
Abstract
A common feature of eukaryotic centromeres is the presence of large tracts of tandemly arranged repeats, known as satellite DNA. However, these centromeric repeats appear to experience rapid evolution under forces such as molecular drive and centromere drive, seemingly without consequence to the integrity of the centromere. Moreover, blocks of heterochromatin within the karyotype, including the centromere, are hotspots for chromosome rearrangements that may drive speciation events by contributing to reproductive isolation. However, the relationship between the evolution of heterochromatic sequences and the karyotypic dynamics of these regions remains largely unknown. Here, we show that a single conserved satellite DNA sequence in the order Rodentia of the genus Peromyscus localizes to recurrent sites of chromosome rearrangements and heterochromatic amplifications. Peromyscine species display several unique features of chromosome evolution compared to other Rodentia, including stable maintenance of a strict chromosome number of 48 among all known species in the absence of any detectable interchromosomal rearrangements. Rather, the diverse karyotypes of Peromyscine species are due to intrachromosomal variation in blocks of repeated DNA content. Despite wide variation in the copy number and location of repeat blocks among different species, we find that a single satellite monomer maintains a conserved sequence and homogenized tandem repeat structure, defying predictions of molecular drive. The conservation of this satellite monomer results in common, abundant, and large blocks of chromatin that are homologous among chromosomes within one species and among diverged species. Thus, such a conserved repeat may have facilitated the retention of polymorphic chromosome variants within individuals and intrachromosomal rearrangements between species—both factors that have previously been hypothesized to contribute towards the extremely wide range of ecological adaptations that this genus exhibits.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In most eukaryotic genomes, large arrays of tandem repeats are often confined to functionally defined subregions within chromosomes, such as the centromere and telomere. Exemplars are the virtually ubiquitous satellite DNA sequences that are present in tandem arrays in the centromeres of many eukaryotes. While maintaining a functional centromere is necessary to sustain chromosome stability by mediating proper kinetochore and microtubule attachment, the satellite DNA sequences that underlie this region are surprisingly among the most rapidly evolving portions of the eukaryotic genome (Henikoff et al. 2001), largely affected by processes such as molecular drive (Dover 1982) and centromere drive (Henikoff and Malik 2002; Malik and Henikoff 2002).
Satellites and other centromeric repeats in the mammalian genome evolve via concerted evolution, a non-independent process of molecular drive by which a species or population maintains an unusually high intraspecific repeat homogeneity and, concomitantly, a high interspecific heterogeneity (Dover 1982). Several mechanisms, including nonhomologous and/or unequal crossing over (Smith 1976), replication slippage (Walsh 1987), gene conversion (Shi et al. 2010), and rolling circle amplification and subsequent reinsertion (Bertelsen et al. 1982; reviewed in Gaubatz 1990), allow specific satellite DNA sequences to quickly spread throughout the genome and replace other repetitive sequences, despite their inherent inability to directly transpose themselves. Centromere drive may also affect the rapid satellite divergence between species as it is hypothesized that satellites and their DNA binding partners at the centromere, such as the centromere-specific histone centromere protein A (CENP-A), are in conflict with one another that manifests during female meiosis (Malik and Henikoff 2002). Further challenging the presumption that the centromere would be selected to remain a static region of the genome in order to support genome stability is the finding that it often is a hotspot for chromosome rearrangements, including translocations, inversions, and fusions (reviewed in Brown et al. 2012). Thus, the centromere is thus a host to both rapid nucleotide and karyotypic variation.
The “library hypothesis” (Salser et al. 1976) provides another explanation for how satellite DNA content at the centromere may diverge rapidly among closely related species. According to this hypothesis, extant but distinct centromeric repeats—which comprise the satellite “library”—may independently expand or contract in copy number in different evolutionary lineages. These alterations in the relative proportions of sequences at the centromere may result in production of divergent or unique centromere profiles for different species, and even different chromosomes within species. For example, human and chimpanzee alpha satellite HORs were found to share ~ 75% sequence identity and phylogenetic analyses of primate alpha satellites indicated each primate species examined carries evolutionarily distinct satellites (Alkan et al. 2007). Several studies support this hypothesis across widespread eukaryotic taxa, including the discovery of four distinct centromere satellites in four species of Palorus (beetle) (Mestrovic et al. 1998). More recently, species-specific satellite DNA amplification has also been observed in Leporinus elongatus (tropical freshwater fish) (da Silva et al. 2013), Aotus azarae (Azara’s owl monkey) (Prakhongcheep et al. 2013), and Oryza brachyantha (wild African rice) (Lee et al. 2005). Moreover, divergent satellite sequences also define the centromeres of specific chromosomes in Cercopithecus pogonias and Cercopithecus solatus (Old World monkeys) (Cacheux et al. 2018), Bactrocera oleae (olive fruit fly) (Tsoumani et al. 2013), Cricetulus griseus (Chinese hamster) (Faravelli et al. 1998), and Solanum tuberosum (potato) (Gong et al. 2012). Hence, rapid satellite DNA divergence is a common feature across eukaryotic lineages and swift changes in centromere DNA content can contribute to reproductive isolation that may drive speciation (Ferree and Barbash 2009).
Following the premise of the library hypothesis, it would reason that chromosome rearrangements affecting centromeric heterochromatin would cause a divergence in centromere satellite content as these events create an opportunity for divergent, or less abundant, repeats present in the library to become amplified within the rearranged chromosome and, through the mechanisms of molecular drive, the entire genome. Thus, this model predicts that alterations in the relative copy numbers of extant, variant satellite sequences should accompany karyotypic evolution. In support of this hypothesis, it has been found that dramatic expansions and contractions of three separate satellites in the Macropus genus (wallabies) are concurrent with specific chromosome rearrangements, notably translocations and fusions—both involving the centromere (Bulazel et al. 2007). A similar phenomenon has been reported in the family Bovidae, which includes cattle, goat, and sheep. Karyotypic evolution of these genera primarily progresses via fusions of two acrocentric chromosomes at the centromere, which is accompanied by a change in a relative copy of the centromeric satellite sequences on the resulting biarmed chromosome (Chaves et al. 2003; Chaves et al. 2000; D’Aiuto et al. 1997). These data support hypothesis that chromosome rearrangements, while not exclusively necessary for the rapid evolution of centromeric DNA, may serve as a means by which centromere satellite evolution is facilitated.
One genus that is particularly useful for studying dynamic centromere and heterochromatin restructuring, chromosome evolution, and rapid speciation in parallel is Peromyscus. This genus encompasses at least 56 species and serves as a valuable model system for a broad set of applications, including studies in behavior, ecology, evolution, and disease systems (reviewed in Bedford and Hoekstra 2015; Shorter et al. 2012). All species share a 2n = 48 karyotype (Arakaki and Sparkes 1967; Cross 1938), although both interspecific and intraspecific variations among chromosomes are typified by pericentric inversions and heterochromatic additions at common, known sites of rearrangement (Duffey 1972; Pathak et al. 1973). As a result, the fundamental number (number of chromosome arms) ranges from 56 to 96 across the genus (Pathak et al. 1973). Thus, while large-scale chromosome rearrangements do not distinguish diverse karyotypes within this genus, changes in heterochromatin content and pericentromeric arrangement characterize chromosome divergence across Peromyscus species.
Here, we present both cytogenetic and sequence analyses of satellite DNA within seven species of Peromyscus representing each of the major clades within the genus (Bradley et al. 2007). Previous work by Louzada et al. (2015) led to the identification of a Peromyscus satellite sequence, Peromyscus maniculatus satellite repeat (PMsat), that is present in high copy number in Peromyscus eremicus. Moreover, this study employed a computational approach to show that PMsat is found in tandem arrays in Peromyscus maniculatus, likely representing a centromeric repeat in this species (McNulty and Sullivan 2018; Plohl et al. 2012). Extending this earlier work, we set out to test whether molecular drive characterizes satellite repeats across a species complex that is exemplified by large-scale changes in the copy number and distribution of satellite-rich heterochromatin. We find that a single, defined centromeric satellite is a key component of heterochromatin additions and rearrangements across the Peromyscus genus and that this satellite displays a remarkable level of evolutionary conservation, contrary to predictions of the various forms of drive that may act on centromeric DNAs. We propose that the conservation of this element may contribute to the nearly unparalleled level of intraspecific and interspecific karyotypic variations exhibited in the Peromyscus genus. While the satellite DNA itself may not be actively participating in these chromosome rearrangements, the conservation of homogenous tandem arrays may facilitate the evolution of diverse karyotypes.
Methods
Cell culture, cytogenetics, and genomic DNA extractions
One ear punch for an individual from the following species was obtained from the Peromyscus Genetic Stock Center at the University of South Carolina: Peromyscus leucopus (LL), Peromyscus aztecus hylocetes (AM), Peromyscus melanophrys xenerus (XZ), Peromyscus californicus insignis (IS), P. eremicus (EP), P. maniculatus bairdii (BW), P. maniculatus sonoriensis (SM2), Peromyscus polionotus subgriseus (PO), and a BW × PO hybrid individual. Ear fibroblast cell lines were established using standard collagenase methods. An ear fibroblast cell line from Cricetulus griseus (Chinese hamster) was utilized as a closely related outgroup species as in Mlynarski et al. (2010). Metaphase chromosomes were prepared from primary fibroblasts as per Brown et al. (2002), and slides were dropped as per standard protocols. High molecular weight genomic DNA was obtained by phenol/chloroform extraction and ethanol precipitation using standard protocols from cell pellets for all species, with the exception of Peromyscus leucocephalus-subgriseus (LP), from which DNA was isolated from liver tissue.
PCR amplification
To determine if the satellite DNA sequence first identified by Louzada et al. (2015) in P. eremicus was also present in the genome of P. leucopus, the following PCR primers were designed with Primer3 (Untergasser et al. 2012): 5′-ACAGGAGCTTCTCTTCAGTCC-3′ and 5′-AAGCAGAGTGTTTTGGGTGT-3′. PCRs were performed using gDNA from P. leucopus, products were subcloned using the StrataClone PCR cloning kit (Agilent) as per the manufacturer’s protocol, and recombinant colonies were sequenced using BigDye® Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher) on an Agilent 3130 Genetic Analyzer.
Fluorescence in situ hybridization
Slides were pretreated with 0.1 mg/ml RNase A and 2× saline sodium citrate (SSC) at 37 °C for 15 min and rinsed four times for 2 min in 2× SSC, followed by a protease treatment as follows: CytoZyme Stabilized Pepsin for 10 min at 37 °C (for P. aztecus hylocetes species hybridization) or 0.2 N HCl for 20 min at room temperature (all other species). The slides were then dehydrated, air-dried, and denatured for 2 min in 70% formamide and 2× SSC at 72 °C before hybridization with the denatured probe.
Target DNA was amplified and labeled with biotin-16-dUTP (Roche) via PCR from P. leucopus genomic DNA using the aforementioned primers. The labeled probe from P. leucopus was used in fluorescence in situ hybridization (FISH) for all species with the following exceptions. Probes were prepared from P. aztecus hylocetes, P. melanophrys xenerus, and C. griseus genomic DNA using the same primers as above and were hybridized conspecifically to metaphase chromosomes as hybridization signal was not detected reliably across different metaphase spreads of these species using the P. leucopus probe. In all cases, hybridization probes were prepared by precipitating 500 ng of labeled DNA and 25 μg salmon sperm DNA and resuspending the DNA in Hybrisol VII (MP Biomedicals). Probes were denatured at 80 °C for 10 min and subsequently hybridized to the denatured slides for about 18 h at 37 °C.
Post-hybridization washes were performed for 5 min in 2× SSC at 45 °C followed by washing two times for 5 min in 1× SSC at 72 °C for all species, except P. aztecus hylocetes (0.5× SSC at 72 °C) due to nonspecific background. Slides were blocked in 4× SSC, 0.2% Tween 20, and 5% bovine serum albumin for 30 min at 37 °C, and probes were detected using a 1:400 dilution of avidin Texas Red (Invitrogen) in 4× SSC, 0.2% Tween 20, and 5% bovine serum albumin for 20 min. Excess detection reagents were removed by washing three times for 5 min in 4× SSC and 0.2% Tween 20 at 45 °C. Metaphases were counterstained with a dilution of 1:4 DAPI:Vectashield (Vector Laboratories). Images were captured using an Olympus AX-70 microscope equipped with Genus imaging software (Applied Imaging). A minimum of 20 cells was assessed for each species. Inverted DAPI images were used to identify chromosome pairs within each karyotype.
Southern blotting
Approximately 10 μg of high molecular weight DNA was digested overnight at 37 °C in separate reactions for each species with MspI (New England Biolabs). Restriction digests were electrophoresed on a 0.8% agarose gel overnight at 30 V and transferred to a Hybond N+ nylon membrane (Amersham) by standard methods. The satellite probe was amplified as above using P. leucopus genomic DNA, the product was labeled with 32P-dCTP using random primers and hybridized to the membrane in Church’s solution (500 mM Na2HPO4, 7% SDS, 1 mM EDTA) at 60 °C overnight. Post hybridization, the membrane was washed in 2× SSC and 0.1% SDS at room temperature for 10 min, 2× SSC and 0.1% SDS at 60 °C for 15 min, and 1× SSC and 0.1% SDS 60 °C for 15 min. The membrane was exposed to X-ray film for 18 h at − 80 °C and then developed.
Sequencing and data analysis
The satellite repeat monomer was amplified via PCR in all species with primers designed to obtain the following complete monomer sequence: 5′-CGCGTCTGTTCCCAGCAA-3′ and 5′-AGTGCTATTTGCACTGTKTAT-3′. The PCR reaction was run on an agarose gel, and DNA from the second largest band (~ 680 bp), representing multiple monomers, was extracted using the QIAquick Gel Extraction Kit (Qiagen) as per the manufacturer’s protocol. The PCR products were subcloned using the StrataClone PCR cloning kit (Agilent) as per the manufacturer’s protocol, and recombinant colonies were sequenced using BigDye® Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher) on an ABI 3130 Genetic Analyzer. At least 10 clones were sequenced per species; sequences were trimmed of vector and primer sequences and a single monomer within the primers used for PCR was selected using Geneious 8.1 (Biomatters) and aligned using MUSCLE (Edgar 2004) and ClustalW (Larkin et al. 2007) implemented in Geneious 8.1. Sequences were searched for functional motifs using the MEME Suite (Bailey et al. 2009). All satellite monomer clones are reported in GenBank under accession numbers KX555281–KX555350.
Acquisition and filtering of next-generation sequencing libraries
Low-coverage next-generation sequencing (NGS) data available for four species of Peromyscus (P. maniculatus SRR1012309, P. polionotus SRR545676, P. californicus SRR545682, and P. leucopus SRR545963) was downloaded using fastq-dump (with parameters “--split-3 --gzip -W --read-filter pass”) and the paired reads were merged using pandaseq (default parameters). The files were then masked using RepeatMasker and the previously identified PMsat sequences (with parameters “-nolow -no_is -norna -gff -e ncbi -lib clones.fasta”). Only reads that contained 99% repetitive sequences were retained in an effort to focus on tandemly arrayed sequences rather than solo, and likely more divergent and noncentromeric monomers. The remaining reads were then mapped using BWA mem (default parameters) against the clone library derived from Sanger sequencing, and a 25-bp sliding window was used to find the region where the highest number of merged reads start mapping.
Clustering and quantification of NGS reads: repeatConnector
We developed a tool, repeatConnector, to mine existing next-generation sequencing data in the SRA database to generate a minimum spanning tree of clusters of repetitive sequences. The tool is designed to compare similar repeats across different datasets, in this case different species. To accomplish this, it first merges paired end reads using pandaSeq (Masella et al. 2012) and then filters the merged reads using a user-supplied fasta file containing the repeat of interest. The filtering is done to reduce the computational time required to cluster the merged reads. Using BWA (Li 2013) and RepeatMasker, the script removes any read that has no similarity to the user-supplied repeat library and then uses cd-hit-est (Fu et al. 2012) for clustering the reads that passed the filtering steps clustered at each of the following rates: 95%, 97%, and 99% similarity (with parameters “-M 50000 -s 0.8 -d -g 1”). The similarity between all clusters was calculated using a matcher from the EMBOSS suite (Rice et al. 2000). To calculate the composition of every cluster, the best hit from BWA to the initial fasta file was used to identify the most similar source sequence. The raw counts, counts normalized to the initial number of reads, and counts normalized to the number of reads passing the quality filters were also calculated. Shannon’s H was calculated for each cluster and for each method of counting merged reads. The above information about each cluster was then used to generate and display the minimum spanning tree in R using the igraph package (Csardi and Nepusz 2006). Scripts and background information on the repeatConnector can be found at https://gitlab.com/roneill_lab/repeatConnector.
Results
Identification of a conserved heterochromatic satellite repeat
A sequence sharing identity to PMsat, a satellite previously identified in P. eremicus (Louzada et al. 2015), was isolated by PCR in P. leucopus and confirmed via Sanger sequencing (Fig. 1). A total of 10 clones aligned to the previously annotated PMsat from P. eremicus (accession numbers KC351943.1, KC351942.1, and KC351938.1) and a single monomer A (KC351939) with an overall pairwise identity of 94.9% to a region slightly smaller than a full monomer (Fig. 1a, b). Given our finding that this P. eremicus satellite is conserved in P. leucopus and previous work demonstrating PMsat is found in divergent species of Rodentia (Cricetus cricetus) (Louzada et al. 2015), we set out to test whether the sequence was a primary component of heterochromatin blocks and/or centromeric regions in other Peromyscus species via fluorescence microscopy using the complete monomer (monomer B).

P. leucopus shares the PMsat satellite with P. eremicus. a Schematic of the relationship between monomer A of PMsat identified from P. eremicus relative to monomer B (targeted herein) and the shorter sequence initially identified in P. leucopus. b Sequence identity of the P. leucopus clones compared to partial and complete monomers previously identified from P. eremicus (accession numbers KC351943.1, KC351942.1, KC351938.1, and KC351939) (Louzada et al. (2015). Similarities among sequences are coded by colors as follows: A is green, C is blue, G is black, T is red, and white is a disagreement among the sequences
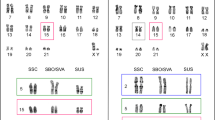
The satellite sequence hybridized to the centromeres of all chromosomes of P. leucopus, in addition to the axial telomeres (i.e., the telomere of the short arm of a chromosome) of some of the biarmed chromosomes (Fig. 2). To qualitatively compare the conservation of PMsat across the Peromyscus genus, its chromosomal location was visualized through FISH using cells from seven additional species as well as an outgroup species, the Chinese hamster (C. griseus). Across all Peromyscus samples, the PMsat sequence was found at the centromeres of all chromosomes, although the variation among species was observed in both copy number and presence at noncentromeric heterochromatic regions, such as the telomere (Fig. 2).

Satellite probe hybridizes to centromeres of all species of Peromyscus but varies at telomeric locations. A fluorescent probe was prepared and hybridized to metaphase spreads, and the genomic distribution of the satellite on each chromosome was recorded in each species. A representative FISH image is shown for each species (DAPI-stained chromosomes are gray, and probe signal is red), and the results are summarized in the ideogram. No hybridization was observed in C. griseus. Green-shaded chromosomes represent those that have diverged from the ancestral karyotype (shown at the base of the tree, centromeres are denoted by black-filled areas), indicating that it has experienced interspecific rearrangement. Asterisks denote chromosomes known to exist in polymorphic forms within an individual of the respective species. Phylogenetic relationships among species shown to the left and based on Bradley et al. (2007)
For each species, the following positive hybridization signal was observed: in P. leucopus, centromeric signal across all chromosomes with four chromosomes showing signal at both the centromere and the axial telomere (chr. 18, 21–23); in P. aztecus, P. melanophrys, and P. californicus, all chromosomes with signal restricted to the centromere; and in P. eremicus, 24 biarmed chromosome pairs, two autosomal pairs with signal restricted to the centromere (chr. 1, 23), two autosomal pairs with signal at both the centromere and the axial telomere (chr. 6, 22), and 19 autosomal pairs with signal extending from the centromere to the axial telomere (chr. 2–5, 7–21). The X chromosome displayed signal both at the centromere and at two other blocks along a single arm, and the Y chromosome exhibited signal at the centromere only. In P. maniculatus, all chromosomes carry signal at the centromere, three pairs with signal at both the centromere and the axial telomere (chr. 18, 21, 22), and two pairs with signal extending from the centromere to the axial telomere (chr. 16, 17). In P. polionotus, all chromosomes carry signal at the centromere and three with signal at both the centromere and the axial telomere (chr. 16, 18, 19). In C. griseus, no specific hybridization to chromosomes was observed.
Repeat organization in the Peromyscus genome
To characterize the length and arrangement of this heterochromatic repeat, Southern blotting was performed across the species that showed high copy numbers of PMsat in defined heterochromatic blocks in the karyotype, thus excluding the outgroup C. griseus, which lacked hybridization signal via FISH. Two additional samples were included in this analysis: LP and a P. maniculatus × P. polionotus hybrid. A ladder-like hybridization pattern was observed across all individuals with a conserved periodicity and monomer size of about 350 bp (Fig. 3), banding patterns that are consistent with a satellite DNA sequence present in a homogenized tandem array (reviewed in Garrido-Ramos 2017). This data suggests that the PMsat satellite is a conserved tandem repeat across all Peromyscus species assayed, with remarkable conservation of restriction site positions and periodicity of the satellite monomer.

Southern analysis confirms tandem arrangement of satellite in all species of Peromyscus. Genomic DNA was digested with MspI, electrophoresed, transferred to a membrane, and probed with the satellite sequence. A ladder-like hybridization pattern observed in all species confirms the tandem arrangement of the satellite. Lanes are as follows from the left: P. californicus, P. eremicus, P. melanophrys, P. aztecus, P. leucopus, P. maniculatus bairdii, P. maniculatus sonoriensis, P. polionotus subgriseus, P. leucocephalus-subgriseus, and P. maniculatus × P. polionotus hybrid. Marker sizes are shown to the right
To quantitatively characterize the nucleotide identity across Peromyscus species and test for evidence of molecular drive, the intraspecific identity of the satellite was compared to interspecific identity in two different ways: first, a set of PCR amplicons for each species was used to determine inter- vs intraspecific identity. This approach is based on the assumption that the most predominant satellite variant within a species is more likely to reach exponential amplification within a single PCR, thus biasing these data towards assessing sequences with lower levels of intraspecific variation. Indeed, in most cases, the intraspecific sequence identity was greater than the interspecific identity of closely related species, indicating that concerted evolution of the satellite may be taking place, at least on common variants (Fig. 4). The monomer size and pairwise identity within each species were as follows: in P. aztecus, 79.2% (346 bp); in P. melanophrys, 95.1% (346 bp); in P. californicus, 93.0% (345 bp); in P. eremicus, 90.1% (345 bp); in P. leucopus, 95.3% (345 bp); in P. maniculatus, 95.7% (344 bp); and in P. polionotus, 97.3% (344 bp) (Fig. 4a). All species-specific consensus sequences share between 80% (P. leucopus) and 96% (P. eremicus) identity to three clones sequenced in P. eremicus by Louzada et al. (2015) (accession numbers KC351938, KC351942, and KC351943). To estimate the interspecific degree of conservation of the satellite to facilitate comparisons to known phylogenetic relationships among species, each intraspecific sequence was subject to multiple pairwise alignments to each PMsat sequence for every species (Fig. 4b, c).

Sequence of satellite monomer does not exhibit species-specific patterns of nucleotide identity. a The intraspecific identity of the satellite and the predicted monomer length were calculated by aligning multiple clones within each respective species. b The interspecific identity of the satellite was calculated by aligning the consensus sequences generated from each species. Asterisks indicate instances where interspecific homology is greater than intraspecific homology, suggesting that the influence of concerted evolution is limited. c Alignment of monomer consensus sequences from each species shows identity throughout the entire satellite, with no specific region of the sequence displaying higher or lower levels of sequence conservation
Several species showed notable exceptions to predictions that interspecific differences would be greater than intraspecific differences. Copies of P. aztecus PMsat showed much lower intraspecific satellite identity than observed in either intra- or interspecific comparisons among all other species. Additionally, the interspecific identity of the satellite between P. maniculatus and P. polionotus is higher than the intraspecific identity of either of the species, perhaps reflective of their recent divergence (estimated at 1.5–3 million years ago) (Platt et al. 2015).
Previously developed bioinformatic tools afford the opportunity to mine next-generation sequencing data for repeats and the relationship of repeat variants within a species (e.g., Repeat Explorer) (Novak et al. 2013) or to detect satellites and derive estimates for intragenomic variation (e.g., satMiner) (Ruiz-Ruano et al. 2016). However, we required a tool that would facilitate the screening of next-generation sequencing datasets for specific satellite sequences and the subsequent analyses of phylogenetic relationships among these satellite repertoires across different species. To further examine the relationship of sequence identity of the PMsat satellite within and between species, we employed our newly developed tool, repeatConnector, using available next-generation sequencing datasets for species included in our study (P. maniculatus, P. polionotus, P. californicus, and P. leucopus) and representing all but one clade within the genus (no data were available for the P. melanophrys-aztecus clade). Paired end, whole-genome shotgun Illumina reads were trimmed for quality, merged, and masked against a de novo repeat database including the PMsat sequences obtained from all species via PCR. Reads with a minimum of 99% repeat content were retained to narrow our focus to those repeats present in tandem arrays and thus more likely to be within the CENP-A-delimited region of the centromere rather than pericentric and degenerated variants (reviewed in Brown and O’Neill 2014; Garrido-Ramos 2017; Plohl et al. 2012). Surviving reads were clustered using cd-hit-est at 95%, 97%, and 99% similarity. To generate the similarity score between each cluster, a matcher was used to calculate the percent similarity among the sequences for each cluster. The sequence identity of satellites within each cluster was calculated using the best hit from BWA alignments among libraries to identify the most similar clone and thus define cluster relationships.
Models of molecular drive predict that satellite variants, represented by nodes, within each species would cluster together and separately from clusters of satellites from other species. However, we find that this is not the case, at least for the clades assessed; clusters from different species representing different clades within Peromyscus are intermingled, as seen by different colored circles within the same network, show high sequence identities between nodes of different colors, most often between 95 and 100% identity, and clear species-specific clusters were not identifiable. Moreover, in many cases, cluster nodes were comprised of satellites from different species (pie graphs at nodes), indicating that interspecific variation is lower than intraspecific variation. Across all clades assessed, we find this pattern to hold true when using low similarity thresholds (97% and 95%, Fig. 5a–d), which would pick up more divergent satellites as well as variants that are more likely to have signatures of concerted evolution within any given dataset. Thus, the ancestor to the P. maniculatus-polionotus, P. leucopus, and P. californicus-eremicus clades, representing all but one of the major clades within Peromyscus, likely carried a limited (i.e., low diversity) satellite library. While the clone-based data for the P. melanophrys/aztecus clade indicates a similar pattern of limited diversity would be expected from deep sequencing of satellite libraries for species within this clade, we lack next-generation sequencing data for these species and thus can only infer that the low diversity of satellites across all clades is an ancestral characteristic.

Satellite cluster analyses across Peromyscus lack structure. RepeatConnector cluster analysis of satellites isolated from P. maniculatus, P. polionotus, P. californicus, and P. leucopus shotgun sequencing data. Reads containing at least 99% satellite sequence identity to centromeric satellites were clustered by sequence identity across all reads. Clusters (shown as nodes) representing 97% identity (a, b) and 95% identity (c, d) were annotated based on the originating sequencing library (colors). Clusters containing reads from more than one species-specific library are represented by pie graphs, with the colors therein denoting the species library. The size of each node indicates the number of sequences therein. The sequence identity among clusters is indicated by line thickness, encompassing a range of identities from 85 to 100%, with most falling between 95 and 100%. a, c The clusters with connections represented by lines whose thickness is represented by the percent divergence. b, d The same clusters with connections represented by lines whose thickness is proportionally reduced for visual clarity
Given the conservation of this satellite as a tandemly arrayed monomer and a component of the centromere region of each chromosome across all species examined, we tested whether this satellite carries detectable signals of centromere protein binding activity, demarcating PMsat as a component of chromatin involved in kinetochore assembly. Using FIMO (Bailey et al. 2009), we searched for evidence of a centromere protein B (CENP-B) binding box, a feature that is prevalent in many functional centromeric satellites, including the human centromeric alpha satellite (Masumoto et al. 1989). Despite the conservation of this satellite in large arrays at centromeres, we did not detect the canonical CENP-B motif. The strongest alignment of the CENP-B motif (p = 0.00133, q = 0.686) was found in the same location of each satellite but yielded an alignment in which only six of the nine bases that are critical for pairing (reviewed in Masumoto et al. 2004) are conserved in four species (P. aztecus, P. melanophrys, P. californicus, P. eremicus), only five of the nine conserved in P. polionotus, and four of the nine conserved in P. leucopus and P. maniculatus (Fig. 6). Based on the consensus CENP-B binding activity known for human and mouse, this motif is predicted to be nonfunctional. However, Peromyscus-specific binding activity for CENP-B is currently unknown as CENP-B antibodies for this species are not available; it is possible that a diverged consensus sequence may be required for functional activity in this lineage.

Divergent CENP-B DNA-binding motifs are present within Peromyscus satellite monomers. Alignment of the 17-bp consensus CENP-B DNA binding motif and consensus satellite monomers across Peromyscus. The nine bases required for CENP-B binding in human are shaded in yellow; sites conserved among all sequences are shown in bold
Discussion
Chromosome rearrangements involving centromeric or telomeric heterochromatin are predicted to lead to divergence in satellite monomer sequence as these DNA breaks can create an opportunity for mutated or less abundant repeats to become amplified in the new chromosome and, eventually—through the mechanisms of concerted evolution—the entire genome. Given the conservation of this satellite sequence across all major clades within the Peromyscus genus, the predominant satellite at Peromyscus centromeres does not follow the predictions of the “library model” of satellite DNA evolution (Salser et al. 1976)—a hypothesis that would explain high centromere sequence divergence between closely related but karyotypically distinct species, such as that observed in the Peromyscine species. Rather than different repetitive sequences being selectively amplified in various Peromyscus genomes through the process of molecular drive (Dover 1982), the same sequence is found at the centromeres of all Peromyscus species studied with little variation in monomer sequence (Figs. 4 and 5). Furthermore, this same sequence localizes to telomeric heterochromatin created as a result of pericentric inversions and heterochromatic additions; these karyotypic variations typify Peromyscus speciation, and this dynamic process of chromosome evolution would be expected to alter the relative proportions of heterochromatic repeats in each genome. Instead, we report a perpetuation of the same repeat despite extensive heterochromatic repatterning (Figs. 2, 4c, and 5).
It does not seem that the degree of intraspecific homogenization of the satellite sequence is correlated with the number of chromosome rearrangements (Figs. 2, 4a, and 5); in this study, P. californicus differs from the primitive Peromyscus karyotype by a single pericentric inversion and has an intraspecific satellite identity of 93.0%, while P. maniculatus differs by 15 pericentric inversions and two heterochromatic additions and P. eremicus differs by at least 19 heterochromatic additions and two pericentric inversions, with intraspecific homologies of 95.7% and 90.1%, respectively. Conversely, P. aztecus diverged from the ancestral Peromyscus karyotype by five pericentric inversions and yet has an intraspecific identity of only 79.2% while it shares higher interspecific homology to all species. According to the theory of molecular drive (Dover 1982), concerted evolution (intraspecific homogenization and interspecific diversity) would be expected to occur more rapidly in species with more diverged karyotypes; this is not observed in Peromyscus.
In addition to the conservation of this single satellite sequence throughout the Peromyscus genus, another unusual characteristic exists regarding Peromyscus chromosome evolution; while most other Rodentia lineages primarily consist of fusions, fissions, and translocations (reviewed in Romanenko et al. 2012), Peromyscus karyotypic evolution is characterized by a high frequency of pericentric inversions and heterochromatic additions, which do not change the chromosome number but result in varying fundamental numbers between species. These same chromosome rearrangements also typify different cytotypes within the same species. Several species show polymorphisms for one or more chromosomes; both acrocentric and biarmed chromosomes, which differ by a pericentric inversion or heterochromatic addition, are present within the same species and often as polymorphisms within a single individual (Ohno et al. 1966; Sparkes and Arakaki 1966; Te and Dawson 1971).
The number of chromosome rearrangements observed in Peromyscus has been shown to correlate with the extent of ecological adaptation (Arakaki et al. 1970; Bradshaw and Hsu 1972; Dixon et al. 1984; Greenbaum and Baker 1978; Loudenslager 1978; Macey and Dixon 1987; Ohno et al. 1966; Sparkes and Arakaki 1966). Pericentric inversions and heterochromatic additions result in both heterochromatic and euchromatic repatterning that would normally be deleterious to the cell, yet they are found abundantly in Peromyscus. Furthermore, the same karyotypic rearrangements involving heterochromatin repatterning have occurred multiple times within independent lineages of Peromyscus. Thus, it has been postulated that heterochromatin amplification and/or repositioning could be evolutionarily beneficial (Ohno et al. 1966; Sparkes and Arakaki 1971). Specifically, it has been previously proposed that heterochromatic repatterning (including both pericentric inversions and heterochromatic additions) may serve four hypothetical advantageous purposes by providing the following: new raw material for novel genes to emerge, spaces for safe breakpoints within euchromatic regions, the rearrangement of linkage groups, and more noncoding sequences to function in gene expression (Dixon et al. 1984). Under these models, individuals heterozygous for these repatterned chromosomes would experience an evolutionary advantage because of their genetic diversity (Sparkes and Arakaki 1971). Our data leads us to hypothesize that the homology of the satellite might allow for such beneficial rearrangements and polymorphisms to occur without gross consequence to chromosome stability. This is evidenced in the interspecific hybrid (Fig. 3), wherein we find no evidence of disruption to satellite arrays as would be predicted by studies of centromere instability in interspecific hybrids (Metcalfe et al. 2007; O’Neill et al. 1998, 2001).
A number of studies have examined the unique meiotic behavior of Peromyscus chromosomes, including those that are polymorphic within an individual. In autosomes, synapsis begins at the telomere of the q arm and proceeds towards the centromere (Greenbaum et al. 1986). While autosomal chromosomes that are homozygous in their heterochromatic patterning undergo usual synapsis, the heterochromatic blocks do not form chiasmata and, as such, do not partake in recombination—an observation first predicted to be true by Ohno et al. (1966). Similarly, the heterochromatic regions of autosomes polymorphic for pericentric inversions participate in “direct heterosynapsis” of the alleged nonhomologous chromatin without the formation of an inversion loop and thus do not participate in recombination. As a result, the polymorphic condition is nondeleterious in that the homologous chromosome pair may undergo meiosis without the lethal recombination of nonhomologous sequences and the subsequent production of unbalanced gametes, a finding that could account for the widespread karyotypic variation in Peromyscus.
From our observations, we propose a model where PMsat arrays serve as the raw material where the direct heterosynapsis between polymorphic chromosomes can occur. The satellite arrays at the centromere and pericentric region would allow for homologous synapsis to occur at these regions, regardless of the presence of pericentric inversions and/or heterochromatic additions. The same principle may be at work in hybrid or intercross Peromyscine species in which homologous chromosomes pair with each other via the alignment of the conserved satellite sequence. While the satellite itself may not be driving the speciation process, it allows for possible advantageous rearrangements to take place without mitotic or meiotic deficiencies. Thus, the karyotypic rearrangements would serve ecological and/or evolutionary purposes; as others have noted, P. maniculatus, which has the most rearrangements from the proposed primitive karyotype of Peromyscus (Fig. 2) and the highest frequency of intraspecific polymorphic chromosomes, is the most numerous, widespread, and ecologically adaptive of all species (Ohno et al. 1966; Sparkes and Arakaki 1966).
Several questions regarding satellite DNA conservation and chromosome evolution in Peromyscus remain elusive. First, what role—if any—the conserved satellite sequence may have in the negative selection against interchromosomal rearrangements, which are notably absent in Peromyscus evolution, is unknown. While the homogenous heterochromatin may be evolutionarily beneficial and allow for intrachromosomal rearrangements and polymorphic variations to exist, it is unknown what prevents the same mechanisms from causing rearrangements between nonhomologous chromosomes. Moreover, while it is possible this satellite may be conserved simply due to a lack of a diverse library of satellites present in the heterochromatin pool of the ancestor of Peromyscus, models of neutral evolution predict there would be a higher level of detectable change to satellite sequences simply due to drift, evidence of which is lacking in this genus. Thus, the sequence itself may serve a biological function and thus is actively maintained within the genome of each species. This observation is similar to that of a conserved satellite originally identified in cat genomes that was found to retain sequence conservation in divergent Bilateria genomes; however, unlike PMsat, the “frozen” satellite was not strictly retained at all centromeres across karyotypes nor in high-copy number tandem arrays in every species (Chaves et al. 2017). We propose that molecular drive alone does not account for the evolution of satellite DNA in Peromyscus and suggest that homologous arrays of satellite DNA may, either directly or indirectly, play a role in Peromyscus chromosome evolution.
Abbreviations
- AM:
-
Peromyscus aztecus hylocetes
- BP:
-
Base pair
- BW:
-
Peromyscus maniculatus bairdii
- CENP-A:
-
Centromere protein A
- CENP-B:
-
Centromere protein B
- DAPI:
-
4′,6-Diamidino-2-phenylindole dihydrochloride
- dCTP:
-
Deoxycytidine triphosphate
- dUTP:
-
Deoxyuridine triphosphate
- EDTA:
-
Ethylenediaminetetraacetic acid
- EP:
-
Peromyscus eremicus
- FISH:
-
Fluorescence in situ hybridization
- HCl:
-
Hydrochloric acid
- HOR:
-
Higher-order repeat
- IS:
-
Peromyscus californicus insignis
- LL:
-
Peromyscus leucopus
- LP:
-
Peromyscus leucocephalus-subgriseus
- Na2HPO4 :
-
Sodium orthophosphate
- NGS:
-
Next-generation sequencing
- PCR:
-
Polymerase chain reaction
- PMsat:
-
Peromyscus maniculatus satellite repeat
- PO:
-
Peromyscus polionotus subgriseus
- SDS:
-
Sodium dodecyl sulfate
- SM2:
-
Peromyscus maniculatus sonoriensis
- SSC:
-
Saline sodium citrate
- XZ:
-
Peromyscus melanophrys xenerus
References
Alkan C, Ventura M, Archidiacono N, Rocchi M, Sahinalp SC, Eichler EE (2007) Organization and evolution of primate centromeric DNA from whole-genome shotgun sequence data. PLoS Comput Biol 3:1807–1818. https://doi.org/10.1371/journal.pcbi.0030181
Arakaki DT, Sparkes RS (1967) The chromosomes of Peromyscus maniculatus hollesteri (deer mouse). Cytologia (Tokyo) 32:180–183
Arakaki DT, Veomett I, Sparkes RS (1970) Chromosome polymorphism in deer mouse siblings (Peromyscus maniculatus). Experientia 26:425–426
Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37:W202–W208. https://doi.org/10.1093/nar/gkp335
Bedford NL, Hoekstra HE (2015) Peromyscus mice as a model for studying natural variation. Elife 4. https://doi.org/10.7554/eLife.06813
Bertelsen AH, Humayun MZ, Karfopoulos SG, Rush MG (1982) Molecular characterization of small polydisperse circular deoxyribonucleic acid from an African green monkey cell line. Biochemistry 21:2076–2085
Bradley RD, Durish ND, Rogers DS, Miller JR, Engstrom MD, Kilpatrick CW (2007) Toward a molecular phylogeny for peromyscus: evidence from mitochondrial cytochrome-b sequences. J Mammal 88:1146–1159. https://doi.org/10.1644/06-MAMM-A-342R.1
Bradshaw WN, Hsu TC (1972) Chromosomes of Peromyscus (Rodentia, Cricetidae). Cytogenet Genome Res 11:436–451
Brown JD, Mitchell SE, O’Neill RJ (2012) Making a long story short: noncoding RNAs and chromosome change. Heredity 108:42–49. https://doi.org/10.1038/hdy.2011.104
Brown JD, O’Neill RJ (2014) The evolution of centromeric DNA sequences. In: eLS. John Wiley & Sons, Ltd, Chichester. https://doi.org/10.1002/9780470015902.a0020827.pub2
Brown JD, Strbuncelj M, Giardina C, O’Neill RJ (2002) Interspecific hybridization induced amplification of Mdm2 on double minutes in a Mus hybrid. Cytogenet Genome Res 98:184–188 doi:69806
Bulazel KV, Ferreri GC, Eldridge MD, O’Neill RJ (2007) Species-specific shifts in centromere sequence composition are coincident with breakpoint reuse in karyotypically divergent lineages. Genome Biol 8:R170. https://doi.org/10.1186/gb-2007-8-8-r170
Cacheux L, Ponger L, Gerbault-Seureau M, Loll F, Gey D, Richard FA, Escude C (2018) The targeted sequencing of alpha satellite DNA in Cercopithecus pogonias provides new insight into the diversity and dynamics of centromeric repeats in Old World monkeys. Genome Biol Evol 10:1837–1851. https://doi.org/10.1093/gbe/evy109
Chaves R, Adega F, Heslop-Harrison JS, Guedes-Pinto H, Wienberg J (2003) Complex satellite DNA reshuffling in the polymorphic t(1;29) Robertsonian translocation and evolutionarily derived chromosomes in cattle. Chromosome Res 11:641–648
Chaves R, Ferreira D, Mendes-da-Silva A, Meles S, Adega F (2017) FA-SAT is an old satellite DNA frozen in several Bilateria genomes. Genome Biol Evol 9:3073–3087. https://doi.org/10.1093/gbe/evx212
Chaves R, Guedes-Pinto H, Heslop-Harrison J, Schwarzacher T (2000) The species and chromosomal distribution of the centromeric alpha-satellite I sequence from sheep in the tribe Caprini and other Bovidae. Cytogenet Cell Genet 91:62–66 doi:56820
Cross JC (1938) Chromosomes of the genus Peromysus (deer mouse). Cytologia 8:408–419
Csardi G, Nepusz T (2006) The igraph software package for complex network research Inter J Complex Systems:1695
D’Aiuto L, Barsanti P, Mauro S, Cserpan I, Lanave C, Ciccarese S (1997) Physical relationship between satellite I and II DNA in centromeric regions of sheep chromosomes. Chromosome Res 5:375–381
da Silva EL, Busso AF, Parise-Maltempi PP (2013) Characterization and genome organization of a repetitive element associated with the nucleolus organizer region in Leporinus elongatus (Anostomidae: Characiformes). Cytogenet Genome Res 139:22–28. https://doi.org/10.1159/000342957
Dixon LK, Nelson BA, Priest RL (1984) Chromosome differences in Peromyscus maniculatus populations at different altitudes in Colorado. Genetica 52:63–68. https://doi.org/10.1007/bf00121815
Dover G (1982) Molecular drive: a cohesive mode of species evolution. Nature 299:111–117
Duffey PA (1972) Chromosome variation in Peromyscus: a new mechanism. Science 176:1333–1334
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Faravelli M, Moralli D, Bertoni L, Attolini C, Chernova O, Raimondi E, Giulotto E (1998) Two extended arrays of a satellite DNA sequence at the centromere and at the short-arm telomere of Chinese hamster chromosome 5. Cytogenet Cell Genet 83:281–286 doi:15171
Ferree PM, Barbash DA (2009) Species-specific heterochromatin prevents mitotic chromosome segregation to cause hybrid lethality in Drosophila. PLoS Biol 7:e1000234. https://doi.org/10.1371/journal.pbio.1000234
Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152. https://doi.org/10.1093/bioinformatics/bts565
Garrido-Ramos MA (2017) Satellite DNA: an evolving topic genes (Basel) 8:pii: E230 https://doi.org/10.3390/genes8090230
Gaubatz JW (1990) Extrachromosomal circular DNAs and genomic sequence plasticity in eukaryotic cells. Mutat Res 237:271–292
Gong Z, Wu Y, Koblizkova A, Torres GA, Wang K, Iovene M, Neumann P, Zhang W, Novak P, Buell CR, Macas J, Jiang J (2012) Repeatless and repeat-based centromeres in potato: implications for centromere evolution. Plant Cell 24:3559–3574. https://doi.org/10.1105/tpc.112.100511
Greenbaum IF, Baker RJ (1978) Determination of the primitive karyotype for Peromyscus. J Mammal 59:820–834
Greenbaum IF, Hale DW, Fuxa KP (1986) The mechanism of autosomal synapsis and the substaging of zygonema and pachynema from deer mouse spermatocytes. Chromosoma 93:203–212
Henikoff S, Ahmad K, Malik HS (2001) The centromere paradox: stable inheritance with rapidly evolving DNA. Science 293:1098–1102. https://doi.org/10.1126/science.1062939
Henikoff S, Malik HS (2002) Centromeres: selfish drivers. Nature 417:227. https://doi.org/10.1038/417227a
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. https://doi.org/10.1093/bioinformatics/btm404
Lee HR, Zhang W, Langdon T, Jin W, Yan H, Cheng Z, Jiang J (2005) Chromatin immunoprecipitation cloning reveals rapid evolutionary patterns of centromeric DNA in Oryza species. Proc Natl Acad Sci U S A 102:11793–11798. https://doi.org/10.1073/pnas.0503863102
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM arXiv:13033997v2
Loudenslager EJ (1978) Variation in the genetic structure of Peromyscus populations. I. Genetic heterozygosity—its relationship to adaptive divergence. Biochem Genet 16:1165–1179
Louzada S, Vieira-da-Silva A, Mendes-da-Silva A, Kubickova S, Rubes J, Adega F, Chaves R (2015) A novel satellite DNA sequence in the Peromyscus genome (PMSat): evolution via copy number fluctuation. Mol Phylogenet Evol 92:193–203. https://doi.org/10.1016/j.ympev.2015.06.008
Macey M, Dixon LK (1987) Chromosomal variation in Peromyscus maniculatus populations along an elevational gradient. Evolution 41:676–678
Malik HS, Henikoff S (2002) Conflict begets complexity: the evolution of centromeres. Curr Opin Genet Dev 12:711–718
Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD (2012) PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics 13:31. https://doi.org/10.1186/1471-2105-13-31
Masumoto H, Masukata H, Muro Y, Nozaki N, Okazaki T (1989) A human centromere antigen (CENPB) interacts with a short specific sequence in alphoid DNA, a human centromeric satellite J Cell Biol 109:1963-1973
Masumoto H, Nakano M, Ohzeki J (2004) The role of CENP-B and alpha-satellite DNA: de novo assembly and epigenetic maintenance of human centromeres Chromosome Res 12:543-556
McNulty SM, Sullivan BA (2018) Alpha satellite DNA biology: finding function in the recesses of the genome. Chromosom Res 26:115–138. https://doi.org/10.1007/s10577-018-9582-3
Mestrovic N, Plohl M, Mravinac B, Ugarkovic D (1998) Evolution of satellite DNAs from the genus Palorus—experimental evidence for the “library” hypothesis. Mol Biol Evol 15:1062–1068
Metcalfe CJ, Bulazel KV, Ferreri GC, Schroeder-Reiter E, Wanner G, Rens W, Obergfell C, Eldridge MDB, O’Neill RJ (2007) Genomic instability within centromeres of interspecific marsupial hybrids. Genetics 177:2507–2517. https://doi.org/10.1534/genetics.107.082313
Mlynarski EE, Obergfell CJ, O’Neill MJ, O’Neill RJ (2010) Divergent patterns of breakpoint reuse in Muroid rodents. Mamm Genome 21:77–87. https://doi.org/10.1007/s00335-009-9242-1
Novak P, Neumann P, Pech J, Steinhaisl J, Macas J (2013) RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 29:792–793. https://doi.org/10.1093/bioinformatics/btt054
O’Neill RJ, Eldridge MD, Graves JA (2001) Chromosome heterozygosity and de novo chromosome rearrangements in mammalian interspecies hybrids. Mammalian Genome: official journal of the International Mammalian Genome Society 12:256–259. https://doi.org/10.1007/s003350010270
O’Neill RJ, O’Neill MJ, Graves JA (1998) Undermethylation associated with retroelement activation and chromosome remodelling in an interspecific mammalian hybrid. Nature 393:68–72. https://doi.org/10.1038/29985
Ohno S, Weiler C, Poole J, Christian L, Stenius C (1966) Autosomal polymorphism due to pericentric inversions in the deer mouse (Peromyscus maniculatus), and some evidence of somatic segregation. Chromosoma 18:177–187
Pathak S, Hsu TC, Arrighi FE (1973) Chromosomes of Peromyscus (Rodentia, Cricetidae). IV. The role of heterochromatin in karyotypic evolution. Cytogenet Cell Genet 12:315–326
Platt RN 2nd, Amman BR, Keith MS, Thompson CW, Bradley RD (2015) What is Peromyscus? Evidence from nuclear and mitochondrial DNA sequences suggests the need for a new classification. J Mammal 96:708–719. https://doi.org/10.1093/jmammal/gyv067
Plohl M, Mestrovic N, Mravinac B (2012) Satellite DNA evolution. Genome Dyn 7:126–152. https://doi.org/10.1159/000337122
Prakhongcheep O, Hirai Y, Hara T, Srikulnath K, Hirai H, Koga A (2013) Two types of alpha satellite DNA in distinct chromosomal locations in Azara’s owl monkey. DNA Research: an international journal for rapid publication of reports on genes and genomes 20:235–240. https://doi.org/10.1093/dnares/dst004
Rice P, Longden I, Bleasby A (2000) EMBOSS: the European molecular biology open software suite. Trends Genet 16:276–277
Romanenko SA, Perelman PL, Trifonov VA, Graphodatsky AS (2012) Chromosomal evolution in Rodentia. Heredity 108:4–16. https://doi.org/10.1038/hdy.2011.110
Ruiz-Ruano FJ, Lopez-Leon MD, Cabrero J, Camacho JP (2016) High-throughput analysis of the satellitome illuminates satellite DNA evolution. Sci Rep 6:28333. https://doi.org/10.1038/srep28333
Salser W, Bowen S, Browne D, el-Adli F, Fedoroff N, Fry K, Heindell H, Paddock G, Poon R, Wallace B, Whitcome P (1976) Investigation of the organization of mammalian chromosomes at the DNA sequence level. Fed Proc 35:23–35
Shi J, Wolf SE, Burke JM, Presting GG, Ross-Ibarra J, Dawe RK (2010) Widespread gene conversion in centromere cores. PLoS Biol 8:e1000327. https://doi.org/10.1371/journal.pbio.1000327
Shorter KR, Crossland JP, Webb D, Szalai G, Felder MR, Vrana PB (2012) Peromyscus as a Mammalian epigenetic model. Genet Res Int 2012:179159. https://doi.org/10.1155/2012/179159
Smith GP (1976) Evolution of repeated DNA sequences by unequal crossover. Science 191:528–535
Sparkes RS, Arakaki DT (1966) Intrasubspecific and intersubspecific chromosomal polymorphism in Peromyscus maniculatus (deer mouse). Cytogenet Genome Res 5:411–418
Sparkes RS, Arakaki DT (1971) Chromosome polymorphism in interbred subspecies of Peromyscus maniculatus (deer mouse). Can J Genet Cytol 13:277–282
Te GA, Dawson WD (1971) Chromosomal polymorphism in Peromyscus polionotus. Cytogenet Genome Res 10:225–234
Tsoumani KT, Drosopoulou E, Mavragani-Tsipidou P, Mathiopoulos KD (2013) Molecular characterization and chromosomal distribution of a species-specific transcribed centromeric satellite repeat from the olive fruit fly, Bactrocera oleae. PLoS One 8:e79393. https://doi.org/10.1371/journal.pone.0079393
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG (2012) Primer3—new capabilities and interfaces. Nucleic Acids Res 40:e115. https://doi.org/10.1093/nar/gks596
Walsh JB (1987) Persistence of tandem arrays: implications for satellite and simple-sequence DNAs. Genetics 115:553–567
Acknowledgements
The authors would like to thank Dr. Judy Brown for offering her microscopy expertise and assistance with planning FISH experiments. We also thank Brittany Brassell for her time spent examining microscopy images and Chris McCann for her help with sequencing. Funding was provided to RJO under NSF Award 1244146 and to BMS through the UConn Honors Program and the UConn Office of Undergraduate Research.
Author information
Authors and Affiliations
Contributions
BMS performed the clone sequencing, MEME analyses, and FISH experiments and wrote the manuscript. TNH wrote the script for extracting and analyzing the satellite sequences from SRA data. BLF assisted in the chromosome preparation and FISH experiments. RJO had oversight over the project, data analyses, figures, and final manuscript presentation.
Corresponding author
Additional information
Responsible Editor: Beth A. Sullivan
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Smalec, B.M., Heider, T.N., Flynn, B.L. et al. A centromere satellite concomitant with extensive karyotypic diversity across the Peromyscus genus defies predictions of molecular drive. Chromosome Res 27, 237–252 (2019). https://doi.org/10.1007/s10577-019-09605-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-019-09605-1