
Abstract
Ectosomes are small heterogeneous membrane vesicles generated by budding from the plasma membrane in a variety of cell types and, more frequently, in tumor cells. They are shed into the extracellular space and are proposed as a novel form of intracellular communication in which information is transmitted from the originating cell to recipient cells without direct cell-to-cell contact. This review focuses on a single population of extracellular vesicles—ectosomes. We summarize recent studies of tumor-derived ectosomes which examine their biogenesis and protein cargo, and their influence on different aspects of cancer progression. We discuss possible clinical implications involving ectosomes as potential biomarkers, diagnostic tools and treatment targets in oncology. The unique composition of the molecules (cargo) that ectosomes carry, and their functional role, depends largely on the state of their originating cell. Through horizontal transfer of a variety of biologically active molecules (including proteins, lipids and nucleic acids) between donor and recipient cells, tumor-derived ectosomes may play functional roles in oncogenic transformation, tumor progression, invasion, metastasis, angiogenesis promotion, escape from immune surveillance, and drug resistance, thereby facilitating disease progression. The presence of tumor-derived ectosomes in body fluids such as the blood and urine of cancer patients makes them potentially useful prognostic and predictive biomarkers. Tumor-derived ectosomes also offer possible targets for multiple therapeutic strategies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cancer development and progression are multistep processes in which a series of changes in the tumor microenvironment and in intercellular communication occur. Those highly specific alterations are driven mainly by endogenous molecular factors with proven oncogenic potential. Most bioactive molecules involved in carcinogenesis are directly secreted and act in an auto-, para- or endocrine manner, but another mechanism of their delivery to target cells has also been described. A variety of cell types, including cancer cells, are known to release extracellular vesicles (EVs)—small, membrane-enclosed particles which can mediate the transfer of different signaling factors, structural proteins, nucleic acids or lipids [1]. Following their in vivo release to the intercellular space, EVs typically are detected in a wide spectrum of body fluids such as blood (plasma or serum), urine, cerebrospinal fluid (CSF), bile, ascites, saliva, amniotic fluid, milk or semen; they can also be isolated in vitro from conditioned media of cultured cells. Such accessibility contributes to their prognostic, diagnostic and therapeutic value for particular health conditions [2].
Over the years, successive studies have revealed striking diversity within EV populations. Across this diversity they can be classified into several distinct populations based on their size, density, cellular origin, release mechanism and marker proteins [3]. In the EV-related nomenclature, one dominant naming convention of definitions has emerged recently [4–6], as outlined below.
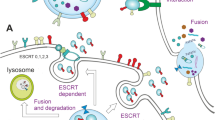
Ectosomes represent a fairly heterogeneous population of vesicles ranging in diameter from 0.1 to 1 µm (Fig. 1a). As a result of loss of calcium-dependent membrane phospholipid asymmetry and rearrangement of the cytoskeleton, ectosomes are formed by outward membrane budding and then are shed from the cell surface [1, 3, 7, 8]. Another population of smaller (30–100 nm) EVs called exosomes can be released by fusion of multivesicular bodies with the cell membrane, followed by exocytosis (Fig. 1b) [1, 3, 7, 8]. Finally, cells undergoing apoptosis and fragmentation also release vesicles formed by membrane protrusion. Unlike ectosomes, however, apoptotic bodies may contain cytosolic organelles or nuclear fragments, and they are considerably larger (up to 5 µm) (Fig. 1c) [3, 8]. Table 1 briefly characterizes these three populations of EV. Table 2 gives examples of molecular markers associated with ectosomes. Recently the existence of a new type of EVs termed sphereosomes has been postulated [9]. The presence of these structures, between 40 and 125 nm in size, was first described in gastrointestinal stromal tumor cells. Sphereosomes are believed to be formed through a newly found mechanism of shedding from multivesicular spheres.

Representative images of extracellular vesicles released from tumor cells. a Exosomes from urine of diabetic patients (transmission electron microscopy, TEM), b ectosomes from human melanoma WM1205Lu cells (TEM), c apoptotic bodies from human acute lymphoblastic leukemia MOLT-4 cells (May-Grűnwald-Giemsa staining)
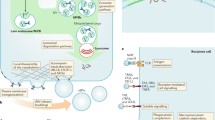
It is well established that EVs can be released under both physiological and pathological conditions. The particular function of an EV arises from the cargo it carries, which is highly dependent on the cell type from which a given EV originates [33]. Examples of ectosome cargo are shown in Fig. 2. The biological processes that may be controlled or modulated by vesicular content include coagulation, local inflammation, cell differentiation, vascular senescence and remodeling [4, 5, 7, 34, 35]. Cancer cells are known to release increased amounts of EVs, often described under a collective name: tumor-derived microvesicles (TMVs). The specific cargo that is horizontally transferred within a TMV affects a variety of cellular events during the respective stages of cancer progression. TMVs contain molecules directly stimulating invasion, metastasis and angiogenesis. Other components promote the acquisition of an aggressive phenotype or influence changes in the tumor microenvironment within the primary site and metastatic niche. TMVs may also facilitate transfer of the commonly used chemotherapeutics out of the cell, thereby contributing to multi-drug resistance. Finally, fusion of TMVs with immune cells often leads to inhibition or alteration of the immune response to cancer cells [1, 5, 36].

Examples of ectosome cargo. ARF6: ADP-ribosylation factor 6, CD40: cluster of differentiation 40, EGFR: epidermal growth factor receptor, EMMPRIN: extracellular matrix metalloproteinase inducer, IL-1β: interleukin 1β, LAMP-1: lysosomal-associated membrane protein 1, MMP: matrix metalloproteinase, uPA: urokinase plasminogen activator, VAMP-3: vesicle-associated membrane protein 3, VEGF: vascular epithelium growth factor
Our knowledge of the role of EVs in carcinogenesis is increasing vastly. This review focuses on a single population of EVs—ectosomes. We give the main points of recent studies of tumor-derived ectosomes which examine their biogenesis, proteome cargo, and the influence of that cargo on the different aspects of cancer progression. We discuss the clinical implications of the potential use of ectosome proteomes as biomarkers, diagnostic tools and treatment targets in oncology.
Biogenesis of ectosomes
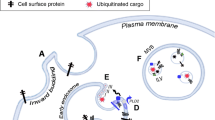
Better knowledge of EV biogenesis will be critical to an understanding of their role in intercellular communication, and eventually may allow this process to be regulated in different cell types, including cancer cells. Recent reports suggest that several types of EVs can be released from a single donor cell [29]. The formation of EVs entails the accumulation of their components in particular domains within the membranes of origin, which subsequently undergo budding (Fig. 3). While this initial assembly mechanism is similar in exosomes and ectosomes, the processes by which these two populations are released to the intercellular space differ significantly. In both cases the release of EVs is highly regulated and proceeds under the control of numerous molecular modulators [1, 7].

Mechanisms responsible for ectosome blebbing and release. Ectosomes are generated by outward budding and fusion of the plasma membrane, but their membrane composition is distinct from that of parental cells. Alterations of phospholipid symmetry are governed by aminophospholipid translocases (flippase and floppase) and Ca2+-dependent scramblase. Ectosomes are enriched in cholesterol, whereas phosphatidylserine (PS) is exposed on the extracellular leaflet of shed ectosomes. Cytoskeletal reorganization upon ectosome release is induced by calpain and gelsolin, although alternative mechanisms in cancer cells have been described, including RhoA [37] or ARF6 [14, 15] and their effectors. ARF6: ADP-ribosylation factor 6, ERK: extracellular signal-regulated kinase, LIMK: LIM domain kinase, MLCK: myosin light chain kinase, RhoA: Ras homolog gene family member A, ROCK: Rho-associated protein kinase
Since Wolf’s first description of ectosome-like particles originated from platelets [38], numerous studies have investigated the possible mechanisms of ectosome formation. Over the years it has become obvious that, unlike in exosomes, the release of ectosomes does not require exocytosis. It involves the formation of outward buds in specific regions of the cell membrane, followed by direct shedding and immediate release of the vesicle to the intercellular space [39]. These processes are initiated mainly via activation of a given cell by different agonists (e.g. ATP, growth factors, cytokines) and subsequently an increase in the level of intracellular Ca2+. Once additional Ca2+ is released from the endoplasmic reticulum, the cell membrane undergoes significant molecular rearrangement at the sites of ectosome origin, including changes in lipid and protein composition as well as in cytoskeleton structure [40].
Free calcium ions act as cytosolic secondary messengers, leading to the recruitment and activation/inhibition of several enzymes engaged in maintaining membrane asymmetry. Prior to ectosome release, phosphatidylserine (PS) and phosphatidylethanolamine (PE) are exposed on the outer leaflet of the cell membrane as a result of floppase and scramblase activation in response to strong Ca2+-mobilizing agents. At the same time, another translocase–flippase—is inhibited, thus preventing relocation of the aforementioned phospholipids [1, 3, 39]. A deficiency of Ca2+-dependent phospholipid scrambling activity, caused by mutation of the transmembrane protein anoctamin-6 gene (ANO6, also known as TMEM16F), reduces platelet activation and PS exposure on platelets and other cells, and it up-regulates a number of cytoskeleton, lysosome/peroxisome-related and phospholipid regulatory proteins [41]. PS-positive ectosomes can be distinguished easily from other circulating EVs with the use of flow cytometry by annexin V or lactadherin binding [21, 34, 42]. Increased Ca2+ levels may also facilitate degradation of the cytoskeleton structure and subsequent vesiculation by activating calcium-dependant proteases such as calpain and gelsolin [3]. Recent studies have identified RhoA, a member of the Ras protein superfamily of small GTPases, another regulator of ectosome release. By acting through its downstream effectors—a Rho-associated coiled-coil containing protein kinase (ROCK), LIM kinase (LIMK) and coffilin-RhoA altered the structure of actin–myosin filaments, leading to increased ectosome release by different cancer cell lines [37]. The release of ectosomes from invasive cells can be regulated by a GTP-binding protein, ARF6 [14, 15]. By activating extracellular signal-regulated kinases (ERK) and myosin light-chain kinase (MLCK), ARF6 regulates actin polymerization and phosphorylation of myosin light chains, leading to ectosome release [14].
Biological role of ectosomes in carcinogenesis
The biological and clinical significance of ectosome secretion has been a subject of sustained research. The majority of evidence for the role of ectosomes in cancer is based on correlative studies in clinical and preclinical settings and on experiments done in vitro. In the first report confirming the release of ectosomes by cancer cells, from 1978, they were identified in cultures of spleen nodules and lymph nodes from a patient with Hodgkin lymphoma [43]. Soon thereafter, different melanoma cell lines were shown to release ectosomes [44, 45] which displayed the ability to boost metastatic potential when fused with less invasive tumor cells [44].
The particular effects exerted by ectosomes during cancer progression depend on their interactions with recipient cells. As described in a recent review, binding of ectosomes is most likely determined by different adhesion molecules, such as integrins [2]. Upon binding, ectosomes stimulate target cells by delivering surface receptors, or by directly inducing receptor-mediated signal transduction via transported ligands, or by delivering bioactive molecules (proteins, lipids, nucleic acids) after fusion with the cell membrane or after endocytosis [33, 46]. Regardless of the mechanism, an ectosomal cargo can modulate essential processes in associated cancer cells, as well as functions of fibroblasts [47], endothelial cells [48] and immune cells [49]. The described disruption of homeostasis within the tumor microenvironment, changes in the structure of the extracellular matrix (ECM) and transfer of oncogenic phenotypes all testify to the participation of ectosome release in different stages of cancer progression.
Transfer of oncogenic phenotypes
Cancer development was long considered a consequence of multistep mutations of genetic material. Nowadays there is a shift from those strictly genocentric explanations for the transformation from normal to malignant cells, towards epigenetic and other non-genetic interpretations. Non-genetic mechanisms of phenotypic transformation may involve the transfer, via ectosomes, of membrane receptors for growth factors, RNA molecules or even lipids [50]. For instance, the oncogenic mutant of an epidermal growth factor receptor (EGFRvIII) was shown to be horizontally transferred within ectosomes released by glioma cells and taken up by a non-invasive population of tumor cells in vitro [17, 18]. Cells that acquired EGFRvIII exhibited activation of the MAPK and Akt signaling pathways, along with altered expression of genes regulating cell survival (BclxL) and proliferation (p27), resulting in a series of morphological changes and stimulation of anchorage-independent growth [17]. Ectosomes containing mRNA for EGFRvIII isolated from human glioblastoma sections consistently stimulated in vitro proliferation of malignant glioma cells (U87) [48].
Other findings highlight the role of ectosomes in the transformation of normal cells within the tumor microenvironment. Antonyak et al. [47] demonstrated that ectosomes released by breast cancer and glioblastoma cell lines (MDAMB231 and U87) contain tissue transglutaminase (tTG) and its tTG-binding partner and cross-linking substrate, fibronectin (FN). Once transferred to the recipient fibroblasts via ectosomes, tTG and FN acted cooperatively to induce their transformation by activating mitogenic signaling (phosphorylation of FAK and ERK kinases), leading to increased cell survival and aberrant growth [47].
Tumor cell invasion and metastasis
The metastatic cascade begins with local invasion of primary tumor cells within the surrounding tissue. Once the cells pass the barrier of the basement membrane, they are able to penetrate the lumen of the blood vessels (intravasation) and then travel with the blood stream to predetermined metastatic sites. Subsequent migration of tumor cells from the blood to the target tissue (extravasation) starts the process of secondary tumor growth. The results of numerous studies support the suggestion that various molecules of the ectosomal cargo play a significant role at each stage of the metastatic cascade [36].
The invasive and migratory properties of tumor cells are highly dependent on the activity of different proteases such as matrix metalloproteinases (MMPs). By degrading particular components of the ECM, this group of enzymes promotes the mobility of migrating cells and creates a path of less resistance. MMPs are also responsible for proteolysis within the basement membrane, which allows tumor cells to penetrate the lumen of blood and lymphatic vessels and metastasize [36]. The presence as well as the proteolytic activity of MMP-2 and MMP-9 have been identified in ectosomes released in vitro by fibrosarcoma (HT1080) [51], ovarian (CABA1, A2780) [23, 28], breast (8701-BC) [12], prostate (PC3, LnCaP) [24, 25] and lung (HTB 177, CCL 185) [26] cancer cell lines.
It is well established that the activity of MMPs is regulated at several levels, including transcription, enzyme activation/inhibition, complex formation, and compartmentalization [52]. Supporting this, studies using fibrosarcoma and prostate cancer cell lines showed that urokinase plasminogen activator (uPA) is associated with ectosomes released in vitro. Addition of plasminogen to the ectosomal fraction resulted in activation of zymogens, indicating a role of the urokinase-plasmin system in MMP-2 and MMP-9 activation [24, 25, 51]. The activity of vesicle-associated proteases may also be influenced by the pH of the tumor microenvironment. In solid tumors the extracellular pH is acidic due to elevated anaerobic glycolysis and impaired clearance of metabolic waste products. It has been found that low pH may promote the invasiveness of ectosomes by cathepsin B-mediated activation of gelatinases. Exposing vesicles released by ovarian cancer cells (CABA1) to acidic medium increased MMP-2 and MMP-9 activities; this effect was abolished by the specific inhibitor of cystein protease or by silenced expression of cathepsin B in CABA1 cells [53]. CD147/extracellular MMP inducer (EMMPRIN), a membrane glycoprotein, may also be involved in the progression of malignancies via regulation of the expression of MMPs in tumor cells. CD147/EMMPRIN-bearing ectosomes derived from breast cancer cell lines (MCF-7, SK-BR-3, MDA-MB231) induced invasion of both autologous and heterologous cells. This effect was not mediated by matrix metalloproteinases, which were absent in the released ectosomes, but rather by activation of the p38/MAPK signaling pathway in tumor cells [27].
Proteolysis within the ECM can also result from the activity of adamalysin metalloproteinases with disintegrin and thrombospondin domains (ADAMTSs). Ectosomes shed by oligodendroglioma cells exhibited aggrecanase activity in vitro, cleaving aggrecan at sites previously identified as targets for different ADAMTSs: ADAMTS1, ADAMTS4 and ADAMTS5. Immunodetection of the cleaved fragments showed one or more of these enzymes to be responsible for the ectosome activity [54].
Ectosomes may promote cancer invasion and metastasis indirectly by altering normal cell function. Through interactions with cancer ectosomes, stromal cells such as fibroblasts contribute to the creation of a favorable niche for cancer development. In studies by Castellana et al. [24], fibroblasts were activated in vitro by ectosomes derived from a highly metastatic prostate cancer cell line (P3C). After incubation, the fibroblasts exhibited ERK phosphorylation and up-regulation of MMP-9. Moreover, the activated fibroblasts themselves released vesicles which in turn were able to boost the invasiveness and migration of P3C cells. A similar observation was reported in yet another study [55] confirming the role of tumor-derived ectosomes in activating stromal cells within the tumor microenvironment in vivo and in vitro. Ectosomes released by cancer stem cell populations (isolated from renal carcinoma cells) activated mesenchymal stromal cells (MCSs) and up-regulated the expression of MMP-1 and MMP-3 in MSCs. Mice inoculated with renal carcinoma cells (K1) previously cultured with activated MSCs showed increased tumor growth versus the control group. Finally, ectosomes released in vitro by MDA-MB-231 carcinoma cells enhanced collagenase activity in fibroblasts (MCF10a cell line). In that case, reorganization of 3D collagen matrices by ectosome-stimulated fibroblasts was associated with increased FAK phosphorylation [56].
Matrix degradation and subsequent tumor invasion have also been correlated with elevated expression of urokinase and other components of the plasminogen activation system. Schroder et al. [57] recently demonstrated that SerpinB2 (plasminogen activator inhibitor type 2/PAI2) is expressed on the surface of ectosomes released by B16 melanoma cells. SerpinB2-expressing ectosomes significantly reduced urokinase (uPA) activity in vitro as compared with vesicles obtained from control cells that did not produce SerpinB2. This mechanism was later used to explain the decreased migratory properties of SerpinB2-expressing B16 cells in Transwell assays, suggesting that ectosome-associated SerpinB2 may also inhibit uPA-mediated cancer invasion, migration and metastasis in vivo [57].
Angiogenesis
In the course of carcinogenesis, enlargement of the tumor requires expansion of the vascular network. Neovascularization allows effective delivery of substances essential for survival of tumors and facilitates their subsequent migration to metastatic sites. Initiation of angiogenesis in tumor lesions is associated primarily with activation of various signaling pathways in tumor cells, leading to proliferation and migration of vascular endothelial cells or their precursors.
Ectosomes released by different cancer cells have been shown to facilitate the transfer of several proangiogenic factors or to up-regulate their expression in endothelial cells. Studies by Taraboletti et al. [28] demonstrated that ectosomes isolated from two ovarian cancer cell lines (CABA1, A2780) contained significant amounts of matrix-degrading metalloproteinases (MMP-2, MMP-9) and vascular epithelium growth factor (VEGF), and that these factors stimulated the motility and invasion of endothelial cells into Matrigel. In other work, human umbilical cord vein cells (HUVECs) showed up-regulated expression of autocrine VEGF upon uptake of EGFRvIII from ectosomes released by two human epithelial carcinoma cell lines (A431, A549) [18]. In another study, induction of the angiogenic phenotype in HUVECs was attributed to the presence of CD147/EMMPRIN in ectosomes derived from three ovarian cancer cell lines (OVCAR3, SKOV3, A2780). CD147/EMMPRIN-positive vesicles stimulated in vitro proliferation, invasiveness and expression of MMP-2 and MMP-9 in HUVECs [27]. Another proangiogenic factor is IL-6, whose production (along with VEGF) by the EA.hy926 endothelial cell line was increased upon incubation with ectosomes derived from human multiple myeloma cells. Ectosomes from myeloma cells induced proliferation and invasion of EA.hy926 cells, as determined in Transwell cell invasion assays [58].
The crucial role of ectosomes in tumor angiogenesis has also been confirmed in animal studies. Munster et al. [59] obtained two distinct populations of ectosomes from EMT/6 breast cancer cells exposed or not exposed to anti-VEGF antibody (B20). As expected, mice inoculated with ectosomes released by B20-untreated cells showed higher mobilization of endothelial precursor cells and their colonization in growing tumors, as well as increased microvessel density [59]. These data suggest that cancer-cell-derived ectosomes stimulate the paracrine mechanism of endothelial cell proliferation in both a VEGF-dependent and a VEGF-independent manner. However, the most important role in cancer neovascularization may well be recognized in the endothelial-to-mesenchymal transition (EMT), a newly identified type of cellular transdifferentiation responsible for vascular system development and repair [60, 61].
At present, much less is known about the role of membrane lipids transported within ectosomes in tumor angiogenesis. Kim et al. [22] identified sphingomyelin in ectosomes shed from HT1080 fibrosarcoma cells as the active component for ectosome-induced endothelial cell migration, in vitro tube formation (Matrigel tube formation assay), and ex ovo neovascularization of the chick chorioallantoic membrane; comparable effects on endothelial cell migration and angiogenesis were exerted by lipid extracts from ectosomes and purified sphingomyelin, but were not observed in the case of lipid extracts previously treated with sphingomyelinase [22]. Besides growth factors, metalloproteinases, cytokines and lipids, ectosomes may supply endothelial cells with proangiogenic miRNAs. Transfer of pro- and anti-angiogenic miRNA from cancer to endothelial cells via ectosomes may promote the formation of blood vessels by altering the translation of particular proangiogenic factors, or it may cause down-regulation of VEGF expression in a microRNA-specific manner [47, 62]. Among the different microRNA and protein cargos identified in human collateral cancer ectosomes, miR-1246 and TGF-β have been demonstrated to exert their pro-angiogenic effects by activating Smad 1/5/8 signaling in HUVECs [63].
Another important example of pro-angiogenic cancer ectosomes and cell interactions is the contribution of platelet-derived microvesicles (PMVs) in carcinogenesis and neovascularization [6]. Below we cover what is currently known about this.
Cancer-induced thrombosis
Since cancer progression is often associated with increased platelet activation and aggregation, PMVs are thought to be mediators in platelet-tumor interactions. Tumor cells activate the production of thrombin, a common agonist of platelets, which induces ectosome shedding [64]. For example, the supernatant obtained from a human neuroblastoma cell line (NCG) induced platelet aggregation via thrombin-induced procoagulant activity [65]. CD41 (GPIIb/IIIa, αIIbβ3) and P-selectin are specific antigens for activated platelets. Their presence on the surface of ectosomes promotes adhesion of cancer cells to the vascular endothelium and facilitates their extravasation [11]. Adhesion of platelets and circulating cancer cells is regulated mostly by the ligand-receptor mechanism of PSGL-1/P-selectin interaction, and the presence of P-selectin on the surface of PMPs may facilitate binding of P-selectin-positive ectosomes to PSGL-1-expressing cancer cells and thereby increase cancer invasiveness [11, 66]. P-selectin- and PSGL-1-dependent accumulation of circulating PMVs in vascular injury foci has been described as an important mechanism of ectosome delivery to thrombi and of tissue-factor-dependent fibrin generation [67].
Among the numerous specific procoagulant molecules, tissue factor (TF) is the major initiator of thrombin activation in blood coagulation pathways. A widely discussed question is whether PMVs contain platelet-originated TF, or if this activator is incorporated into PMVs due to binding of TF-positive EVs derived from extravascular cells and macrophages to PMVs or platelets, or if TF is de novo expressed in activated platelets [68]. It is now commonly accepted that two forms of TF are present in the circulatory system: full length (flTF) and alternatively spliced (asTF) [69]. The extracellular domain of flTF was found to initiate coagulation by binding coagulation factor VII or its activated form (VIIa) to make a membrane-bound complex which activates coagulation factor X. Oncogenic transformation caused by the RAS mutation and loss of p53 resulted in TF up-regulation [70]. Later it was shown that ectosome-mediated transfer of TF between two breast cancer cell lines changed cell TF expression related to their aggressiveness potential [71]. Therefore it is highly likely that PMVs contribute to the transfer of TF-positive ectosomes from macrophages and different populations of cancer cells, and that they can facilitate the propagation of TF-related aggressive phenotypes [11, 16, 71, 72]. TF-bearing microvesicles arise from lipid rafts and then fuse with activated platelets through a PSGL-1-dependent mechanism; their shedding was significantly reduced under conditions of depleted membrane cholesterol [20, 67]. The molecular mechanism for activation of TF and other coagulation factors also involves PS exposure. Presentation of negatively charged PS on the surface of ectosomes is closely related to exposure of binding sites for coagulation factors Va, VIII and X, which leads to their activation and phosphatidylserine-dependent initiation of coagulation pathways [11].
In contrast to the procoagulatory activity of ectosomes, anticoagulation and antimetastatic effects of tumor-derived large microvesicles have been reported. Expression of plasminogen activator inhibitor type 2/PAI-2 (SERPINB2) effectively inhibits urokinase activity, and was associated with favorable prognoses [57]. Work by Mezouar et al. [73] showed that inhibition of platelet activation with clopidogrel (an anti-platelet agent) in an in vivo mouse model prevents P-selectin- and (αvβ1, αvβ3) integrin-mediated accumulation of ectosomes at the site of thrombosis. Upon treatment with clopidogrel, animals bearing pancreatic tumors showed a decrease of tumor growth and metastasis. Taken together, these observations indicate that the use of anti-platelet drugs may improve the efficacy of anticancer therapy and slow the rate of disease progression.
Influence on the immune system
The immune system is well adapted to impede cancer progression, although its role remains inessential until the accumulated genetic changes are fixed. At that point, spontaneous cancer immunity may arise to contain tumor growth in early stages of its progression; however, many types of cancer cells have developed a number of mechanisms allowing them to evade immune surveillance. Ectosomes and other MVs are widely associated with suppression of the immune response to transformed cells, and several theories explaining their role have been proposed [36, 74–76].
Numerous studies indicate that tumor-derived ectosomes may induce chemotaxis of blood leukocytes. In vitro, ectosomes released by pancreatic adenocarcinoma (HPC-4), colorectal adenocarcinoma (DeTa) and lung carcinoma (A549) cell lines stimulated the chemotactic activity of granulocytes, lymphocytes and monocytes. Those results were obtained using Transwell chambers and were attributed to several chemokines (particularly IL-8) transferred inside ectosomes [74]. Upon interaction with immune cells, ectosomes can interfere with the T-cell response by altering the differentiation of antigen-presenting cells, or else can inhibit functions of effector cells [36]. For example, Köppler et al. [75] described the immunosuppressive properties of ectosomes released in vitro by the Kato gastric carcinoma cell line. Incubation with isolated vesicles interfered with the activation of monocytes by lipopolysaccharide (LPS), resulting in decreased release of tumor necrosis factor α (TNF-α) and granulocyte macrophage colony-stimulating factor (GM–CSF). Another mechanism was shown for ectosomes from melanoma and colon carcinoma cell lines [76] that inhibited the differentiation of monocytes to antigen-presenting dendritic cells. Moreover, the remaining population of monocytes released an immunosuppressive cytokine—transforming growth factor β (TGF-β)—which inhibited T-cell cytolytic activity.
The immunosuppressive effects exerted by ectosomes do not appear to be constant and universal. Vesicles isolated from pancreatic adenocarcinoma (HPC-4), colorectal adenocarcinoma (DeTa) and lung carcinoma (A549) cell lines induced the production of proinflammatory cytokines (TNF-α and IL-12) and reactive oxygen intermediates (ROIs) by monocytes [77]. As a result, stimulated monocytes showed significantly increased cytotoxic/cytostatic effects on cancer cells in vitro, as compared with control monocytes. Despite the enhancement of the antitumor response, increased production of the anti-inflammatory cytokine IL-10 was also observed [77]. An explanation for such a discrepancy has recently been suggested by another study. Ectosomes released by colon cancer cell lines (Caco-2, SW480, SW620, LoVo) were shown to influence the differentiation of monocytes to macrophages, resulting in their variable polarization status (M1/M2, proinflammatory/anti-inflammatory) [78]. The impact of ectosomes on macrophage differentiation and cytokine production depended on the timing of the monocytes’ contact with isolated vesicles. Immediate exposure resulted in the highest release of IL-10, whereas monocytes incubated with ectosomes on the sixth day of culture exhibited the strongest secretion of IL-12 and TNF-α (corresponding to the increased fraction of proinflammatory M1 monocyte-derived macrophages). Macrophages that differentiated after prolonged exposure to vesicles (days 0, 3 and 6) secreted the lowest amounts of IL-12 and TNF-α, probably due to deactivation of monocytes/macrophages by ectosomal hyaluronan fragments [78, 79]. Observations made in the last-mentioned time regime most likely reflect the immunosuppressive effect of prolonged ectosome exposure during cancer progression in vivo.
Interactions of tumor-derived ectosomes with immune cells may also indirectly regulate different aspects of cancer progression, including angiogenesis. In studies by Baj-Krzyworzeka et al. [74], ectosomes released by pancreatic adenocarcinoma (HPC-4), colorectal adenocarcinoma (DeTa) and lung carcinoma (A549) cell lines increased in vitro secretion of proangiogenic cytokine IL-8 by monocytes. Later the proangiogenic potential of ectosome-treated monocytes was tested in vivo in a mouse model. Matrigel matrixes containing stimulated or not stimulated monocytes were implanted into NOD-SCID mice and excised 7 days later. Hemoglobin content in excised Matrigel matrixes showed stronger proangiogenic activity of monocytes previously incubated with ectosomes released by cancer cells [74].
Finally, it is important to note that the majority of comprehensive studies have focused on the exosomal fraction of microvesicles, so the data on the effects of ectosomes on the immune system are still very limited. It is very likely that effects attributed to exosomes, such as induction of effector cell apoptosis, stimulation of suppressor cell differentiation, or loss of antigens essential for recognition by NK cells and cytotoxic T-cells [80, 81], can also be exerted by ectosomes. Additional studies are needed to shed light on this question.
The role of ectosomes of non-cancer origin
As described in previous sections, ectosomes released by cancer cells exert multiple regulatory effects during different stages of cancer progression. An increasing number of studies also implicate vesicles derived from non-transformed cells (such as fibroblasts and immune cells) or platelets in disease development. For instance, ectosomes released by immune cells (isolated from mouse spleen) induced migration of hepatocarcinoma (H22) and melanoma (B16) cell lines in vitro and hepatic cancer metastasis in mouse in vivo. These observations were related to the transfer of integrin αMβ2 (CD11b/CD18) from immune cells to ectosomes and then to tumor cells. The use of antibodies against either CD11b or CD18 led to significant decreases in ectosome-mediated tumor cell migration and metastasis [82]. In other work, ectosomes released in vitro by cancer-associated fibroblasts increased the proliferation of pancreatic cancer cells (DU145 cell line). Moreover, the utilization of lactate in anabolic processes was higher when DU145 cells received fibroblast proteins via ectosomes in Transwell or coculture conditions, suggesting that the acquisition of enzymes of the second step of glycolysis (subsequently identified in isolated ectosomes) may contribute to the metabolic shift of DU145 cells towards a reverse Warburg phenotype, more efficient in highly proliferative conditions [83].
Other reports support the suggestion that ectosomes released by platelets and megakaryocytes also contribute to cancer progression. They are constitutively produced in physiological conditions, though elevated levels of them have been observed in patients with various types of cancer, frequently correlated with disease stage, the presence of metastases, or survival [11]. In vivo and in vitro studies confirmed the involvement of platelet-derived vesicles (PMVs) in tumor growth, invasion and angiogenesis through interactions with cancer or endothelial cells. Janowska-Wieczorek et al. [10] showed that incubation with platelet-derived ectosomes resulted in higher expression of mRNA for several proangiogenic factors (MMP-9, VEGF, IL-8, HGF) by different lung cancer cell lines, and their increased adhesion to endothelial cells in vitro. Up-regulation of MMP-2 expression and activation of selected proliferative signaling pathways was also observed in cells cultured in the presence of vesicles. After injection into mice, cells previously incubated with ectosomes induced more lung cancer metastatic foci than control cancer cells [10]. A complete characterization of circulating EVs in colorectal cancer patients revealed that microvesicles obtained by 15,000×g centrifugation (ectosomes) are mostly CD41- and CD61-positive (platelet origin) and may act as conveyors of cancer-derived smaller vesicles [13].
Potential clinical applications
The presence of ectosomes in various body fluids points to their potential use as biomarkers or prognostic indicators of cancer development and progression. Tumor-specific markers exposed on the surface of ectosomes might serve as confirmatory tools during the diagnostic process, whereas specific changes in the number of released vesicles or in their molecular composition appear to be highly indicative of disease stage and treatment efficacy.
Ectosomes are also being studied for possible improvement of clinical outcomes, mainly with regard to inhibition of the vesiculation process or for drug delivery; both strategies may benefit cancer management [8]. The clinical applications of ectosomes are still in development; their full potential is yet to be realized.
Diagnostic value
Ectosomes generally contribute to the pathogenesis of cancer, but some of their unique characteristics can be exploited as diagnostic, prognostic and surveillance indicators for cancer patients. Elevated levels of ectosomes as compared with those of healthy controls have been detected in peripheral blood samples from patients with glioblastoma [84], non-small-cell lung carcinoma [85] and multiple myeloma [86]. On the other hand, ectosome levels were found to be significantly lower in colorectal carcinoma patients; thus, increased vesiculation may not be a universal characteristic of all types of cancer. The variation of ectosome release levels has been used to distinguish benign tumors from malignant breast [87] and prostate [88] cancers. Such differences were not observed between benign colorectal disease and colorectal carcinoma [89]. Increased plasma levels of ectosome-bearing tissue factor may indicate a highly invasive and poorly differentiated type of pancreatic cancer more able to infiltrate peripancreatic vessels [90].
A thorough determination of ectosomal molecular status may allow detection of specific cancer biomarkers. Ectosomes obtained from patients with malignant breast cancer exhibited elevated expression of several surface antigens (CD66; human epidermal growth factor receptor 2, Her2/neu; breast cancer resistance protein, BRCP; Hsp27) as compared with benign tumors, suggesting their potential use as relevant diagnostic markers for malignancy [87]. In other work, ectosomes released during progression of colorectal and pancreatic cancers were found to express surface glycoproteins such as mucine1 (MUC1), carcinoembryonic antigen (CEA) and carbohydrate antigen 19-9 (CA19-9). The number of MUC1- and CA19-9-positive vesicles differed significantly between the two cancer types, with MUC1 expression higher in colorectal cancer and CA19-9 expression higher in pancreatic cancer. Since MUC1 and CA19-9 are already being used in histopathology as differential markers for digestive system cancers, isolation of ectosomes from peripheral blood may obviate the need for invasive biopsy procedures in the future [88]. Ectosomes found in the blood could also furnish a novel prognostic tool to monitor malignant cells in multiple myeloma, where elevated numbers of CD138-positive vesicles have been correlated with the tumor burden [86].
The diagnostic and prognostic uses of elevated numbers of ectosomes, or of the presence of vesicles bearing certain molecules, depend on the establishment of proper isolation protocols. To gain valid information for clinical practice, optimal concentrations of uncontaminated vesicle populations are required. The several methods of obtaining such samples are based on vesicle size or density, or on marker expression. In general, ectosomes can be selected by differential centrifugation, immunoaffinity isolation (adsorption to magnetic/non-magnetic microbeads) or size exclusion chromatography [91, 92]. Unfortunately, so far no single isolation protocol can guarantee complete recovery of ectosomes from samples, nor ensure maintenance of their native form and function.
Management of multidrug resistance
A number of reports suggest that ectosomes are among the critical factors in multidrug resistance (MDR), which remains an obstacle in cancer chemotherapy. MDR is associated with several mechanisms responsible for compromising the effectiveness of different chemotherapeutics. The mechanisms include changes in the rate of drug uptake and efflux, altered drug metabolism, decreased drug-target complex formation, and enhanced DNA repair [93]. MDR cancer cells have already been shown to overexpress different transporter proteins involved in the efflux of anticancer drugs including P-glycoprotein (Pgp), multidrug resistance-associated protein 1 (MRP1) and breast cancer resistance protein (BCRP) [94]. Recent in vitro studies demonstrated transference of functional Pgp [95, 96] or MRP1 [97] and the mRNAs for both proteins [97] via ectosomes released by MDR chronic/acute myeloid leukemia cells to drug-sensitive cells which subsequently acquired MDR phenotypes. Some of these transporter proteins are transferred alongside CD44, ERM (ezrin, radixin, moesin) protein family and cytoskeleton proteins within the ectosomal cargo [31]. Ezrin is known to determine Pgp membrane localization through cytoskeletal association, as shown in leukemic and breast cancer cells [31, 32]. de Souza et al. [96] found that upon incubation with ectosomes bearing different inhibitors of apoptosis proteins (IAPs), drug-sensitive human breast adenocarcinoma (MCF7) and human lung carcinoma (A595) cells were more resistant to apoptosis when treated with cisplatin or paclitaxel.
Apart from delivering transporter proteins, ectosomes may also directly facilitate the expulsion of chemotherapeutics from tumor cells and promote their survival. Doxorubicin-treated MCF7 human breast adenocarcinoma cells accumulated and released the drug in shed microvesicles [98]. This observation implied that modulatory interventions in the vesiculation process may offer a solution for various MDR cases, so studies were carried out later to verify that suggestion. Jorfi et al. [93]. described in vitro sensitization of pancreatic cancer cells (PC3 cell line) to docetaxel upon treatment with vesiculation-inhibiting calpeptin (calpain inhibitor). As a result, 20-fold lower concentrations of docetaxel used in the presence of calpeptin induced the same degree of apoptosis in PC3 cells as docetaxel alone. Inhibition of ectosome budding similarly improved the effectiveness of a combination chemotherapy (docetaxel and methotrexate) and also reduced the docetaxel dose required to limit tumor growth in mouse in vivo. Another study pointed to the therapeutic potential of peptidylarginine deiminases (PADs), a family of enzymes responsible for post-translational conversion of protein-bound arginine to citrulline [99]. PADs have been associated with deimination of cellular actin, which in turn rearranges the actin cytoskeleton and may facilitate vesiculation. Treatment of PC3 cells with chloramidine (PAD inhibitor) significantly reduced ectosome release and increased the sensitivity of PC3 cells to the cytotoxic effect of methotrexate.
In contrast to the presented findings, clinical outcomes may be improved by stimulation of the vesiculation process in certain types of cancer. Different ectosome-releasing agents are considered as potential alternative drugs in, for example, differentiation therapy against acute myeloid leukemia. Anso-Adda et al. [100] found that promonocytic leukemia cells (THA-1) released increased amounts of ectosomes upon stimulation with phorbol myristate acetate, all-trans retinoic acid and histamine. Isolated vesicles containing TGF-β1 inhibited the proliferation of THA-1 cells, and they induced differentiation of those cells to monocytes/macrophages.
Therapeutic use
Ectosomes can carry a multitude of bioactive molecules. Potentially they offer a unique carrier system to deliver different therapeutic agents to cancer cells. Obvious advantages include easy preparation and manipulation, no restriction regarding the physicochemical properties of drugs, and the lack of autoimmune reaction due to the autochthonous origin of isolated vesicles [101]. Since ectosomes carry a variety of cancer-related surface receptors and adhesion molecules, they might be easily transported to specific tumor sites, enabling local drug delivery.
Recent preclinical and clinical trials have already exploited synthetic liposomes to successfully deliver chemotherapeutics such as doxorubicin or daunorubicin to in vitro cultures of melanoma cells [102] or in leukemic patients [103, 104]. The observed anticancer effects (decreased tumor growth [101] or prolonged survival after leukemia relapse [103, 104]) suggest that refinement and modification of natural vesiculation processes may allow ectosomes to be used as novel therapeutic vehicles. Tang et al. [101] demonstrated that malignant hepatocarcinoma cells (H22 cell line) incubated with doxorubicin, cisplatin or methotrexate subsequently released drug-containing ectosomes. Isolated vesicles had a cytotoxic effect on tumor cells in vitro and reduced hepatocarcinoma and ovarian cancer growth in mouse in vivo. Moreover, the ectosome-encapsulated chemotherapeutic agents showed higher efficacy and less adverse effects than drugs administered directly.
Ectosomes can also be used as vaccines in cancer management. In studies by Zhang et al. [105], 50% of mice immunized with ectosomes isolated from hepatocarcinoma (H22), melanoma (B16) and colon carcinoma (CT26) cell lines remained tumor-free after injection of cancer cells. Vesicles from the vaccines were taken up by dendritic cells and induced the expression of type-I IFN by activating the pathway of the cyclic GMP-AMP synthase/stimulator of interferon genes (cGAS/STING). Type-I IFN enhanced the maturation of dendritic cells that activated tumor-specific T-cells, leading to cytolysis of cancer cells [105, 106]. Only 12.5% of mice immunized with exosomes released by the same cell lines did not develop tumors, suggesting that ectosomes are more immunogenic and are a better option for developing cancer vaccines [105].
Ran et al. [107] demonstrated that ectosomes can also deliver an oncolytic adenovirus into the nucleus of tumorogenic cells and thus are potential agents for virotherapy. The applied vesicles were fatal to tumor cells cultured in vitro and also reduced tumor growth in vivo in adenocarcinoma mice. Moreover, the cytolytic activity of virus-containing ectosomes was more efficient than that of a free virus, because they appeared to be resistant to virus antibodies. Finally, ectosomes have also been shown to transfer miRNAs that regulate target gene expression and functions of recipient cells [108, 109]. The use of vesicles containing particular miRNAs in gene therapy clearly holds great promise, but so far no studies involving different models of cancer disease have been carried out.
Concluding remarks
In this review we summarized the recent literature on the properties and biogenesis of tumor-derived ectosomes, as well as their potential roles in cancer growth. Revealing their biological roles is only the beginning. This branch of research has entered a phase of rapid progress. Tumor-derived ectosomes are now recognized to play significant roles in tumor development, facilitating the spread and release of cancer cells to generate metastases. Work on ectosomes has shed new light on the pathogenesis of malignant disease. They are a reservoir of biological information and a vital component of the specific microenvironment of the cell to the tumor microenvironment and to recipient cells, tumor-derived ectosomes provide a unique means of cellular export and cell-to-cell transport of insoluble bioactive molecules such as nucleic acids, membrane receptors and signaling molecules which affect recipient cell metabolism, mRNA processing, cell growth and motility, as well as angiogenesis and immune system functioning (Fig. 4). Elevated levels of tumor-derived ectosomes are associated with a variety of cancers, including brain [84], lung [85], breast [87], prostate [88], pancreatic [90] and gastric [108] cancers, as well as acute promyelocytic leukemia [110]. As it is well established that the number of tumor-derived ectosomes increases with cell invasiveness or disease progression [14], potentially these vesicles can serve as prognostic biomarkers of disease stages and of treatment efficacy, and can be effective targets for anticancer therapies.

Role of tumor ectosomes in key pathways promoting cancer progression through interaction with local and distant cells
Abbreviations
- ADAMT:
-
Adamalysin metalloproteinase with disintegrin and thrombospondin domains
- ARF6:
-
ADP-ribosylation factor 6
- BCRP:
-
Breast cancer resistance protein
- CSF:
-
Cerebrospinal fluid
- ECM:
-
Extracellular matrix
- EGFR:
-
Epidermal growth factor receptor
- EMMPRIN:
-
Extracellular matrix metalloproteinase inducer
- EMT:
-
Endothelial to mesenchymal transition
- ERK:
-
Extracellular signal-regulated kinase
- EV:
-
Extracellular vesicle
- FAK:
-
Focal adhesion kinase
- FN:
-
Fibronectin
- HGF:
-
Hepatocyte growth factor
- LIMK:
-
LIM kinase
- MDR:
-
Multidrug resistance
- MLCK:
-
Myosin light-chain kinase
- MMP:
-
Matrix metalloproteinase
- MRP1:
-
Multidrug resistance-associated protein 1
- PAD:
-
Peptidylarginine deiminase
- PE:
-
Phosphatidylethanolamine
- PMV:
-
Platelet-derived microvesicle
- PS:
-
Phosphatidylserine
- PSGL:
-
P-selectin glycoprotein ligand
- ROCK:
-
Rho-associated coiled coil containing protein kinase
- ROS:
-
Reactive oxygen species
- TF:
-
Tissue factor
- TGF-β:
-
Transforming growth factor β
- TNF-α:
-
Tumor necrosis factor α
- TMV:
-
Tumor-derived microvesicle
- tTG:
-
Tissue transglutaminase
- uPA:
-
Urokinase plasminogen activator
- VEGF:
-
Vascular epithelium growth factor
References
Minciacchi VR, Freeman MR, Di Vizio D (2015) Extracellular vesicles in cancer: exosomes, microvesicles and the emerging role of large oncosomes. Semin Cell Dev Biol 40:41–51. doi:10.1016/j.semcdb.2015.02.010
Raposo G, Stoorvogel W (2013) Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 200:373–383. doi:10.1083/jcb.201211138
Mause SF, Weber C (2010) Microparticles: protagonists of a novel communication network for intercellular information exchange. Circ Res 107:1047–1057.doi: 10.1161/CIRCRESAHA.110.226456
Hargett LA, Bauer NN (2013) On the origin of microparticles: from “platelet dust” to mediators of intercellular communication. Pulm Circ 3:329–340. doi:10.4103/2045-8932.114760
Van der Pol E, Böing AN, Harrison P, Sturk A, Nieuwland R (2012) Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev 64:676–705. doi:10.1124/pr.112.005983
Rak J (2010) Microparticles in cancer. Semin Thromb Hemost 36:888–906. doi:10.1055/s-0030-1267043
Cocucci E, Meldolesi J (2015) Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol 25:364–372. doi:10.1016/j.tcb.2015.01.004
Gong J, Jaiswal R, Dalla P, Luk F, Bebawy M (2015) Microparticles in cancer: a review of recent developments and the potential for clinical application. Semin Cell Dev Biol 40:35–40. doi:10.1016/j.semcdb.2015.03.009
Junquera C, Castiella T, Muńoz G, Fernandez-Pacheco R, Luesma MJ, Monzón M (2016) Biogenesis of a new type of extracellular vesicles in gastrointestinal stromal tumors: ultrastructural profiles of spheresomes. Histochem Cell Biol 146:557–567. doi:10.1007/s00418-016-1460-5
Janowska-Wieczorek A, Wysoczynski M, Kijowski J, Marquez-Curtis L, Machalinski B, Ratajczak J, Ratajczak MZ (2005) Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int J Cancer 113:752–760. doi:10.1002/ijc.20657
Mezouar S, Mege D, Darbousset R, Farge D, Debourdeau P, Dignat-George F, Panicot-Dubois L, Dubois C (2014) Involvement of platelet-derived microparticles in tumor progression and thrombosis. Semin Oncol 41:346–358. doi:10.1053/j.seminoncol.2014.04.010
Dolo V, Ginestra A, Cassarŕ D, Violini S, Lucania G, Torrisi MR, Nagase H, Canevari S, Pavan A, Vittorelli ML (1998) Selective localization of matrix metalloproteinase 9, beta1 integrins, and human lymphocyte antigen class I molecules on membrane vesicles shed by 8701-BC breast carcinoma cells. Cancer Res 58:4468–4474
Stec M, Baj-Krzyworzeka M, Baran J, Węglarczyk K, Zembala M, Barbasz J, Szczepanik A, Zembala M (2015) Isolation and characterization of circulating micro(nano)vesicles in the plasma of colorectal cancer patients and their interactions with tumor cells. Oncol Rep 34:2768–2775. doi:10.3892/or.2015.4228
Muralidharan-Chari V, Clancy J, Plou C, Romao M, Chavrier P, Raposo G, D’Souza-Schorey C (2009) ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr Biol 19:1875–1885. doi:10.1016/j.cub.2009.09.059
D’Souza-Schorey C, Chavrier P (2006) ARF proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell Biol 7:347–358. doi:10.1038/nrm1910
Tesselaar ME, Romijn FP, Van der Linden IK, Prins FA, Bertina RM, Osanto S (2007) Microparticle-associated tissue factor activity: a link between cancer and thrombosis. J Thromb Haemost 5:520–527. doi:10.1111/j.1538-7836.2007.02369.x
Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, Rak J (2008) Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol 10:619–624. doi:10.1038/ncb1725
Al-Nedawi K, Meehan B, Kerbel RS, Allison AC, Rak J (2009) Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc Natl Acad Sci USA 106:3794–3799. doi:10.1073/pnas.0804543106
Berchem G, Noman MZ, Bosseler M, Paggetti J, Baconnais S, Le Cam E, Nanbakhsh A, Moussay E, Mami-Chouaib F, Janji B, Chouaib S (2015) Hypoxic tumor-derived microvesicles negatively regulate NK cell function by a mechanism involving TGF-β and miR23a transfer. Oncoimmunology. doi:10.1080/2162402X.2015.1062968
Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA (2005) Tissue factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 106:1604–1611. doi:10.1182/blood-2004-03-1095
Shi J, Heegaard CW, Rasmussen JT, Gilbert GE (2004) Lactadherin binds selectively to membranes containing phosphatidyl-L-serine and increased curvature. Biochim Biophys Acta 1667:82–90. doi:10.1016/j.bbamem.2004.09.006
Kim CW, Lee HM, Lee TH, Kang C, Kleinman HK, Gho YS (2002) Extracellular membrane vesicles from tumor cells promote angiogenesis via sphingomyelin. Cancer Res 62:6312–6317
Dolo V, D’Ascenzo S, Violini S, Pompucci L, Festuccia C, Ginestra A, Vittorelli ML, Canevari S, Pavan A (1999) Matrix-degrading proteinases are shed in membrane vesicles by ovarian cancer cells in vivo and in vitro. Clin Exp Metastasis 17:131–140
Castellana D, Zobairi F, Martinez MC, Panaro MA, Mitolo V, Freyssinet JM, Kunzelmann C (2009) Membrane microvesicles as actors in the establishment of a favorable prostatic tumoral niche: a role for activated fibroblasts and CX3CL1-CX3CR1 axis. Cancer Res 69:785–793. doi:10.1158/0008-5472.CAN-08-1946
Angelucci A, D’Ascenzo S, Festuccia C, Gravina GL, Bologna M, Dolo V, Pavan A (2000) Vesicle-associated urokinase plasminogen activator promotes invasion in prostate cancer cell lines. Clin Exp Metast 18:163–170. doi:10.1023/A:1006778000173
Wysoczynski M, Ratajczak MZ (2009) Lung cancer secreted microvesicles: underappreciated modulators of microenvironment in expanding tumors. Int J Cancer 125:1595–1603. doi:10.1002/ijc.24479
Millimaggi D, Mari M, D’Ascenzo S, Carosa E, Jannini EA, Zucker S, Carta G, Pavan A, Dolo V (2007) Tumor vesicle-associated CD147 modulates the angiogenic capability of endothelial cells. Neoplasia 9:349–357. doi:10.1593/neo.07133
Taraboletti G, D’Ascenzo S, Giusti I, Marchetti D, Borsotti P, Millimaggi D, Giavazzi R, Pavan A, Dolo V (2006) Bioavailability of VEGF in tumor-shed vesicles depends on vesicle burst induced by acidic pH. Neoplasia 8:96–103. doi:10.1593/neo.05583
Bianco F, Perrotta C, Novellino L, Francolini M, Riganti L, Menna E, Saglietti L, Schuchman EH, Furlan R, Clementi E, Matteoli M, Verderio C (2009) Acid sphingomyelinaseactivity triggers microparticle release from glial cells. EMBO J 28:1043–1054. doi:10.1038/emboj.2009.45
Charras GT, Yarrow JC, Horton MA, Mahadevan L, Mitchison TJ (2005) Non-equilibration of hydrostatic pressure in blebbing cells. Nature 435:365–369. doi:10.1038/nature03550
Jaiswal R, Luk F, Dalla PV, Grau GE, Bebawy M (2013) Breast cancer-derived microparticles display tissue selectivity in the transfer of resistance proteins to cells. PLoS ONE. doi:10.1371/journal.pone.0061515
Pokharel D, Padula MP, Lu JF, Tacchi JL, Luk F, Djordjevic SP, Bebawy M (2014) Proteome analysis of multidrug-resistant, breast cancer-derived microparticles. J Extracell Vesicles 3:1–10. doi:10.3402/jev.v3.24384
Choi DS, Kim DK, Kim YK, Gho YS (2015) Proteomics of extracellular vesicles: exosomes and ectosomes. Mass Spectrom Rev 34:474–490. doi:10.1002/mas.21420
Stępień E, Kabłak-Ziembicka A, Czyż J, Przewłocki T, Małecki M (2012) Microparticles, not only markers but also a therapeutic target in the early stage of diabetic retinopathy and vascular aging. Expert Opin Ther Targets 16:677–688. doi:10.1517/14728222.2012.691471
Alexandru N, Badila E, Weiss E, Cochior D, Stępień E, Georgescu A (2016) Vascular complications in diabetes: Microparticles and microparticle associated microRNAs as active players. Biochem Biophys Res Commun 472:1–10. doi:10.1016/j.bbrc.2016.02.038
Muralidharan-Chari V, Clancy JW, Sedgwick A, D’Souza-Schorey C (2010) Microvesicles: mediators of extracellular communication during cancer progression. J Cell Sci 123:1603–1611. doi:10.1242/jcs.064386
Li B, Antonyak MA, Zhang J, Cerione RA (2012) RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene 31:4740–4749. doi:10.1038/onc.2011.636
Wolf P (1967) The nature and significance of platelet products in human plasma. Br J Haematol 13:269–288. doi:10.1111/j.1365-2141.1967.tb08741.x
Morel O, Jesel L, Freyssinet JM, Toti F (2011) Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler Thromb Vasc Biol 31:15–26. doi:10.1161/ATVBAHA.109.200956
Pap E, Pállinger E, Pásztói M, Falus A (2009) Highlights of a new type of intercellular communication: microvesicle-based information transfer. Inflamm Res 58:1–8. doi:10.1007/s00011-008-8210-7
Solari FA, Mattheij NJ, Burkhart JM, Swieringa F, Collins PW, Cosemans JM, Sickmann A, Heemskerk JW, Zahedi RP (2016) Combined quantification of the global proteome, phosphoproteome and proteolytic cleavage to characterize altered platelet functions in the human Scott syndrome. Mol Cell Proteom 15:3154–3169. doi:10.1074/mcp.M116.060368
Otzen DE, Blans K, Wang H, Gilbert GE, Rasmussen JT (2012) Lactadherin binds to phosphatidylserine-containing vesicles in a two-step mechanism sensitive to vesicle size and composition. Biochim Biophys Acta 1818:1019–1027. doi:10.1016/j.bbamem.2011.08.032
Friend C, Marovitz W, Henie G, Henie W, Tsuei D, Hirschhorn K, Holland JG, Cuttner J (1978) Observations on cell lines derived from a patient with Hodgkin’s disease. Cancer Res 38:2581–2591
Poste G, Nicolson GL (1980) Arrest and metastasis of blood-borne tumor cells are modified by fusion of plasma membrane vesicles from highly metastatic cells. Proc Natl Acad Sci USA 77:399–403
Poutsiaka DD, Schroder EW, Taylor DD, Levy EM, Black PH (1985) Membrane vesicles shed by murine melanoma cells selectively inhibit the expression of Ia antigen by macrophages. J Immunol 134:138–144
Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ (2006) Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia 20:1487–1495. doi:10.1038/sj.leu.2404296
Antonyak MA, Li B, Boroughs LK, Johnson JL, Druso JE, Bryant KL, Holowka DA, Cerione RA (2011) Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc Natl Acad Sci USA 108:4852–4857. doi:10.1073/pnas.1017667108
Skog J, Würdinger T, Van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry MT Jr, Carter BS, Krichevsky AM, Breakefield XO (2008) Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 10:1470–1476. doi:10.1038/ncb1800
Szajnik M, Czystowska M, Szczepanski MJ, Mandapathil M, Whiteside TL (2010) Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS ONE. doi:10.1371/journal.pone.0011469
Ogorevc E, Kralj-Iglic V, Veranic P (2013) The role of extracellular vesicles in phenotypic cancer transformation. Radiol Oncol 47:197–205. doi:10.2478/raon-2013-0037
Ginestra A, Monea S, Seghezzi G, Dolo V, Nagase H, Mignatti P, Vittorelli ML (1997) Urokinase plasminogen activator and gelatinases are associated with membrane vesicles shed by human HT1080 fibrosarcoma cells. J Biol Chem 272:17216–17222. doi:10.1074/jbc.272.27.17216
Hadler-Olsen E, Fadnes B, Sylte I, Uhlin-Hansen L, Winberg JO (2010) Regulation of matrix metalloproteinase activity in health and disease. FEBS J 278:28–45. doi:10.1111/j.1742-4658.2010.07920.x
Giusti I, D’Ascenzo S, Millimaggi S, Taraboletti G, Carta G, Franceschini N, Pavan A, Dolo V (2008) Cathepsin B mediates the pH-dependent proinvasive activity of tumor-shed microvesicles. Neoplasia 10:481–488. doi:10.1593/neo.08178
Lo Cicero A, Majkowska I, Nagase H, Di Liegro I, Troeberg L (2012) Microvesicles shed by oligodendroglioma cells and rheumatoid synovial fibroblasts contain aggrecanase activity. Matrix Biol 31:229–233. doi:10.1016/j.matbio.2012.02.005
Lindoso RS, Collino F, Camussi G (2015) Extracellular vesicles derived from renal cancer stem cells induce a pro-tumorigenic phenotype in mesenchymal stromal cells. Oncotarget 6:7959–7969. doi:10.18632/oncotarget.3503
Bordeleau F, Chan B, Antonyak MA, Lampi MC, Cerione RA, Reinhart-King CA (2016) Microvesicles released from tumor cells disrupt epithelial cell morphology and contractility. J Biomech 49:1272–1279. doi:10.1016/j.jbiomech.2015.10.003
Schroder WA, Major LD, Le TT, Gardner J, Sweet MJ, Janciauskiene S, Suhrbier A (2014) Tumor cell-expressed SerpinB2 is present on microparticles and inhibits metastasis. Cancer Med 3:500–513. doi:10.1002/cam4.229
Liu Y, Zhu XJ, Zeng C, Wu PH, Wang HX, Chen ZC, Li QB (2014) Microvesicles secreted from human multiple myeloma cells promote angiogenesis. Acta Pharmacol Sin 35:230–238. doi:10.1038/aps.2013
Munster M, Fremder E, Miller V, Ben-Tsedek N, Davidi S, Scherer SJ, Shaked Y (2014) Anti-VEGF-A affects the angiogenic properties of tumor-derived microparticles. PLoS ONE. doi:10.1371/journal.pone.0095983
Kovacic JC, Mercader N, Torres M, Boehm M, Fuster V (2012) Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: from cardiovascular development to disease. Circulation 125:1795–1808. doi:10.1161/CIRCULATIONAHA.111.040352
Bobis-Wozowicz S, Kmiotek K, Kania K, Karnas E, Labedz-Maslowska A, Sekula M, Kedracka-Krok S, Kolcz J, Boruczkowski D, Madeja Z, Zuba-Surma EK (2016) Diverse impact of xeno-free conditions on biological and regenerative properties of hUC-MSCs and their extracellular vesicles. J Mol Med. doi:10.1007/s00109-016-1471-7
Zhang H, Bai M, Deng T, Liu R, Wang X, Qu Y, Duan J, Zhang L, Ning T, Ge S, Li H Zhou L, Liu Y, Huang D, Ying G, Ba Y (2016) Cell-derived microvesicles mediate the delivery of miR-29a/c to suppress angiogenesis in gastric carcinoma. Cancer Lett 375:331–339. doi:10.1016/j.canlet.2016.03.026
Yamada N, Tsujimura N, Kumazaki M, Shinohara H, Taniguchi K, Nakagawa Y, Naoe T, Akao Y (2014) Colorectal cancer cell-derived microvesicles containing microRNA-1246 promote angiogenesis by activating Smad 1/5/8 signaling elicited by PML down-regulation in endothelial cells. Biochim Biophys Acta 1839:1256–1272. doi:10.1016/j.bbagrm.2014.09.002
Goubran H, Sabry W, Kotb R, Seghatchian J, Burnouf T (2015) Platelet microparticles and cancer: an intimate cross-talk. Transfus Apher Sci 53:168–172. doi:10.1016/j.transci.2015.10.014
Esumi N, Todo S, Imashuku S (1987) Platelet aggregating activity mediated by thrombin generation in the NCG human neuroblastoma cell line. Cancer Res 47:2129–2135
Trummer A, De Rop S, Stadler M, Ganser A, Buchholz S (2011) P-selectin glycoprotein ligand-1 positive microparticles in allogeneic stem cell transplantation of hematologic malignancies. Exp Hematol 39:1047–1055. doi:10.1016/j.exphem.2011.08.007
Vandendries ER, Furie BC, Furie B (2004) Role of P-selectin and PSGL-1 in coagulation and thrombosis. Thromb Haemost 92:459–466. doi:10.1160/TH04-05-0306
Mackman N, Luther T (2013) Platelet tissue factor: to be or not to be. Thromb Res 132:3–5. doi:10.1016/j.thromres.2013.05.009
Gardiner C, Harrison P, Belting M, Böing A, Campello E, Carter BS, Collier ME, Coumans F, Ettelaie C, Van Es N, Hochberg FH, Mackman N, Rennert RC, Thaler J, Rak J, Nieuwland R (2015) Extracellular vesicles, tissue factor, cancer and thrombosis—discussion themes of the ISEV 2014 Educational Day. J Extracell Vesicles. doi:10.3402/jev.v4.26901
Yu JL, May L, Lhotak V, Shahrzad S, Shirasawa S, Weitz JI, Coomber BL, Mackman N, Rak JW (2005) Oncogenic events regulate tissue factor expression in colorectal cancer cells: implications for tumor progression and angiogenesis. Blood 105:1734–1741. doi:10.1182/blood-2004-05-2042
Lima LG, Leal AC, Vargas G, Porto-Carreiro I, Monteiro RQ (2013) Intercellular transfer of tissue factor via the uptake of tumor-derived microvesicles. Thromb Res 132:450–456. doi:10.1016/j.thromres.2013.07.026
Hron G, Kollars M, Weber H, Sagaster V, Quehen-Berger P, Eichinger S, Kyrle PA, Weltermann A (2007) Tissue factor-positive microparticles: cellular origin and association with coagulation activation in patients with colorectal cancer. Thromb Haemost 97:119–123. doi:10.1160/TH06-03-0141
Mezouar S, Darbousset R, Dignat-George F, Panicot-Dubois L, Dubois C (2015) Inhibition of platelet activation prevents the P-selectin and integrin-dependent accumulation of cancer cell microparticles and reduces tumor growth and metastasis in vivo. Int J Cancer 136:462–475. doi:10.1002/ijc.28997
Baj-Krzyworzeka M, Weglarczyk K, Mytar B, Szatanek R, Baran J, Zembala M (2011) Tumour-derived microvesicles contain interleukin-8 and modulate production of chemokines by human monocytes. Anticancer Res 31:1329–1335
Köppler B, Cohen C, Schlöndorff D, Mack M (2006) Differential mechanisms of microparticle transfer to B cells and monocytes: anti-inflammatory properties of microparticles. Eur J Immunol 36:648–660. doi:10.1002/eji.200535435
Valenti R, Huber V, Filipazzi P, Pilla L, Sovena G, Villa A, Corbelli A, Fais S, Parmiani G, Rivoltini L (2006) Human tumor-released microvesicles promote the differentiation of myeloid cells with transforming growth factor-beta-mediated suppressive activity on T lymphocytes. Cancer Res 66:9290–9298. doi:10.1158/0008-5472.CAN-06-1819
Baj-Krzyworzeka M, Szatanek R, Weglarczyk K, Baran J, Zembala M (2007) Tumour-derived microvesicles modulate biological activity of human monocytes. Immunol Lett 113:76–82. doi:10.1016/j.imlet.2007.07.014
Baj-Krzyworzeka M, Mytar B, Szatanek R, Surmiak M, Węglarczyk K, Baran J, Siedlar M (2016) Colorectal cancer-derived microvesicles modulate differentiation of human monocytes to macrophages. J Transl Med 14:1–15. doi:10.1186/s12967-016-0789-9
Kuang DM, Wu Y, Chen N, Cheng J, Zhuang SM, Zheng L (2007) Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood 110:587–595. doi:10.1182/blood-2007-01-068031
Cocucci E, Racchetti G, Meldolesi J (2009) Shedding microvesicles: artefacts no more. Trends Cell Biol 19:43–51. doi:10.1016/j.tcb.2008.11.003
Sadallah S, Eken C, Schifferli JA (2011) Ectosomes as modulators of inflammation and immunity. Clin Exp Immunol 163:26–32. doi:10.1111/j.1365-2249.2010.04271.x
Ma J, Cai W, Zhang Y, Huang C, Zhang H, Liu J, Tang K, Xu P, Katirai F, Zhang J, He W, Ye D, Shen GX, Huang B (2013) Innate immune cell-derived microparticles facilitate hepatocarcinoma metastasis by transferring integrin α(M)β2 to tumor cells. J Immunol 191:3453–3461. doi:10.4049/jimmunol.1300171
Santi A, Caselli A, Ranaldi F, Paoli P, Mugnaioni C, Michelucci E, Cirri P (2015) Cancer associated fibroblasts transfer lipids and proteins to cancer cells through cargo vesicles supporting tumor growth. Biochim Biophys Acta 1853:3211–3223. doi:10.1016/j.bbamcr.2015.09.013
Reynés G, Vila V, Fleitas T, Reganon E, Font de Mora J, Jordá M, Martínez-Sales V (2013) Circulating endothelial cells and procoagulant microparticles in patients with glioblastoma: prognostic value. PLoS ONE. doi:10.1371/journal.pone.0069034
Fleitas T, Martínez-Sales V, Vila V, Reganon E, Mesado D, Martín M, Gómez-Codina J, Montalar J, Reynés G (2012) Circulating endothelial cells and microparticles as prognostic markers in advanced non-small cell lung cancer. PLoS ONE. doi:10.1371/journal.pone.0047365
Krishnan SR, Luk F, Brown RD, Suen H, Kwan Y, Bebawy M (2016) Isolation of human CD138(+) microparticles from the plasma of patients with multiple myeloma. Neoplasia 18:25–32. doi:10.1016/j.neo.2015.11.011
Liebhardt S, Ditsch N, Nieuwland R, Rank A, Jeschke U, Von Koch F, Friese K, Toth B (2010) CEA-, Her2/neu-, BCRP- and Hsp27-positive microparticles in breast cancer patients. Anticancer Res 30:1707–1712
Tavoosidana G, Ronquist G, Darmanis S, Yan J, Carlsson L, Wu D, Conze T, Ek P, Semjonow A, Eltze E, Larsson A, Landegren UD, Kamali-Moghaddam M (2011) Multiple recognition assay reveals prostasomes as promising plasma biomarkers for prostate cancer. Proc Natl Acad Sci USA 108:8809–8814. doi:10.1073/pnas.1019330108
Mege D, Panicot-Dubois L, Ouaissi M, Robert S, Sielezneff I, Sastre B, Dignat-George F, Dubois C (2016) The origin and concentration of circulating microparticles differ according to cancer type and evolution: a prospective single-center study. Int J Cancer 138:939–948. doi:10.1002/ijc.29837
Thaler J, Ay C, Mackman N, Metz-Schimmerl S, Stift J, Kaider A, Müllauer L, Gnant M, Scheithauer W, Pabinger I (2013) Microparticle-associated tissue factor activity in patients with pancreatic cancer: correlation with clinicopathological features. Eur J Clin Invest 43:277–285. doi:10.1111/eci.12042
Böing AN, Van der Pol E, Grootemaat AE, Coumans FAW, Sturk A, Nieuwland R (2014) Single-step isolation of extracellular vesicles by size-exclusion chromatography. J Extracell Vesicles. doi:10.3402/jev.v3.23430
Rupert DL, Claudio V, Lässer C, Bally M (2016) Methods for the physical characterization and quantification of extracellular vesicles in biological samples. Biochim Biophys Acta. doi:10.1016/j.bbagen.2016.07.028
Jorfi S, Ansa-Addo EA, Kholia S, Stratton S, Valley S, Lange S, Inal J (2015) Inhibition of microvesiculation sensitizes prostate cancer cells to chemotherapy and reduces docetaxel dose required to limit tumor growth in vivo. Sci Rep 5:1–13. doi:10.1038/srep13006
De Souza PS, Faccion RS, Bernardo PS, Maia RC (2016) Membrane microparticles: shedding new light into cancer cell communication. J Cancer Res Clin Oncol 142:1395–1406. doi:10.1007/s00432-015-2029-8
Bebawy M, Combes V, Lee E, Jaiswal R, Gong J, Bonhoure A, Grau GE (2009) Membrane microparticles mediate transfer of P-glycoprotein to drug sensitive cancer cells. Leukemia 23:1643–1649. doi:10.1038/leu.2009.76
De Souza PS, Cruz AL, Viola JP, Maia RC (2015) Microparticles induce multifactorial resistance through oncogenic pathways independently of cancer cell type. Cancer Sci 106:60–68. doi:10.1111/cas.12566
Lu JF, Luk G, Gong J, Jaiswal R, Grau GE, Bebawy M (2013) Microparticles mediate MRP1 intercellular transfer and the re-templating of intrinsic resistance pathways. Pharmacol Res 76:77–83. doi:10.1016/j.phrs.2013.07.009
Shedden K, Xie XT, Chandaroy P, Chang YT, Rosania GR (2003) Expulsion of small molecules in vesicles shed by cancer cells: association with gene expression and chemosensitivity profiles. Cancer Res 63:4331–4337
Kholia S, Jorfi S, Thompson PR, Causey CP, Nicholas AP, Inal JM, Lange S (2015) A novel role for peptidylarginine deiminases in microvesicle release reveals therapeutic potential of PAD inhibition in sensitizing prostate cancer cells to chemotherapy. J Extracell Vesicles 4:1–16. doi:10.3402/jev.v4.26192
Ansa-Addo EA, Lange S, Stratton D, Antwi-Baffour S, Cestari I, Ramirez MI, McCrossan MV, Inal JM (2010) Human plasma membrane-derived vesicles halt proliferation and induce differentiation of THP-1 acute monocytic leukemia cells. J Immunol 185:5236–5246. doi:10.4049/jimmunol.1001656
Tang K, Zhang Y, Zhang H, Xu P, Liu J, Ma J, Lv M, Li D, Katirai F, Shen GX, Zhang G, Feng ZH, Ye D, Huang B (2012) Delivery of chemotherapeutic drugs in tumour cell-derived microparticles. Nat Commun 3:1–11. doi:10.1038/ncomms2282
Yu KF, Zhang WQ, Luo LM, Song P, Li D, Du R, Ren W, Huang D, Lu WL, Zhang X, Zhang Q (2013) The antitumor activity of a doxorubicin loaded, iRGD-modified sterically-stabilized liposome on B16-F10 melanoma cells: in vitro and in vivo evaluation. Int J Nanomed 8:2473–2485. doi:10.2147/IJN.S46962
Latagliata R, Breccia M, Fazi P, Iacobelli S, Martinelli G, Di Raimondo F, Sborgia M, Fabbiano F, Pirrotta MT, Zaccaria A, Amadori S, Caramatti C, Falzetti F, Candoni A, Mattei D, Morselli M, Alimena G, Vignetti M, Baccarani M, Mandelli F (2008) Liposomal daunorubicin versus standard daunorubicin: long term follow-up of the GIMEMA GSI 103 AMLE randomized trial in patients older than 60 years with acute myelogenous leukaemia. Br J Haematol 143:681–689. doi:10.1111/j.1365-2141.2008.07400.x
Kaspers GJ, Zimmermann M, Reinhardt D, Gibson BE, Tamminga RY, Aleinikova O, Armendariz H, Dworzak M, Ha SY, Hasle H, Hovi L, Maschan A, Bertrand Y, Leverger GG, Razzouk BI, Rizzari C, Smisek P, Smith O, Stark B, Creutzig U (2013) Improved outcome in pediatric relapsed acute myeloid leukemia: results of a randomized trial on liposomal daunorubicin by the International BFM Study Group. J Clin Oncol 31:599–607. doi:10.1200/JCO.2012.43.7384
Zhang H, Tang K, Zhang Y, Ma R, Ma J, Li Y, Luo S, Liang X, Ji T, Gu Z, Lu J, He W, Cao X, Wan Y, Huang B (2015) Cell-free tumor microparticle vaccines stimulate dendritic cells via cGAS/STING signaling. Cancer Immunol Res 3:196–205. doi:10.1158/2326-6066.CIR-14-0177
Zhang H, Huang B (2015) Tumor cell-derived microparticles: a new form of cancer vaccine. Oncoimmunology. doi:10.1080/2162402X.2015.1017704
Ran L, Tan X, Li Y, Zhang H, Ma R, Ji T, Dong W, Tong T, Liu Y, Chen D, Yin X, Liang X, Tang K, Ma J, Zhang Y, Cao X, Hu Z, Qin X, Huang B (2016) Delivery of oncolytic adenovirus into the nucleus of tumorigenic cells by tumor microparticles for virotherapy. Biomaterials 89:56–66. doi:10.1016/j.biomaterials.2016.02.025
Akao Y, Iio A, Itoh T, Noguchi S, Itoh Y, Ohtsuki Y, Naoe T (2011) Microvesicle-mediated RNA molecule delivery system using monocytes/macrophages. Mol Ther 19:395–399. doi:10.1038/mt.2010.254
Kim HK, Song KS, Park YS, Kang YH, Lee YJ, Lee KR, Kim HK, Ryu KW, Bae JM, Kim S (2003) Elevated levels of circulating platelet microparticles, VEGF, IL-6 and RANTES in patients with gastric cancer: possible role of a metastasis predictor. Eur J Cancer 39:184–191. doi:10.1016/S0959-8049(02)00596-8
Ma G, Liu F, Lv L, Gao Y, Su Y (2013) Increased promyelocytic-derived microparticles: a novel potential factor for coagulopathy in acute promyelocytic leukemia. Ann Hematol 92:645–652. doi:10.1007/s00277-013-1676-6
Acknowledgements
We are grateful to Dr. Małgorzata Opydo-Chanek (Department of Experimental Hematology, Jagiellonian University, Krakow, Poland) for providing photographs showing apoptotic bodies. Michael Jacobs line-edited the paper for submission. Due to length limitations we could not cite all of the valuable studies done in this area of research, and for this we apologize. This work was supported by a grant from the Polish National Science Centre (2013/11/B/NZ4/04315).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Surman, M., Stępień, E., Hoja-Łukowicz, D. et al. Deciphering the role of ectosomes in cancer development and progression: focus on the proteome. Clin Exp Metastasis 34, 273–289 (2017). https://doi.org/10.1007/s10585-017-9844-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-017-9844-z