K (K = 6). Colors are not coordinated between panels
Bottlenecks and effective population size
Mean estimates of effective population size were consistently low, ranging from Ne = 11 to 64, with an average across the ten populations of Ne = 30 (Table 1). We also found consistent evidence for population bottlenecks in all of our populations. Bottleneck indicated significant signs of historical reductions in population size in all ten populations under the infinite alleles (IAM) and two-phase mutational (TPM) models and in one population under the stepwise mutational model (SMM) as well (Table 3). M-Ratio also indicated significant signals of population bottlenecks in all ten populations for θ = 0.05 and in six populations for θ = 0.10 (Table 3). We also detected a significant signal for population bottlenecks when we combined samples from all 10 populations using M-Ratio when the pre-bottleneck effective population size was estimated as 1,000 individuals (P = 0.046) and a signal that approached significance using Bottleneck under the IAM (P = 0.062). When we combined sets of 9 populations, we found a significant signal from all sets using M-Ratio for pre-bottleneck Ne = 900 (P < 0.05 in all cases). We also found a significant signal for the set that excluded population OEV using M-Ratio for pre-bottleneck Ne = 900 (P = 0.042) and in Bottleneck under the IAM (P = 0.039) and the TPM (P = 0.049). Thus, we found strong signals of population bottlenecks in all populations except for OEV, which has a moderate signal under some analyses.
Gene flow
BayesAss+ indicated that the confidence interval for gene flow estimates that might result from uninformative data was fairly large. We recovered three rates that fell outside this range, and therefore indicated strong evidence for recent gene flow: FMC to OEV = 0.261 (0.184–0.317), VS3 to VS2 = 0.197 (0.075–0.304), and BCS to SCR = 0.157 (0.086–0.228). That is, for these pairs of ponds, 15.7–26.1% of individuals sampled had parents that were inferred to have come from another breeding site.
Regressions with breeding pond area
Linear regression on log-transformed breeding pond area values revealed no significant statistical relationship with allelic richness,  (Fig. 3b; F (1,8) = 0.924; P = 0.365; R 2 = 0.10), average pairwise F ST (Fig. 3d; F (1,8) = 0.625; P = 0.452; R 2 = 0.07), or heterozygosity, HO (Fig. 3c; F (1,8) = 0.773; P = 0.773; R 2 = 0.01). However, we found a positive relationship between breeding pond area and Ne (Fig. 3a; F (1,8) = 8.013; P = 0.022; R 2 = 0.50). When the vernal and modified perennial ponds were analyzed separately, we found that this relationship between Ne and pond area was entirely due to vernal pools (Fig. 3a; F (1,3) = 30.580; P = 0.012; R 2 = 0.91), with no evidence of a similar relationship for modified perennial ponds (Fig. 3a; F (1,3) = 0.003; P = 0.962; R 2 = 0.00). We also detected a significant negative relationship between pond area and  in permanent ponds (Fig. 3c; F (1,3) = 11.980; P = 0.041; R 2 = 0.80).

Results of regression analysis between log-transformed pond area and effective population size (a), allelic richness (b), observed heterozygosity (c), and F ST (d). Error bars denote 95% confidence intervals. Black regression line is for the full 10-pond data set; red and blue lines are for perennial ponds and vernal pools, respectively
Discussion
The levels of population structure and gene flow that we detected in our study are in line with estimates from this endangered salamander in other regions. Molecular estimates of gene flow from a network of A. californiense breeding ponds in Monterey County, California (Fort Ord Natural Reserve), found levels of gene flow nearly as high as those from this study. Estimates of gene flow in that system, also based on BayesAss+ and microsatellite data, ranged from 0.105 to 0.199 across a more complex landscape containing three diverse habitat classes instead of just one (Wang et al. 2009). A long-term mark–recapture study of A. californiense also indicated that interpond dispersal occurs regularly. In a network of breeding ponds from a topographically and ecologically complex landscape in central California (Hastings Reservation) that is similar to that at Fort Ord, approximately 30% of first-time breeders moved to different breeding pond from their birth pond, and about 30% of salamanders that bred in two different years switched ponds between breeding events (Trenham et al. 2001), suggesting that a salamander that breeds twice has a roughly 50% probability of moving from its birth pond for at least one of those breeding events. These numbers, from very different sites with very different ecological conditions, suggest that movement among breeding sites is quite common in this species, and that the precise philopatry that is often ascribed to Ambystoma salamanders (Gamble et al. 2007) may not hold for A. californiense.
Interestingly, we found no evidence for isolation-by-distance, which often occurs in amphibian systems (Funk et al. 2005; Spear et al. 2005; Wang et al. 2009; Wang and Summers 2010). However, pairwise estimates of F ST among our ponds are consistent with estimates of gene flow and genetic clustering. The pairs of populations in which we detected high levels of recent gene flow (FMC + OEV; VS2 + VS3; BCS + SCR) all had relatively low values of F ST (from 0.037, 0.057, and 0.078). Additionally, Structure output suggested that FMC + OEV and VS2 + VS3 could share membership in the same genetic clusters. On the other hand, BAPS identified our breeding pond sampling localities as independent genetic clusters, and the discrepancy in clustering between BAPS and Structure is likely due to the underlying model of admixture implemented by each program. Taken together, these results indicate that there is moderate genetic differentiation among most breeding ponds, with signatures of some admixture between several pairs of populations. Given the high probabilities of interpond dispersal found by Trenham et al. (2001), the genetic distinctiveness of breeding sites both here and at Fort Ord (Wang et al. 2009) may indicate that migrants are less successful breeders than resident salamanders, that different landscapes promote different levels of interpond movement, or that movement between ponds is spatially restricted to breeding sites that are in very close proximity only, as was found by Trenham et al. (2001).
We estimated relatively low effective population sizes (Ne) in all of our breeding ponds, ranging from Ne = 11 to 64. These estimates are similar to estimates of Ne in other salamanders (Funk et al. 1999; Jehle and Arntzen 2002; Savage et al. 2010) and other amphibians (Scribner et al. 1997; Driscoll 1999; Rowe and Beebee 2004; Wang 2009), all of which describe effective population sizes well below 100 individuals per population and often as small as Ne = 10–20. Additionally, a recent review of estimates of Ne across animal systems has indicated that effective population sizes in wild animal populations are generally smaller than previously thought (Frankham 2009). These results suggest that conservation biology in general would benefit from a greater understanding of Ne and its relationship to census population size, in nature. A key question, particularly for pond-breeding amphibians, is whether values of Ne < 100 indicate a reduction in population fitness, decreased population viability, or both, as has sometimes been argued (Lande 1998).
Small effective population sizes can occur for a variety of reasons, including population bottlenecks, genetic isolation, low thresholds for total population sizes supported by particular habitat patches, reproductive skew among breeding adults, and sex-ratio asymmetry (Hartl and Clark 1997; Waples 2002; Wang 2009). Our analyses of population bottlenecks indicates that historical reductions in population size likely contributed to our observation of low Ne in all ten of our sampled populations (Table 3). Both Bottleneck and M-Ratio indicated strong support for population bottlenecks under most model conditions, and this demographic history could influence estimates of Ne in current populations through reductions in allelic diversity and time to coalescence between sampled genotypes (Bohonak 1999; Gillespie 2004; Waples 2002). This result was robust to additional analyses that combined data from all populations except OEV, and combining data from all 10 populations produced a significant signal of a population bottleneck under some analyses as well (see Results), suggesting that these results are not an artifact resulting from gene flow among subdivided populations (Chikhi et al. 2010; Peter et al. 2010).
While population bottlenecks may explain the low estimates of Ne overall, the suitability of different breeding habitats also appears to have contributed to variation in Ne among populations (Table 1). Our analysis of the correlations between pond size and allelic diversity, heterozygosity, and Ne provides some of the first quantitative evidence that variation in breeding pond size can influence effective population size. We found a strong correlation between pond area and Ne (Fig. 3), particularly for natural vernal pools, and this results is robust in light of other variables that could affect Ne. For example, effective population size can be influenced by the overall level of genetic isolation of a population, but we found no evidence of isolation-by-distance (Wright 1943) and no significant correlation between pond area and average pairwise F ST (Fig. 3). Thus, smaller ponds did not support smaller populations because they were more genetically isolated than larger ponds. Rather, effective population size seems to be associated with pond area alone.
The correlation between pond area and Ne was even stronger when we considered only vernal pools, raising the correlation coefficient to R 2 = 0.91 (P = 0.012) from R 2 = 0.50 (P = 0.022). This may be due to the rate at which vernal pools dry, which is a function of their surface area and volume. Larval salamanders must either metamorphose before a pool dries or perish, and smaller pools that dry more quickly may often experience lower survivorship to metamorphosis. Artificial ponds, which are often relatively deep and more permanent, may lack this relationship between pond size and larval mortality, and do not show a relationship between pond surface area and Ne. We also found a significant negative relationship between pond area and allelic richness for artificial ponds, which could potentially indicate that these ponds are acting as sink populations, but additional studies would be necessary to test this. Interestingly, the correlation we detected between Ne and pond area was logarithmic, suggesting that Ne begins to plateau with increasing pond area and that some threshold for population size may be reached in very large ponds. Although smaller ponds supported smaller populations, we did not find any evidence for a reduction in allelic diversity or heterozygosity based on pond area (Table 1; Fig. 3). So, although smaller ponds have smaller Ne, these populations do not appear to have lower genetic diversity relative to larger ponds with larger Ne, suggesting they may not be experiencing the deleterious fitness effects sometimes associated with small population size and with population bottlenecks (Table 3). A better understanding of the minimum threshold for Ne necessary to maintain population viability will require additional empirical studies, such as this, of Ne and genetic diversity in natural populations.
Our results have clear conservation implications. While most conservation genetics studies have focused on the importance of maintaining connectivity among populations, few have considered the ecological conditions necessary to maintain viable effective population sizes. Population connectivity is clearly important, both for sustaining genetic diversity and the complex source-sink dynamics that characterize many natural populations (Trenham et al. 2001; Manel et al. 2003; Storfer et al. 2007; Spear et al. 2005; Wang et al. 2009). However, genetic diversity can also be protected by maintaining adequate effective population sizes (Storfer et al. 2007; Charlesworth and Willis 2009; Wang 2009a), and this may be particularly important as landscapes become fragmented and gene flow is interrupted. Understanding the relationship between key habitat features and Ne is thus another key component of a comprehensive conservation effort. In the case of endangered California tiger salamanders, our findings indicate that large vernal pools may be particularly valuable for ensuring large effective population sizes in this species and that natural vernal pools and artificial perennial ones may play very different roles in the maintenance of large effective populations. Other systems may also benefit from similar conservation genetics studies that identify important features of breeding habitats for safeguarding effective population size.
References
Andersen LW, Fog K, Damgaard C (2004) Habitat fragmentation causes bottlenecks and inbreeding in the European tree frog (Hyla arborea). Proc R Soc Lond B 271:1293–1302
Beebee TJC (2005) Conservation genetics of amphibians. Heredity 95:423–427
Bohonak AJ (1999) Dispersal, gene flow, and population structure. Q Rev Biol 74:21–45
Charlesworth D, Willis JH (2009) The genetics of inbreeding depression. Nat Rev Genet 10:783–796
Chikhi L, Sousa VC, Luisi P, Goossens B, Beaumont MA (2010) The confounding effects of population structure, genetic diversity and the sampling scheme on the detection and quantification of population size changes. Genetics 186:983–993
Collins JP, Storfer A (2003) Global amphibian declines: sorting the hypotheses. Divers Distrib 9:89–98
Corander J, Waldmann P, Marttinen P, Sillanpaa MJ (2004) BAPS 2: enhanced possibilities for the analysis of genetic population structure. Bioinformatics 20:2363–2369
Cornuet JM, Piry S, Luikart G, Estoup A, Solignac M (1999) New methods employing multilocus genotypes to select or exclude populations as origins of individuals. Genetics 153:1989–2000
Crawford AJ (2003) Huge populations and old species of Costa Rican and Panamanian dirt frogs inferred from mitochondrial and nuclear gene sequences. Mol Ecol 12:2525–2540
Crnokrak P, Roff DA (1999) Inbreeding depression in the wild. Heredity 83:260–270
Driscoll DA (1999) Genetic neighbourhood and effective population size for two endangered frogs. Biol Conserv 88:221–229
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Fisher RN, Shaffer HB (1996) The decline of amphibians in California’s Great Central Valley. Conserv Biol 10:1387–1397
Fitzpatrick BM, Johnson JR, Kump DK, Shaffer HB, Smith JJ, Voss SR (2009) Rapid fixation of non-native alleles revealed by genome-wide SNP analysis of hybrid tiger salamanders. BMC Evol Biol 9:176
Frankham R (2005) Genetics and extinction. Biol Conserv 126:131–140
Frankham R (2009) Effective population size/adult population size ratios in wildlife: a review. Genet Res 66:95–107
Funk WC, Tallmon DA, Allendorf FW (1999) Small effective population size in the long-toed salamander. Mol Ecol 8:1633–1640
Funk WC, Blouin MS, Corn PS, Maxell BA, Pilliod DS, Amish S, Allendorf FW (2005) Population structure of Columbia spotted frogs (Rana luteiventris) is strongly affected by the landscape. Mol Ecol 14:483–496
Gamble LR, McGarigal K, Compton BW (2007) Fidelity and dispersal in the pond-breeding amphibian, Ambystoma opacum: implications for spatio-temporal population dynamics and conservation. Biol Conserv 139:247–257
Garza JC, Williamson EG (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10:305–318
Gibbs JP (1998) Distribution of woodland amphibians along a forest fragmentation gradient. Landsc Ecol 13:263–268
Gillespie JH (2004) Population genetics: a concise guide. The Johns Hopkins University Press, Baltimore, MD
Giordano AR, Ridenhour BJ, Storfer A (2007) The influence of altitude and topography on genetic structure in the long-toed salamander (Ambystoma macrodactulym). Mol Ecol 16:1625–1637
Goldstein DB, Schlotterer C (1999) Microsatellites: evolution and applications. Oxford University Press, Oxford
Guerry AD, Hunter ML (2002) Amphibian distributions in a landscape of forests and agriculture: an examination of landscape composition and configuration. Conserv Biol 16:745–754
Hartl D, Clark A (1997) Principles of population genetics. Sinauer Associates, Sunderland
Hoffman EA, Schueler FW, Blouin MS (2004) Effective population sizes and temporal stability of genetic structure in Rana pipiens, the northern leopard frog. Evolution 58:2536–2545
Houlahan JE, Findlay CS, Schmidt BR, Meyer AH, Kuzmin SL (2000) Quantitative evidence for global amphibian population declines. Nature 404:752–755
Jehle R, Arntzen JW (2002) Review: microsatellite markers in amphibian conservation genetics. Herpetol J 12:9
Keller LF, Grant PR, Grant BR, Petren K (2002) Environmental conditions affect the magnitude of inbreeding depression in survival of Darwin’s finches. Evolution 56:1229–1239
Kimura M (1968) Evolutionary rate at the molecular level. Nature 217:624–626
Lande R (1998) Anthropogenic, ecological and genetic factors in extinction and conservation. Res Popul Ecol 40:259–269
Leberg PL (2002) Estimating allelic richness: effects of sample size and bottlenecks. Mol Ecol 11:2445–2449
Loredo I, Vuren DV, Morrison ML (1996) Habitat use and migration behavior of the California tiger salamander. J Herpetol 30:282–285
Manel S, Schwartz MK, Luikart G, Taberlet P (2003) Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol 18:189–197
Mills LS (2007) Conservation of wildlife populations: demography, genetics and management. Wiley-Blackwell, London
Nieminen M, Singer MC, Fortelius W, Schöps K, Hanski I (2001) Experimental confirmation that inbreeding depression increases extinction risk in butterfly populations. Am Nat 157:237–244
Palstra FP, Rruzzante DE (2008) Genetic estimates of contemporary effective population size: what can they tell us about the importance of genetic stochasticity for wild population persistence? Mol Ecol 17:3428–3447
Peakall R, Smouse PE (2006) GenAlEx 6: genetic analysis in Excel. Mol Ecol Notes 6:288–295
Pearse DE, Crandall KA (2004) Beyond FST: analysis of population genetic data for conservation. Conserv Genet 5:585–602
Peter BM, Wegmann D, Excoffier L (2010) Distinguishing between population bottleneck and population subdivision by a Bayesian model choice procedure. Mol Ecol 19:4648–4660
Piry S, Luikart G, Cornuet JM (1999) Bottleneck: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90:502–503
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
R Development Core Team (2008) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3
Rittenhouse TAG, Semlitsch RD (2006) Grasslands as movement barriers for a forest-associated salamander: migration behavior of adult and juvenile salamanders at a distinct habitat edge. Biol Conserv 131:14–22
Rowe G, Beebee TJC (2004) Reconciling genetic and demographic estimators of effective population size in the anuran amphibian Bufo calamita. Conserv Genet 5:287–298
Ryan ME, Johnson JR, Fitzpatrick BM (2009) Invasive hybrid tiger salamander genotypes impact native amphibians. Proc Natl Acad Sci USA 106:11166
Savage W (2008) Tandem repeat markers for population genetic studies of the protected California tiger salamander (Ambystoma californiense). Conserv Genet 9:1707–1710
Savage WK, Fremier AK, Shaffer HB (2010) Landscape genetics of alpine Sierra Nevada salamanders reveal extreme population subdivision in space and time. Mol Ecol 19:3301–3314
Schwartz M, Luikart G, Waples R (2007) Genetic monitoring as a promising tool for conservation and management. Trends Ecol Evol 22:25–33
Scribner KT, Arntzen JW, Burke T (1997) Effective number of breeding adults in Bufo bufo estimated from age-specific variation at minisatellite loci. Mol Ecol 6:701–712
Searcy CA, Shaffer HB (2008) Calculating biologically accurate mitigation credits: insights from the California tiger salamander. Conserv Biol 22:997–1005
Shaffer HB, Trenham PC (2005) California tiger salamanders. In: Lannoo M (ed) Amphibian declines: the conservation status of United States species. University of California Press, Berkeley, pp 605–608
Spear SF, Peterson CR, Matocq MD, Storfer A (2005) Landscape genetics of the blotched tiger salamander (Ambystoma tigrinum melanostictum). Mol Ecol 14:2553–2564
Spencer CC, Neigel JE, Leberg PL (2000) Experimental evaluation of the usefulness of microsatellite DNA for detecting demographic bottlenecks. Mol Ecol 9:1517–1528
Storfer A, Murphy MA, Evans JS, Goldberg CS, Robinson S, Spear SF, Dezzani R, Delmelle E, Vierling L, Waits LP (2007) Putting the ‘landscape’ in landscape genetics. Heredity 98:128–142
Stuart SN, Chanson JS, Cox NA, Young BE, Rodrigues ASL, Fischman DL, Waller RW (2004) Status and trends of amphibian declines and extinctions worldwide. Science 306:1783
Trenham PC, Shaffer HB (2005) Amphibian upland habitat use and its consequences for population viability. Ecol Appl 15:1158–1168
Trenham PC, Koenig WD, Shaffer HB (2001) Spatially autocorrelated demography and interpond dispersal in the salamander Ambystoma californiense. Ecology 82:3519–3530
van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-Checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Wang IJ (2009a) Fine-scale population structure in a desert amphibian: landscape genetics of the black toad (Bufo exsul). Mol Ecol 18:3847–3856
Wang J (2009b) A new method for estimating effective population sizes from a single sample of multilocus genotypes. Mol Ecol. doi:10.1111/j.1365-1294X.2009.04175.x
Wang IJ (2010) Recognizing the temporal distinctions between landscape genetics and phylogeography. Mol Ecol 19:2605–2608
Wang IJ, Summers K (2010) Genetic structure is correlated with phenotypic divergence rather than geographic isolation in the highly polymorphic strawberry poison-dart frog. Mol Ecol 19:447–458
Wang IJ, Savage WK, Shaffer HB (2009) Landscape genetics and least-cost path analysis reveal unexpected dispersal routes in the California tiger salamander (Ambystoma californiense). Mol Ecol 18:1365–1374
Waples RS (2002) Definition and estimation of effective population size in the conservation of endangered species. In: Beissinger SR, McCullough DR (eds) Population viability analysis. University of Chicago Press, Chicago, IL
Waples RS, Gaggiotti O (2006) What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol Ecol 15:1419–1439
Williamson-Natesan EG (2005) Comparison of methods for detecting bottlenecks from microsatellite loci. Conserv Genet 6:551–562
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191
Wright S (1938) Size of population and breeding structure in relation to evolution. Science 87:2263–2264
Wright S (1943) Isolation by distance. Genetics 28:114–138
Zamudio KR, Wieczorek AM (2007) Fine-scale spatial genetic structure and dispersal among spotted salamander (Ambystoma maculatum) breeding populations. Mol Ecol 16:257–274
Acknowledgments
We thank B. Fitzpatrick for help with field collections; E. Loomis, N. Norvell and H. Teaford for assistance with lab work; the California Department of Fish and Game for granting scientific collecting permits; and the UC Davis College of Biological Sciences DNA Sequencing Facility and College of Agriculture and Life Sciences Genomics Facility for help with microsatellite genotyping. This research was supported by grants from the Bureau of Reclamation and NSF (DEB 0817042 (to HBS), the UC Davis Agricultural Experiment Station, and by an NSF graduate research fellowship (to IJW).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wang, I.J., Johnson, J.R., Johnson, B.B. et al. Effective population size is strongly correlated with breeding pond size in the endangered California tiger salamander, Ambystoma californiense . Conserv Genet 12, 911–920 (2011). https://doi.org/10.1007/s10592-011-0194-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-011-0194-0