
Abstract
The molecular mechanisms underlying spontaneous neoplastic transformation in cultured mammalian cells remain poorly understood, confounding recognition of parallels with the biology of naturally occurring cancer. The broad use of tumorigenic canine cell lines as research tools, coupled with the accumulation of cytogenomic data from naturally occurring canine cancers, makes the domestic dog an ideal system in which to investigate these relationships. We developed a canine kidney cell line, CKB1-3T7, which allows prospective examination of the onset of spontaneous immortalization and tumorigenicity. We documented the accumulation of cytogenomic aberrations in CKB1-3T7 over 24 months in continuous culture. The majority of aberrations emerged in parallel with key phenotypic changes in cell morphology, growth kinetics, and tumor incidence and latency. Focal deletion of CDKN2A/B emerged first, preceding the onset and progression of tumorigenic potential, and progressed to a homozygous deletion across the cell population during extended culture. Interestingly, CKB1-3T7 demonstrated a tumorigenic phenotype in vivo prior to exhibiting loss of contact inhibition in vitro. We also performed the first genome-wide characterization of the canine tumorigenic cell line MDCK, which also exhibited CDKN2A/B deletion. MDCK and CKB1-3T7 cells shared several additional aberrations that we have reported previously as being highly recurrent in spontaneous canine cancers, many of which, as with CDKN2A/B deletion, are evolutionarily conserved in their human counterparts. The conservation of these molecular events across multiple species, in vitro and in vivo, despite their contrasting karyotypic architecture, is a powerful indicator of a common mechanism underlying emerging neoplastic activity. Through integrated cytogenomic and phenotypic characterization of serial passages of CKB1-3T7 from initiation to development of a tumorigenic phenotype, we present a robust and readily accessible model (to be made available through the American Type Culture Collection) of spontaneous neoplastic transformation that overcomes many of the limitations of earlier studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It has been evident since the 1940s that untreated mammalian cells propagated in culture can evolve during serial passage to express a tumorigenic phenotype, defined as the capacity to form tumors when inoculated into immunocompromised rodents (Gey 1941; Earle et al. 1943). Since these early observations, several immortalized cell lines, established from a variety of different species, have been documented to develop the ability to form tumors in vivo, spontaneously and without chemical or genetic modification (Macpherson and Stoker 1962; Defendi et al. 1963; Aaronson and Todaro 1968; Jarrett and Macpherson 1968; Contreras et al. 1985; Kraemer et al. 1987; Rubin et al. 1990; United States of America Food and Drug Administration 2005; Manohar et al. 2008; Liu et al. 2010; Onions et al. 2010; Omeir et al. 2011). Notable among these are the Madin-Darby canine kidney (MDCK) cell line (Madin and Darby 1958; United States of America Food and Drug Administration 2005; Gregersen 2008; Liu et al. 2010; Onions et al. 2010; Omeir et al. 2011; Brinster et al. 2013) and the VERO line of African green monkey kidney (AGMK) cells (Contreras et al. 1985; Manohar et al. 2008). The MDCK and VERO cell lines are used as substrates for viral vaccine production (Barrett 1980; Montagnon 1989; Medema et al. 2006; Betakova et al. 2013). The use of tumorigenic cell lines in this context has raised concerns over the potential for vaccine-mediated transfer of neoplastic activity by cellular components, such as oncogenic viruses and cellular DNA (Sheng-Fowler et al. 2009), and subcellular components such as exosomes and microRNAs (Fevrier and Raposo 2004; Lujambio and Lowe 2012). A rigorous assessment of these concerns requires a comprehensive understanding of the complex processes that govern the acquisition of a tumorigenic phenotype. Studies in vitro have revealed a multistage series of cellular events evolving from genomic and epigenetic alterations that provide growth advantages to replicating cells (Barrett 1980; Thomassen 1986; Rubin 2005; Buonanno et al. 2011). These investigations have focused heavily on individual loci of specific interest, and the few genome-wide reports have been limited in resolution, confounding the ability to correlate molecular alterations with biological behavior. Furthermore, many prior studies represent retrospective investigations of induced rather than spontaneous transformation, often in rodent cell line models, restricting opportunities for correlation of in vitro observations with in vivo data from naturally occurring tumors within the same species.
Our recent evaluation of the complex tumorigenic phenotype of MDCK cells (Omeir et al. 2011; Brinster et al. 2013) offers the potential for molecular characterization of this widely used cell line in context with the expanding catalog of genomic profiling data arising from naturally occurring tumors of domestic dogs (Breen 2009; Thomas et al. 2009, 2011, 2014; Angstadt et al. 2011; Hedan et al. 2011; Poorman et al. 2015; Shapiro et al. 2015). Unfortunately, the biological characteristics of the MDCK cell line following initiation of the original culture are not well documented (Madin and Darby 1958; Gaush et al. 1966; Leighton et al. 1969; Rindler et al. 1979; Saier 1981), and the event that resulted in transformation of MDCK cells into a continuous cell line is not recorded. Furthermore, several biologically heterogeneous derivatives of the original line are in use today (Dukes et al. 2011). These limitations, coupled with the fact that early nontumorigenic MDCK cell populations are no longer available, preclude the ability to investigate the evolution of this phenotype in a progressive manner.
We describe the development of a new canine kidney cell line, CKB1-3T7, which permits a prospective investigation of emerging neoplastic events in vitro and overcomes many of the limitations of earlier studies. We present a comprehensive cellular and molecular characterization of serial passages of the CKB1-3T7 cell line banked at regular intervals over a 2-year period of continuous culture. These cell banks encompass the spectrum of events leading from cell line initiation to immortalization and the development and progression of tumorigenic activity. We compare these data with biological and cytogenomic profiles we obtained from the MDCK cell line to identify conserved features that support the independent association of specific genomic alterations with cellular phenotype. Finally, we discuss the degree to which these observations recapitulate the profiles of genomic instability evident in naturally occurring cancers. We propose that this series of well-characterized reagents confers a robust and widely accessible system for systematically elucidating the biological intricacies of neoplastic development in mammals.
Materials and methods
Development of the CKB1-3T7 cell line
Healthy canine kidney tissue was provided by the National Institutes of Health (NIH) Division of Veterinary Services. Kidneys were obtained from a healthy 2-year-old male Beagle (Charles River Laboratories, Frederick, MD) used in an Institutional Animal Care and Use Committee (IACUC)-approved study of the effects of intra-aortic balloon counterpulsation on septic shock (Solomon et al. 2009). Histopathological evaluation of formalin-fixed tissue sections from the specimen was unremarkable. Representative fresh tissue was washed three times with phosphate-buffered saline (PBS), minced into 2–3 mm pieces, and disaggregated with 0.25 % trypsin (Mediatech, Manassas, VA) for 1 h at 37 °C with shaking. The disaggregated cells were washed three times with PBS and resuspended in 15 mL Dulbecco’s modified Eagle’s medium (DMEM, Mediatech) supplemented with 10 % fetal bovine serum (FBS, Lot ASL31024; Hyclone, Logan, UT), 2 mM l-glutamine (Mediatech) (DMEM-10), and antibiotics [penicillin 100 U/mL, streptomycin 100 μg/mL, gentamicin 50 μg/mL (Mediatech), and 10 μg/mL tetracycline (Webster Veterinary Supply, Devens, MA)]. Cells were plated into two T75 flasks (Corning Incorporated, Corning, NY) and incubated at 37 °C, 5 % CO2. Beginning at passage (p) 3, cells were cultured in DMEM-10 without antibiotics. Upon reaching confluence, approximately 4 × 105 cells/mL were cryopreserved by resuspension in 1 mL of 7.5 % dimethylsulfoxide (DMSO, Sigma Aldrich, St. Louis, MO) in DMEM-10 and stored in the vapor phase of liquid nitrogen. For cell line establishment, p3 cells (approximately 4 × 105 cells) were thawed, plated in a T25 flask (Corning) in DMEM-10, and incubated at 37 °C, 5 % CO2. The medium was replaced every 3–4 days for 6 weeks by which time the cells were confluent. By p9, cell growth was such that cells plated at 3 × 105 cells/T25 flask were consistently 95–98 % confluent after 7 days in culture, making it possible to initiate serial passaging.
For serial passage (p10–p90), at 7-day intervals, cells from two T25 flasks were washed with PBS-EDTA (Teknova, Hollister, CA), detached by incubation in 0.25 % trypsin (Mediatech) and 0.53 mM EDTA (Quality Biological Inc., Gaithersburg, MD), and pooled. The number of cells was determined using a Cellometer (Nexcelom Bioscience, Lawrence, MA), and the cell suspension was used to seed T25 flasks with 3 × 105 cells (1.2 × 104 cells/cm2) in DMEM-10 for the next passage. During serial passage, cell monolayers, cell morphology, and cell-sheet confluence were monitored daily. Based on the origin of the tissue and the method of culture (Todaro and Green 1963), the cell line was designated CKB1-3T7: canine kidney Beagle1 [CKB1]—3 × 105 cells, transferred [T] every 7 days. The same lot of FBS was used throughout the establishment and propagation of CKB1-3T7 cells. The cell line tested negative for 31 rodent agents (26 viruses and mycoplasma species/pulmonis; IDEXX RADIL, Columbia, MO). Cells banked every ten passages from p10 to p90 will be available from the American Type Culture Collection (ATCC, Manassas, VA).
Cell growth characteristics
The doubling time (DT) of CKB1-3T7 cells in hours was determined at each passage. Every 7 days, cells from two of the T25 flasks plated at 3 × 105 cells/flask for passage were harvested. The number of cells plated at passage initiation and the number of cells after 7 days (168 h) in culture were used to determine the doubling time using the formula: 1 / [((log B − log A) × 3.32) / 168], where A = initial number of cells plated, B = final number of cells in the flask, and 3.32 represents 1/log2. Population doubling levels (PDL) were determined by the formula: PDL = PDLi + 3.32 × (total viable cells at harvest/total viable cells at plating), where PDLi = the PDL of the previous passage.
To evaluate the cell growth characteristics at ten-passage intervals, a 95–98 % confluent monolayer of CKB1-3T7 cells from a T150 flask seeded at 1.2 × 104 cells/cm2 was split into 30 T25 flasks at 3 × 105 cells/flask and the medium replaced every 3–4 days for 20 days (480 h) to 28 days (672 h). Every 2–3 days, cells in two T25 flasks were trypsinized and pooled, and the average number of cells/flask was determined. Assays were performed in duplicate, and the values for the average numbers of cells were used to graph population growth over time.
Cell migration and tumorigenicity assays
Wound-healing assays were undertaken to evaluate potential changes in the migration phenotype of CKB1-3T7 cells at different passage levels. One million cells were plated in triplicate in 60-mm-diameter culture dishes. When cultures reached 90 % confluence, cells were serum starved for 8 h, and the monolayers were wounded with a P200 pipette tip, washed with PBS, and cultured in DMEM-10. Phase-contrast images of cell migration into the wounded area were photographed at 0, 12, 24, and 36 h using an Olympus IX51 microscope with a DP72 camera and a ×20 objective. Cell migration (% of wound closure) was determined at 36 h by the formula [(initial wound size − wound size at time of measurement) / initial wound size] × 100.
Tumorigenicity studies were performed as described previously (Omeir et al. 2011) in both newborn and adult mice due to the difference in sensitivity (newborn mice being more sensitive) to tumor formation. Briefly, newborn (<72 h old) and adult (4–6 weeks old) athymic nude mice (Frederick Cancer Research Facility, National Cancer Institute, NIH) were inoculated subcutaneously in the dorsal region of the thorax above the scapulae with 107 cells in 0.1 mL PBS per mouse. The animals were examined weekly for 12 months for the presence and progression of tumors. Progressive tumor growth was determined by two-dimensional measurements at weekly intervals using a VWR Digital Caliper (VWR International, Radnor, PA). Tumor incidence data, represented by the percent of tumor-free animals, were plotted as Kaplan-Meier survival curves. Mice were euthanized when tumors reached approximately 20 mm in any dimension. All institutional and national guidelines for the care and use of laboratory animals were followed, and the protocols for these assays were approved by the IACUC of the Center for Biologics Evaluation and Research.
Preparation of selected cell line passages for cytogenomic analysis
Cytogenomic analysis of CKB1-3T7 cells was performed initially at p7, p15, and p22 and subsequently at intervals of approximately ten passages (p32, p43, p52, p62, p73, and p92), representing a period of continual propagation over 24 months. Duplicate flasks for each selected passage were allowed to approach confluence, the cells were rinsed with Hanks’ balanced salt solution (Mediatech), disaggregated with 0.05 % trypsin/EDTA (Mediatech), pooled, and split equally into three new T75 flasks in 25 mL of DMEM-10 per flask. Cultures were propagated until approaching confluence, at which time the cells from one flask were cryopreserved in 10 % DMSO/90 % FBS (Mediatech). Cells from the two remaining flasks were arrested at metaphase by exposure to 50 ng/mL Karyomax (Gibco/Life Technologies, Grand Island, NY) for 16 h (flask 1) and 100 ng/mL Karyomax for 4 h (flask 2). Cells from both flasks were recovered by trypsin-EDTA treatment and pooled. The combined cell number was determined using a Cellometer and divided into two equivalent aliquots. The first aliquot was harvested with conventional hypotonic treatment and 3:1 methanol/glacial acetic acid fixation. Metaphase chromosome preparations were dropped onto clean, uncharged glass microscope slides; cured for 3–5 days at room temperature; dehydrated through 70, 90, and 100 % ethanol baths; and stored at −80 °C until required. The second aliquot of cells was rinsed in PBS and used for isolation of total genomic DNA (Qiagen DNeasy Blood and Tissue Kit, Qiagen, Valencia, CA).
Genomic DNA copy number profiling analysis of CKB1-3T7 cells
Oligonucleotide-array comparative genomic hybridization (oaCGH) analysis was performed as described previously (Poorman et al. 2015; Thomas et al. 2014; Shapiro et al. 2015), using a custom 1,000,000-feature microarray (Agilent Technologies, Santa Clara, CA). The array comprises repeat-masked ∼60-mer oligonucleotides distributed at approximately 2.4 kb intervals throughout the domestic dog genome sequence assembly (Lindblad-Toh et al. 2005). The genome-wide DNA copy number profile of each of the nine serial cell line passages (“test” samples) was evaluated in a series of pairwise hybridizations against a common reference sample comprising equimolar quantities of blood-derived DNA acquired from five, young (<3 years), clinically healthy, male Beagles during routine wellness examinations. Array image data were processed using Feature Extraction version 10.10 and Genomic Workbench version 7 (Agilent Technologies). Raw data were filtered to exclude probes exhibiting nonuniform hybridization or signal saturation. Nonrandom, recurrent DNA copy number aberrations (CNAs) were defined using the ADM2 segmentation algorithm in Genomic Workbench with a threshold of six, based on a minimum of three consecutive probes with log2 test:reference values ≥0.2 (copy number gain) or ≤−0.2 (copy number loss), resulting in an effective resolution of ∼4.8 kb (two intervals of ∼2.4 kb). High-amplitude gains and losses were defined using default log2 test:reference values of >1.14 and <−1.1, respectively. Genes and uncharacterized coding sequences within recurrent CNAs were defined using the UCSC canine genome sequence browser (http://genome.ucsc.edu/). Discrete genomic regions are herein denoted according to their cytogenetic location and their megabase (Mb) coordinates within the dog genome sequence assembly (Lindblad-Toh et al. 2005). Dog chromosome Y (Canis familiaris (CFA) Y) was excluded from analysis due to the absence of this chromosome in the female dog genome sequence assembly.
Targeted fluorescence in situ hybridization (FISH) analysis of CKB1-3T7
Key CNAs identified by oaCGH analysis were selected for targeted FISH analysis based on their emergence and/or change in amplitude during cell line propagation. FISH analysis was performed using metaphase chromosome preparations obtained from an aliquot of the same cell population of each passage from which DNA was extracted for use in oaCGH. Two types of FISH probes were used. Fluorescently labeled single-locus probes (SLPs) were generated using cytogenetically validated bacterial artificial chromosome (BAC) clones from the RPCI-81 or CHORI-82 canine BAC libraries (https://bacpac.chori.org) (Thomas et al. 2007, 2008). Whole chromosome paint probes were prepared from bivariate flow-sorted dog chromosomes (Breen et al. 1999b). All FISH probes were first hybridized to metaphase chromosome preparations from clinically healthy dogs to validate signal quality and location (Breen et al. 1999b; Thomas et al. 2008). Pools of differentially labeled FISH probes were then used to assess the range and distribution of both structural and numerical chromosome aberrations in up to 50 cells from each of the passages evaluated by oaCGH analysis.
Cytogenomic evaluation of the MDCK cell line
Cells from three different lots of parental MDCK cells (NBL-2, catalog number CCL-34) were obtained from the ATCC. The biological characteristics of these cells have been described previously (Omeir et al. 2011). Briefly, MDCK vial 1 (V1: lot no. 3563161, frozen January 30, 2004) was acquired at p55 and maintained in DMEM-10. MDCK vial 2 (V2: lot no. 4398972, frozen January 20, 2006) and vial 3 (V3: lot no. 7643577, frozen July 20, 2007) were received at p56 and p55, respectively, and were propagated using ATCC-formulated EMEM supplemented with 10 % FBS. V1 and V2 cells originated from the same token lot established at p52. V3 cells were established from a different token lot that was also established at p52. These different MDCK cell lots exhibit differing tumor-forming capacity in athymic nude mice, with log10 tumor-producing doses at 50 % endpoints [TPD50] of 5.2 (V1), 4.4 (V2), and 5.8 (V3) (Omeir et al. 2011). Cells from all three lots were evaluated by oaCGH and FISH analyses as described above.
Results
Evolution of growth rate, morphology, and tumorigenicity in CKB1-3T7 cells
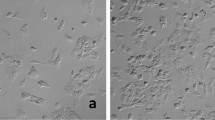
During the first 30 passages after initiation, CKB1-3T7 cells demonstrated a gradual increase in growth rate, with the DT decreasing 1.8-fold from 107.5 h at p14 to 60.3 h at p32 (Fig. 1a). During this period, the cell population exhibited a mixture of cells with epithelial and mesenchymal characteristics (SOM Fig. 1A). By p42, the DT decreased further to 37.3 h (Fig. 1a), and the morphological composition of the cell culture began to evolve, becoming predominantly cuboidal by p50 (SOM Fig. 1B). Both cell morphology and the DT subsequently remained stable for the next 20 passages (30.4 h at p62, 30.9 h at p72, and 31.7 h at p82). Loss of contact inhibition emerged by p90, with the cell sheets exhibiting multilayer areas throughout the flask (SOM Fig. 1C), resulting in a decrease in DT, an increase in cell density, and a sustained population expansion (Fig. 1a).

CKB1-3T7 cell growth characteristics and tumorigenicity. a Population growth, cell doubling times (DT), and population doubling levels (PDL) of CKB1-3T7 cells sampled at approximately ten passage intervals from cell line initiation. b Kaplan-Meier curves of tumor formation in newborn (NB) and adult (AD) athymic nude mice after inoculation with 107 CKB1-3T7 cells. The graphs plot the percent of mice that are tumor-free over time, while the numbers in parentheses indicate the tumor incidence at end of the experiment
Wound-healing and tumor formation assays were performed to assess the relationship between cell migration and tumor-forming ability in CKB1-3T7 cells. At p32 and p42, there was little or no motility observed during wound-healing assays at 36 h, but there was a progressive increase in migratory capacity from p52 (51.4 % wound closure) to p74 (71.4 % wound closure), with the wound being 100 % closed at p94 (SOM Fig. 2). From p50, CKB1-3T7 cells showed a gradual onset of tumor-forming capacity and a decrease in the interval between cellular inoculation and the appearance of a tumor (tumor latency). The incidence of tumor formation in newborn athymic nude mice increased from 1/6 (83 % tumor-free) at p51 to 13/15 (13 % tumor-free) at p90, with tumor latency falling from 26–28 weeks (p51) to 2–3 weeks (p90) (Fig. 1b). In contrast, tumor formation in adult nude mice was first observed at p60 (1/11, 91 % tumor-free), increasing to 5/11 (55 % tumor-free) at p90, with a marked decrease in tumor latency from 52–53 weeks (p60) to 2–3 weeks (p90) (Fig. 1b). Two tumors that formed in newborn athymic nude mice inoculated with p90 cells regressed.
Evolution of genomic profiles during propagation of CKB1-3T7 cells
Genome-wide DNA copy number profiling demonstrated that the evolution in cell growth, morphology, and tumor-forming potential of CKB1-3T7 cells during propagation from p7 to p92 was accompanied by a progressive accumulation of DNA copy number aberrations (Fig. 2 and SOM Fig. 3). Cells at p7 exhibited a grossly balanced DNA copy number profile in oaCGH analysis, and the number and morphology of chromosomes was consistent with that of the normal dog karyotype [2n = 78, (Breen et al. 1999a)]. Apparent regional genomic imbalances at p7, including copy number losses of CFA 9q14:20.7 Mb and CFA 19q13.1:14.4 Mb, coincided with natural DNA copy number polymorphisms that have been described in the domestic dog (Chen et al. 2009; Nicholas et al. 2009) and remained evident in subsequent passages. oaCGH analysis of cells at p15 detected deletions of CFA 27 and CFA 36 and also a low amplitude focal deletion of a 1.04 Mb interval on CFA 11q16:44.17–45.20 Mb, a region containing the full length of the MTAP, CDKN2A/B, and DMRTA1 loci. Targeted FISH analysis demonstrated hemizygous interstitial deletion of this interval on CFA 11q16 in 49 % of cells evaluated at p15 (Figs. 3 and 4). The amplitude (log2 test:reference ratio) of this deletion continued to increase progressively in p22 and p32 cells (Figs. 3 and 4), at which time genomic profiles remained otherwise consistent with that of cells at p15. At p32, FISH analysis demonstrated hemizygous deletion of CFA 11q16:44.17–45.20 Mb in 50 % of cells, with the remainder of the population exhibiting homozygous deletion. The amplitude of the aberration began to plateau at p43, at which time this region of CFA 11q16 was undetectable in 91.5 % of cells by FISH analysis (Figs. 3 and 4). oaCGH analysis of p43 cells also revealed that the copy number status of CFA 27, which was reduced to n = 1 in 24.1 % of cells at p32, had returned to a balanced state. FISH analysis using a chromosome paint probe representing CFA 27 showed that this return to n = 2 coincided with the appearance of two derivative chromosomes, each formed by fusion of one copy of CFA 27 with a second, unidentified chromosome (Fig. 4). These derivatives remained evident throughout continued propagation of CKB1-3T7. oaCGH analysis at p43 also revealed gain of CFA 13 and loss of CFA 16, which FISH analysis showed to be consistent with trisomy and monosomy, respectively (Fig. 4). No additional changes were evident at p52. At p62, gain of CFA 1 became evident along with a low amplitude gain of ∼9.6 Mb at CFA 5q34-q35:76.21–85.77 Mb, followed at p73 by gain of CFA 9 and an increase in the amplitude of gain at CFA 5q34-q35. The discrete gain of CFA 5q34-q35:76.21–85.77 Mb was evident in FISH analysis as a tandem duplication in 23 % of p73 cells evaluated (Fig. 4). By p92, this aberration was detected in only 3.1 % of cells, consistent with return to a grossly balanced state indicated by oaCGH analysis. By p92, CKB1-3T7 cells had also accumulated gain of CFA 14 and 38 and deletion of the central region of CFA 18 (CFA 18q12-q24:13.81–43.74 Mb). The amplitude of the interstitial deletion of CFA 11q16:44.17–45.20 Mb in oaCGH analysis reached a maximum at p92, at which time this region was rarely detectable in cells analyzed by FISH analysis (Figs. 3 and 4). The genomic location of all FISH probes is provided in SOM Table 1.

Integrated summary of the evolution in cellular and cytogenomic characteristics during long-term propagation of CKB1-3T7 cells. CNAs identified by oaCGH analysis are shown against the interval in which they emerged, denoted in blue (gain) or red (loss). The change in breadth of the arrow denoting the CDKN2A/B deletion reflects the progressive increase in the amplitude of this CNA in the successive passages analyzed. Overlaid on these data are key time points associated with the biological phenotype of each passage

Assessment of DNA copy number status at the CDKN2A/B locus on CFA 11q16 in CKB1-3T7 cells. The mean copy number of the region as determined by FISH analysis of serial passages of CKB1-3T7 cells is shown on the primary vertical axis to the left. The secondary vertical axis on the right indicates the corresponding test:reference signal intensity value at this locus as determined by oaCGH analysis. These values are plotted on a linear (rather than logarithmic scale) to aid comparison with FISH data for the same passage


Multicolor FISH analysis of CKB1-3T7 cells over successive passages. a FISH analysis with two five-color probe sets. SLPs were targeted to detect the central region of the chromosome or subchromosomal region of interest. Panel 1 (ai, bi, ci, and di) comprises SLPs for regions of CFA 1:62 Mb (red), CFA 9:20 Mb (magenta), CFA 9:33.5 Mb (gold), CFA 19:15 Mb (green), and CFA 27:22 Mb (aqua), and panel 2 (aii, bii, cii, and dii) comprises SLPs for regions of CFA 5:83 Mb (red), CFA 11:43.8 Mb (green), CFA 11[CDKN2A/B]:44.3 Mb (gold), CFA 13:28 Mb (magenta), and CFA 16:33 Mb (aqua). The BAC clone representing CFA 13 also contains the full length of the MYC gene (Thomas et al. 2007, 2008). The numbers in parentheses adjacent to the probe in each figure refer to the mean SLP copy number derived from counts of up to 50 cells from clinically normal control dog chromosomes (ai, aii) and from CKB1-3T7 cell line chromosomes at p43 (bi, bii), p73 (ci, cii), and p92 (di, dii). Aqua arrowheads in bi, ci, and di indicate use of a CFA 27 whole chromosome paint probe in place of the SLP to permit structural as well as numerical analysis. Pink and aqua asterisks in cii and dii indicate abnormal derivatives containing at least a segment of CFA 13 (pink asterisk) and CFA 16 (aqua asterisk). BAC addresses for clones used to generate each SLP are provided in SOM Table 1. Size bars represent 5 μm. b Graphical representation of the mean copy number of each SLP assessed across all CKB1-3T7 cell passages analyzed. The SLP at CFA 11:44.3 Mb includes the CDKN2A/B locus
Genomic profiling analysis of MDCK cells
oaCGH analysis of MDCK V1, V2, and V3 cells demonstrated relatively limited DNA copy number imbalance (Fig. 5). This was supported by observations from chromosome enumeration (V1 mean = 77, range 76–79; V2 and V3 mean = 80, range 78–82 and 78–83, respectively). Cells from all three lots showed elevated copy number along the length of CFA X relative to the male reference in oaCGH analysis (Fig. 5) and exhibited two bi-armed (submetacentric) chromosomes, both of which hybridized CFA X-specific FISH probes (Fig. 6). All other chromosomes appeared to be acrocentric. No CFA Y signal was detected in FISH analysis (Fig. 6), and each cell lot tested negative for the CFA Y-specific sex-determining region (SRY) using PCR analysis ((Bannasch et al. 2005; data not shown). The natural DNA copy number polymorphisms observed for CFA 9q14:20.7 Mb and CFA 19q13.1:14.4 Mb in CKB1-3T7 cells were also evident in cells from all three lots of MDCK cells. All three showed targeted interstitial deletion of a 60-kb region at CFA 11q16:44.23–44.29 Mb, consistent with homozygous loss of CDKN2A/B and the distal end (exons 7 and 8) of the MTAP gene, and also shared gain along the length of CFA 14 and CFA 17. In contrast to the CKB1-3T7 cell line, FISH analysis of this region in MDCK cells showed a near-balanced copy number status (n ≥ 1.96) in all cells scored (Fig. 7). This was consistent with the smaller interval of deletion around the CDKN2A/B region in MDCK cells (60 kb) versus CKB1-3T7 cells (1.04 Mb), relative to the size of the BAC clone used in FISH analysis of that region (172 kb).

DNA copy number profiles of MDCK V1, MDCK V2, and MDCK V3 cells obtained by oaCGH analysis. The horizontal axis indicates the genomic location of arrayed probes and the vertical axis indicates the log2 test:reference value at that locus for each of the three MDCK lots. Color-matched horizontal bars above and below each CGH profile delineate regions of DNA copy number gain and loss, respectively. Each MDCK profile shows an apparent copy number increase of CFA X, relative to the male reference. Data from CKB1-3T7 cells at p92 are included to aid comparison with data from MDCK cells

Multicolor FISH of MDCK cells using sex chromosome-specific SLPs. SLPs were generated using clones from the CHORI-82 canine BAC library [CFA Xp21 (317O17, gold) and Xp22.3/Yp11.3 (315K06, red)] and the RPCI-81 canine BAC library [CFA Yq11.1 (079A18, pink) and Yp11.1 (183L18, green)]. a Hybridization of each SLP to clinically healthy male dog control chromosomes. The inset shows enlarged and correctly oriented CFA X and CFA Y from this metaphase chromosome spread with each SLP signal identified by the corresponding BAC clone address. Clone 315K06 hybridized to both CFA X and Y, consistent with its location within the canine pseudoautosomal region. The same SLP panel was hybridized to b MDCK V1 cells, c MDCK V2 cells, and d MDCK V3 cells. Size bars represent 5 μm

Targeted FISH analysis of MDCK cells. Metaphase chromosome preparations from a a clinically healthy control dog, b MDCK V1 cells, c MDCK V2 cells, and d MDCK V3 cells were assessed using two SLP panels targeting loci on CFA 11, 21, and 24 (panel i) and CFA 8, 13, 14, 17, and 38 (panel ii). For each panel, the genomic location of the center of the BAC clone used is indicated in the control cells (ai and aii). The numbers in parentheses adjacent to each chromosome ID are the mean copy number of that probe obtained from enumeration of SLP signals in up to 50 cells. BAC addresses for clones used to generate each SLP are provided in SOM Table 1
Other DNA copy number aberrations were evident in oaCGH profiles of only one or two of the MDCK lots. V2 and V3 cells shared common gains of CFA 8, CFA 13, and CFA 38. V3 cells showed gain of CFA 35, which was also evident at low amplitude in V2 cells but fell outside the threshold required for classification as aberrant in oaCGH. V1 cells showed several aberrations that were not evident in the other two cell lots, including partial losses of CFA 4q33-qtel:73.67–91.48 Mb, CFA 21q24.2-qtel:44.37–53.93 Mb, and CFA 24q11.1-q24:3.06–39.45 Mb and a partial gain at CFA 9q22.3:36.29–41.71 Mb. Data from oaCGH were supported by FISH analysis on all three lots of MDCK cells, the results of which are summarized in Fig. 7.
Discussion
The CKB1-3T7 cell line offers a unique tool for the in vitro evaluation of spontaneous neoplastic activity. The generation of cell banks from CKB1-3T7 at regular intervals permits characterization of the progressive evolution of biological and genomic events involved in the pathway from initiation to immortalization and the acquisition of a tumorigenic phenotype. We propose that this series of well-characterized reagents also fills a critical niche in our ability to comprehend the fundamental requirements for expression of a tumorigenic phenotype in vivo. The CKB1-3T7 cell line exhibited several key biological properties that support its use in this context, including a gradual decrease in doubling time accompanied by an increase in growth rate (Fig. 1a), loss of contact inhibition (SOM Fig. 1C), and a gradual increase in tumorigenicity (Fig. 1b). In addition, wound-healing assays revealed a progressive increase in the cell motility of CKB1-3T7 cells over continued passage (SOM Fig. 2). We have shown a similar association between cell migration and tumor-forming ability in the VERO line of AGMK cells (Teferedegne et al. 2010, 2014).
Although key activities associated with the development of a neoplastic phenotype were exhibited by CKB1-3T7 cells, there was a deviation from the generally accepted order of events. For example, there was a lack of association between loss of contact inhibition and tumor formation in CKB1-3T7 cells. It is generally considered that cells need an unrestricted capacity to divide in order to form tumors in vivo. CKB1-3T7 cells, however, expressed the capacity to form tumors in newborn athymic nude mice at p51, and in both newborns and adults at p81, passages when their growth remained inhibited by cell-to-cell contact in culture. From p52 to p82, population DT decreased by 26 % from 42.7 to 31.7 h. At p90, when loss of contact inhibition occurred, the DT declined further only modestly to 28.1 h. We are not aware of prior studies that assessed the effects of DT and contact inhibition on the tumor-forming ability of cells immortalized during spontaneous neoplastic development in vitro. There are two possible explanations for the apparent lack of correlation between the loss of contact inhibition and the acquisition of a tumorigenic phenotype. First, while the presence of a small number of the 107 cells comprising the inoculum is likely sufficient to form a tumor in vivo, loss of contact inhibition in vitro requires that most of the cells have acquired this property. Second, it is not known whether cells that are contact inhibited in vitro are also contact inhibited in vivo, where factors in the microenvironment might allow cells to overcome such inhibition.
Perhaps the most dramatic property exhibited by sequential passages of CKB1-3T7 cells was the progressive reduction in tumor latency, falling from 28 weeks (p51) to 2 weeks (p90) in newborn nude mice and from 53 weeks (p60) to 2 weeks (p90) in adult nude mice (Fig. 1b). From p62 to p92, the DT decreased from 30.4 to 28.1 h (Fig. 1a), which seems disproportionately small compared with the marked reduction in tumor latency, suggesting that these events may not be related directly. Understanding the relationship between DT, tumor incidence, tumor latency, and the loss of contact inhibition requires, in part, knowledge of the genomic alterations occurring in concert with these phenotypic changes. A limited number of studies have attempted to identify genomic alterations that correlate with the spontaneous emergence of immortalization and tumorigenicity in vitro. Certain discrete CNAs have been shown to be conserved between independent clones developed from a common parental cell line, suggestive of a nonrandom etiology [for example (Ray et al. 1986)], but no comprehensive genome-wide studies exist. Despite long-term propagation in culture, the majority of CNAs evident in CKB1-3T7 cells (as late as p92), and also in MDCK cells (beyond p55), were whole chromosome aneuploidies rather than focal imbalances of individual genes or discrete subchromosomal regions (Fig. 5). This suggests that the development of a tumorigenic phenotype may occur within a background of limited genomic instability, at least in terms of DNA copy number. Detailed characterization of structural, mutational, transcriptional, and epigenetic alterations in CKB1-3T7 cells will be required to reveal the relative contribution of these factors in the acquisition of a tumorigenic phenotype.
Among the small number of targeted genomic imbalances detected in CKB1-3T7 cells was focal deletion of a 1.04-Mb region at CFA 11q16:44.17–45.20 Mb, which encodes the MTAP, CDKN2A/B, and DMRTA1 loci. Deletion of CFA 11q16:44.17–45.20 Mb was one of the earliest detectable CNAs, evident first at p15 as a hemizygous deletion in a proportion of the cell population, and increasing dramatically in penetrance over the next 30 passages before plateauing from p43 onwards as a homozygous deletion (Fig. 3). The progressive loss of this locus during sequential passage in vitro paralleled the progressive decline in the DT and an increase in growth rate of CKB1-3T7 cells (Figs. 1a and 2). The genomic boundaries of this deletion remained stable from initial onset at p15 to apparent homozygous deletion by p92, representing an interstitial deletion within the midregion of CFA 11, with the remainder of the chromosome remaining grossly intact. Each of the three MDCK lots also exhibited homozygous deletion of CDKN2A/B, spanning a markedly smaller interval of 60 kb at CFA 11q16:44.23–44.29 Mb that lay below the limits of detection by BAC-based SLP analysis. CDKN2A/B represents a cyclin-dependent kinase inhibitor that binds to cyclin-dependent kinases (CDK4, CDK6) and halts the cell cycle in the G1 phase, thus arresting cell proliferation. Focal deletions involving the MTAP and CDKN2A/B loci on CFA 11q16 are among the most highly recurrent somatic CNAs in a broad range of naturally occurring canine cancers, including appendicular osteosarcoma, histiocytic sarcoma, and non-Hodgkin’s lymphoma (Angstadt et al. 2011; Hedan et al. 2011; Thomas et al. 2011; Karlsson et al. 2013). This region also harbors germline risk factors for both osteosarcoma and histiocytic sarcoma in dogs (Shearin et al. 2012; Karlsson et al. 2013). This association with both somatic and germline aberrations in spontaneous canine cancer, together with the temporal relationship between copy number loss and tumor-forming capacity in both CKB1-3T7 cells and MDCK cells, emphasizes the fundamental role of this locus as a driver of neoplastic activity both in vivo and in vitro. Interstitial deletion of human chromosome 9p21, resulting in loss of CDKN2A/B, is also widely regarded to be a critical event in the development of numerous diverse human cancers (for example, Kohno and Yokota 2006; Schiffman et al. 2010; Sarhadi et al. 2013; LaPak and Burd 2014; Su et al. 2014). Furthermore, we (unpublished observations) and others (Osada et al. 2014) have recently identified the same aberration in both novel and established cell lines of the African green monkey. The conservation of this molecular event in three species not only points to the significance of these genes in cancer development but might also offer new opportunities to elucidate mechanisms of double-strand breakage and rejoining involved in tumorigenesis through a comparative structural analysis of chromatin architecture.
The onset of the focal deletion of CFA 11q16:44.17–45.20 Mb at p15 in CKB1-3T7 cells was accompanied by monosomies of CFA 27 and CFA 36. Aside from the progressively increasing amplitude of the CFA 11q16 deletion, and progressive loss of CFA 27, the overall genome-wide DNA copy number profiles of CKB1-3T7 cells remained stable during the interval between p15 and p32. By p43, several changes to the CNA profiles became evident, which were verified by targeted FISH analysis (Fig. 4). Interestingly, however, the single-copy loss of CFA 27 was no longer evident from p43 onwards. This return to a balanced state was suggestive of an in vitro form of uniparental disomy (UPD) through replication of the one remaining copy of CFA 27. It is tempting to consider that this “monosomy rescue” event may be causally related to the subsequent evolution of a neoplastic phenotype in CKB1-3T7 cells during propagation to p52 through the unmasking of cancer-associated mutations via copy number neutral loss of heterozygosity. The incidence of UPD and the relationship between UPD and cancer remain enigmatic. This is due in large part to the historical challenges associated with its detection, particularly where only a single snapshot of genomic status in vivo is available for evaluation. To our knowledge, there have been no descriptions of this phenomenon in the dog. The series of sequential passages of the CKB1-3T7 cell line thus offers an opportunity to monitor the onset and consequences of UPD in an in vitro system.
CKB1-3T7 cells exhibited a second transient CNA in the form of a targeted gain of 9.6 Mb at CFA 5q34-q35:76.21–85.77 Mb, which manifested as a duplication on one copy of CFA 5 (Fig. 4a). This aberration was evident in 9 % of cells at p62 and persisted in 13 % of cells through p73 before returning to a balanced state by p92 (Fig. 4a). The proximal boundary of this region lies within the canine ortholog of the WWOX gene. WWOX (WW domain containing oxidoreductase) spans one of the two most active fragile sites in the human genome (FRA16D, human chromosome 16q23) and is disrupted frequently in several human cancers (Drusco et al. 2011). There was no evidence for CNA surrounding the canine FHIT gene on CFA 20q14, which lies within the other highly active fragile site in the human genome. FISH analysis revealed that the frequency of the hemizygous duplication at CFA 5q34-q35 fell to a level that was undetectable by CGH analysis at p92. It is possible that this was due to the emergence of an abnormal subclone of CKB1-3T7 that was subsequently diluted out over continued culture. Alternatively, this may represent a second example of apparent UPD, in which the aberrant copy of CFA 5 present at p62 and p73 was lost during culture to p92, perhaps due to some deleterious effect, and the remaining, grossly normal CFA 5 duplicated to result in a return to balanced status. As with the transient loss of CFA 27, there is, however, no clear explanation for this observation.
Gain of CFA 13, which was first detected in CKB1-3T7 cells at p43, was also evident in MDCK V2 and V3 (Fig. 5). In common with deletion of CDKN2A/B, CFA 13 gain is among the most highly recurrent CNAs across the spectrum of spontaneously established tumor cell lines and naturally occurring canine cancers, including appendicular osteosarcoma, histiocytic sarcoma, glioma, non-Hodgkin’s lymphoma, mucosal melanoma, and urothelial carcinoma (Thomas et al. 2009, 2011; Angstadt et al. 2011; Hedan et al. 2011; Seiser et al. 2013; Poorman et al. 2015; Shapiro et al. 2015). Interestingly, CFA 13 gain was also among the first CNAs to evolve in a panel of induced pluripotent stem (iPS) cells generated from normal adult canine fibroblasts, which developed low-level aneuploidy during prolonged culture in conjunction with the ability to produce solid tumors in mice (Koh et al. 2013). The CFA 13 gain therefore appears to be an early event in the genomic evolution of spontaneous tumorigenesis both in vitro and in vivo. CFA 13 harbors several key cancer genes, most notably the c-MYC and c-KIT proto-oncogenes; however, CFA 13 gain manifests primarily as a whole chromosome aneuploidy, in contrast to the recurrent focal deletion of CFA 11q16 (Thomas et al. 2009, 2011; Angstadt et al. 2011; Hedan et al. 2011; Seiser et al. 2013; Poorman et al. 2015; Shapiro et al. 2015). In combination, this suggests that CFA 13 gain may be a secondary event that is more closely related to the architecture of the canine karyotype rather than being fundamental to the onset of a tumorigenic phenotype. The same may apply to loss of CFA 16, which, like CFA 13 gain, was first detected in CKB1-3T7 cells at p43. In common with CFA 13 gain, loss of CFA 16 is also common to several spontaneous canine cancers, particularly soft tissue sarcomas, including appendicular osteosarcoma, hemangiosarcoma, histiocytic sarcoma, and non-Hodgkin’s lymphoma (Angstadt et al. 2011; Hedan et al. 2011; Thomas et al. 2011, 2014; Karlsson et al. 2013). This CNA was not, however, detected in any of the MDCK variants. Conversely, the CKB1-3T7 cell line and all three MDCK variants accumulated gain of CFA 14 (Fig. 5). This aberration became evident in CKB1-3T7 cells only at p92, several passages after the acquisition of a tumorigenic phenotype and once cells had lost contact inhibition and tumor latency fell to 2–3 weeks (SOM Fig. 1B, C). Gain of CFA 14 may therefore represent a secondary event associated with prolonged culture and increasingly dysregulated cell proliferation. This may apply equally to those additional CNAs (gain of CFA 1, CFA 9, and CFA 38 and loss of CFA 18) that occurred in CKB1-3T7 cells only after prolonged culture beyond p62 (Fig. 2).
Despite the extensive use of MDCK cells as a research tool over the past five decades, to our knowledge, this report represents the first genome-wide molecular cytogenetic characterization of this cell line. Our findings concur with prior reports (Cassio 2013) indicating that the total chromosome number and morphology of MDCK cells has remained close to that of the normal dog karyotype (2n = 78), with acrocentric autosomes and submetacentric sex chromosomes (Breen et al. 1999a). Our findings do, however, contradict prior reports suggesting the presence of a Y chromosome and/or origin from a male donor [for example, (Gaush et al. 1966; Noma et al. 1998)]. These contrasting observations in part reflect the challenge of accurate dog chromosome identification using conventional banding analysis. It is, however, becoming increasingly apparent that inconsistencies between studies involving MDCK cells may arise due to the use of different variants of this cell line. Several derivatives of the original MDCK line are in use today, with extensive heterogeneity evident in their cellular morphology and biological behavior (Dukes et al. 2011; Omeir et al. 2011). Furthermore, MDCK cells exhibit additional genomic and phenotypic “drift” during continued propagation (Dukes et al. 2011; Cassio 2013). Provenance is a particular concern when cell lines are sourced from individual laboratories rather than from centralized repositories, and resulting data frequently are reported without detailed description of their origin, culturing conditions, and number of population doublings since initiation. The present study offers a panel of widely accessible and biologically characterized reagents that can serve as a baseline reference for standardization and integration of future studies utilizing the CKB1-3T7 cell line.
The mechanistic association between genomic instability and cancer in humans has been explored in depth (Thompson and Compton 2011), and it is increasingly evident that seemingly limited defects can have substantial consequences. For example, the renowned Philadelphia chromosome rearrangement in chronic myelogenous leukemia is sufficient to drive the disease (Chandra et al. 2011; Frankfurt and Licht 2013), and there is evidence that artificial induction of a single chromosome aberration may drive the onset of in vitro neoplastic transformation in vertebrate cells (Gascoigne and Cheeseman 2013). Our understanding of these dynamic processes remains incomplete, however, particularly in the definition of cause versus effect and the temporal basis of these events. We have shown previously that chromosome aberrations in naturally occurring canine cancers parallel those of their human counterparts (Breen and Modiano 2008; Thomas et al. 2009, 2011, 2014; Angstadt et al. 2011; Karlsson et al. 2013; Poorman et al. 2015; Shapiro et al. 2015) and also coincide with genomic rearrangements that arise during speciation (Becker et al. 2011). The present study highlights a subset of chromosome aberrations that are also common to spontaneous neoplastic development in vitro and reveals their temporal relationships in context with the evolution of biological behavior. Continued assessment of genomic characteristics that are shared between in vitro models and naturally occurring disease across genetically distinct species should aid in the elucidation of driving factors that are fundamentally involved in the onset of these phenotypes. The CKB1-3T7 cell line offers a systematic tool for evaluating safety concerns associated with vaccines developed in immortalized cells and for identification of novel targets for effective modulation of tumorigenesis in spontaneous cancers.
Abbreviations
- AGMK:
-
African green monkey kidney
- ATCC:
-
American Type Culture Collection
- BAC:
-
Bacterial artificial chromosome
- CFA:
-
Canis familiaris
- CNA:
-
Copy number aberration(s)
- CDKN:
-
Cyclin-dependent kinase inhibitor
- CHORI:
-
Children’s Hospital Oakland Research Institute
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- DT:
-
Doubling time
- FBS:
-
Fetal bovine serum
- FISH:
-
Fluorescence in situ hybridization
- IACUC:
-
Institutional Animal Care and Use Committee
- iPS cells:
-
Induced pluripotent stem cells
- MDCK:
-
Madin-Darby canine kidney
- NIH:
-
National Institutes of Health
- oaCGH:
-
Oligonucleotide-array comparative genomic hybridization
- p:
-
Passage
- PAR:
-
Pseudoautosomal region
- PBS:
-
Phosphate-buffered saline
- PDL:
-
Population doubling level
- RPCI:
-
Roswell Park Cancer Institute
- SLP:
-
Single-locus probe
- UCSC:
-
University of California Santa Cruz
- UPD:
-
Uniparental disomy
References
Aaronson SA, Todaro GJ (1968) Basis for the acquisition of malignant potential by mouse cells cultivated in vitro. Science 162:1024–1026
Angstadt AY, Motsinger-Reif A, Thomas R et al (2011) Characterization of canine osteosarcoma by array comparative genomic hybridization and RT-qPCR: signatures of genomic imbalance in canine osteosarcoma parallel the human counterpart. Genes, Chromosomes Cancer 50:859–874
Bannasch DL, Bannasch MJ, Ryun JR, Famula TR, Pedersen NC (2005) Y chromosome haplotype analysis in purebred dogs. Mamm Genome 16:273–280
Barrett JC (1980) A preneoplastic stage in the spontaneous neoplastic transformation of Syrian hamster embryo cells in culture. Cancer Res 40:91–94
Becker SE, Thomas R, Trifonov VA et al (2011) Anchoring the dog to its relatives reveals new evolutionary breakpoints across 11 species of the Canidae and provides new clues for the role of B chromosomes. Chromosome Res 19:685–708
Betakova T, Svetlikova D, Gocnik M (2013) Overview of measles and mumps vaccine: origin, present, and future of vaccine production. Acta Virol 57:91–96
Breen M (2009) Update on genomics in veterinary oncology. Top Companion Anim Med 24:113–121
Breen M, Modiano JF (2008) Evolutionarily conserved cytogenetic changes in hematological malignancies of dogs and humans—man and his best friend share more than companionship. Chromosome Res 16:145–154
Breen M, Bullerdiek J, Langford CF (1999a) The DAPI banded karyotype of the domestic dog (Canis familiaris) generated using chromosome-specific paint probes. Chromosome Res 7:401–406
Breen M, Langford CF, Carter NP et al (1999b) Fish mapping and identification of canine chromosomes. J Hered 90:27–30
Brinster LR, Omeir RL, Foseh GS et al (2013) Failure-to-thrive syndrome associated with tumor formation by Madin-Darby canine kidney cells in newborn nude mice. Comp Med 63:323–330
Buonanno M, De Toledo SM, Azzam EI (2011) Increased frequency of spontaneous neoplastic transformation in progeny of bystander cells from cultures exposed to densely ionizing radiation. PLoS One 6, e21540
Cassio D (2013) Long term culture of MDCK strains alters chromosome content. BMC Res Notes 6:162
Chandra HS, Heisterkamp NC, Hungerford A et al (2011) Philadelphia Chromosome Symposium: commemoration of the 50th anniversary of the discovery of the Ph chromosome. Cancer Genet 204:171–179
Chen WK, Swartz JD, Rush LJ, Alvarez CE (2009) Mapping DNA structural variation in dogs. Genome Res 19:500–509
Contreras G, Bather R, Furesz J, Becker BC (1985) Activation of metastatic potential in African green monkey kidney cell lines by prolonged in vitro culture. In Vitro Cell Dev Biol 21:649–652
Defendi V, Lehman J, Kraemer P (1963) “Morphologically normal” hamster cells with malignant properties. Virology 19:592–598
Drusco A, Pekarsky Y, Costinean S et al (2011) Common fragile site tumor suppressor genes and corresponding mouse models of cancer. J Biomed Biotechnol 2011:984505
Dukes JD, Whitley P, Chalmers AD (2011) The MDCK variety pack: choosing the right strain. BMC Cell Biol 12:43
Earle WR, Nettleship A, Schilling EL et al (1943) Production of malignancy in vitro V. Results of injections of cultures into mice. J Natl Cancer Inst 4:213–227
Fevrier B, Raposo G (2004) Exosomes: endosomal-derived vesicles shipping extracellular messages. Curr Opin Cell Biol 16:415–421
Frankfurt O, Licht JD (2013) Ponatinib—a step forward in overcoming resistance in chronic myeloid leukemia. Clin Cancer Res 19:5828–5834
Gascoigne KE, Cheeseman IM (2013) Induced dicentric chromosome formation promotes genomic rearrangements and tumorigenesis. Chromosome Res 21:407–418
Gaush CR, Hard WL, Smith TF (1966) Characterization of an established line of canine kidney cells (MDCK). Proc Soc Exp Biol Med 122:931–935
Gey G (1941) Cytological and cultural observations on transplantable rat sarcomata produced by the inoculation of altered normal cells maintained in continuous culture. 34th Annual Meeting of American Association for Cancer Research, 1941 Chicago, IL. American Association for Cancer, 737
Gregersen JP (2008) A risk-assessment model to rate the occurrence and relevance of adventitious agents in the production of influenza vaccines. Vaccine 26:3297–3304
Hedan B, Thomas R, Motsinger-Reif A et al (2011) Molecular cytogenetic characterization of canine histiocytic sarcoma: a spontaneous model for human histiocytic cancer identifies deletion of tumor suppressor genes and highlights influence of genetic background on tumor behavior. BMC Cancer 11:201
Jarrett O, Macpherson I (1968) The basis of the tumorigenicity of BHK 21 cells. Int J Cancer 3:654–662
Karlsson E, Sigurdsson S, Ivansson E et al (2013) Genome-wide association in three dog breeds identifies 33 osteosarcoma risk loci and implicates CDKN2A/B regulation as early driver of disease. Genome Biol 14:R132
Koh S, Thomas R, Tsai S et al (2013) Growth requirements and chromosomal instability of induced pluripotent stem cells generated from adult canine fibroblasts. Stem Cells Dev 22:951–963
Kohno T, Yokota J (2006) Molecular processes of chromosome 9p21 deletions causing inactivation of the p16 tumor suppressor gene in human cancer: deduction from structural analysis of breakpoints for deletions. DNA Repair 5:1273–1281
Kraemer PM, Ray FA, Bartholdi MF, Cram LS (1987) Spontaneous in vitro neoplastic evolution: selection of specific karyotypes in Chinese hamster cells. Cancer Genet Cytogenet 27:273–287
LaPak KM, Burd CE (2014) The molecular balancing act of p16(INK4a) in cancer and aging. Mol Cancer Res 12:167–183
Leighton J, Brada Z, Estes LW, Justh G (1969) Secretory activity and oncogenicity of a cell line (MDCK) derived from canine kidney. Science 163:472–473
Lindblad-Toh K, Wade CM, Mikkelsen TS et al (2005) Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438:803–819
Liu J, Mani S, Schwartz R, Richman L, Tabor DE (2010) Cloning and assessment of tumorigenicity and oncogenicity of a Madin-Darby canine kidney (MDCK) cell line for influenza vaccine production. Vaccine 28:1285–1293
Lujambio A, Lowe SW (2012) The microcosmos of cancer. Nature 482:347–355
Macpherson I, Stoker M (1962) Polyoma transformation of hamster cell clones—an investigation of genetic factors affecting cell competence. Virology 16:147–151
Madin SH, Darby NBJ (1958) Established kidney cell strains of normal caprine and canine origin. Technical Progress Report. Naval Biological Laboratory
Manohar M, Orrison B, Peden K, Lewis AM Jr (2008) Assessing the tumorigenic phenotype of VERO cells in adult and newborn nude mice. Biol 36:65–72
Medema JK, Meijer J, Kersten AJ, Horton R (2006) Safety assessment of Madin Darby canine kidney cells as vaccine substrate. Dev Biol (Basel) 123:243–250, discussion 265-6
Montagnon BJ (1989) Polio and rabies vaccines produced in continuous cell lines: a reality for Vero cell line. Dev Biol Stand 70:27–47
Nicholas TJ, Cheng Z, Ventura M et al (2009) The genomic architecture of segmental duplications and associated copy number variants in dogs. Genome Res 19:491–499
Noma K, Kiyotani K, Kouchi H et al (1998) Endogenous protease-dependent replication of human influenza viruses in two MDCK cell lines. Arch Virol 143:1893–1909
Omeir RL, Teferedegne B, Foseh GS et al (2011) Heterogeneity of the tumorigenic phenotype expressed by Madin-Darby canine kidney cells. Comp Med 61:243–250
Onions D, Egan W, Jarrett R, Novicki D, Gregersen JP (2010) Validation of the safety of MDCK cells as a substrate for the production of a cell-derived influenza vaccine. Biologicals 38:544–551
Osada N, Kohara A, Yamaji T et al (2014) The genome landscape of the African green monkey kidney-derived Vero cell line. DNA Res 21(6):673–683
Poorman K, Borst L, Moroff S, et al (2015) Comparative cytogenetic characterization of primary canine melanocytic lesions using array CGH and fluorescence in situ hybridization. Chromosome Res. doi:10.1007/s10577-014-9444-6
Ray FA, Bartholdi MF, Kraemer PM, Cram LS (1986) Spontaneous in vitro neoplastic evolution: recurrent chromosome changes of newly immortalized Chinese hamster cells. Cancer Genet Cytogenet 21:35–51
Rindler MJ, Chuman LM, Shaffer L, Saier MH Jr (1979) Retention of differentiated properties in an established dog kidney epithelial cell line (MDCK). J Cell Biol 81:635–648
Rubin H (2005) Degrees and kinds of selection in spontaneous neoplastic transformation: an operational analysis. Proc Natl Acad Sci U S A 102:9276–9281
Rubin AL, Arnstein P, Rubin H (1990) Physiological induction and reversal of focus formation and tumorigenicity in NIH 3T3 cells. Proc Natl Acad Sci U S A 87:10005–10009
Saier MH Jr (1981) Growth and differentiated properties of a kidney epithelial cell line (MDCK). Am J Physiol 240:C106–C109
Sarhadi VK, Lahti L, Scheinin I et al (2013) Targeted resequencing of 9p in acute lymphoblastic leukemia yields concordant results with array CGH and reveals novel genomic alterations. Genomics 102:182–188
Schiffman JD, Hodgson JG, VandenBerg SR et al (2010) Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res 70:512–519
Seiser EL, Thomas R, Richards KL et al (2013) Reading between the lines: molecular characterization of five widely used canine lymphoid tumour cell lines. Vet Comp Oncol 11:30–50
Shapiro SG, Raghunath S, Williams C, et al (2015) Canine urothelial carcinoma: genomically aberrant and comparatively relevant. Chromosome Res. doi:10.1007/s10577-015-9471-y
Shearin AL, Hedan B, Cadieu E et al (2012) The MTAP-CDKN2A locus confers susceptibility to a naturally occurring canine cancer. Cancer Epidemiol Biomark Prev 21:1019–1027
Sheng-Fowler L, Lewis AM Jr, Peden K (2009) Issues associated with residual cell-substrate DNA in viral vaccines. Biologicals 37:190–195
Solomon SB, Minneci PC, Deans KJ et al (2009) Effects of intra-aortic balloon counterpulsation in a model of septic shock. Crit Care Med 37:7–18
Su CY, Chang YC, Chan YC et al (2014) MTAP is an independent prognosis marker and the concordant loss of MTAP and p16 expression predicts short survival in non-small cell lung cancer patients. Eur J Surg Oncol 40:1143–1150
Teferedegne B, Murata H, Quinones M, Peden K, Lewis AM (2010) Patterns of microRNA expression in non-human primate cells correlate with neoplastic development in vitro. PLoS One 5, e14416
Teferedegne B, Macauley J, Foseh G et al (2014) MicroRNAs as potential biomarkers for VERO cell tumorigenicity. Vaccine 32:4799–4805
Thomas R, Duke SE, Bloom SK et al (2007) A cytogenetically characterized, genome-anchored 10-Mb BAC set and CGH array for the domestic dog. J Hered 98:474–484
Thomas R, Duke SE, Karlsson EK et al (2008) A genome assembly-integrated dog 1 Mb BAC microarray: a cytogenetic resource for canine cancer studies and comparative genomic analysis. Cytogenet Genome Res 122:110–121
Thomas R, Duke SE, Wang HJ et al (2009) ‘Putting our heads together’: insights into genomic conservation between human and canine intracranial tumors. J Neuro Oncol 94:333–349
Thomas R, Seiser EL, Motsinger-Reif A et al (2011) Refining tumor-associated aneuploidy through ‘genomic recoding’ of recurrent DNA copy number aberrations in 150 canine non-Hodgkin lymphomas. Leuk Lymphoma 52:1321–1335
Thomas R, Borst L, Rotroff D et al (2014) Genomic profiling reveals extensive heterogeneity in somatic DNA copy number aberrations of canine hemangiosarcoma. Chromosome Res 22:305–319
Thomassen DG (1986) Role of spontaneous transformation in carcinogenesis: development of preneoplastic rat tracheal epithelial cells at a constant rate. Cancer Res 46:2344–2348
Thompson SL, Compton DA (2011) Chromosomes and cancer cells. Chromosome Res 19:433–444
Todaro GJ, Green H (1963) Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol 17:299–313
United States of America Food and Drug Administration (2005) Proceedings of the FDA vaccines and related biological products advisory committee meeting. [Cited 16 November 2005]. Internet references. Retrieved from http://www.Fda.Gov/ohrms/dockets/ac/05/slides/2005-4188s1_final.Htm. 18 Nov 2014
Acknowledgments
This study was supported in part by a contract from the Division of Microbiology and Infectious Diseases of the National Institute of Allergy and Infectious Diseases through an Interagency Agreement with CBER/FDA (contract number YI-AI-4893-02NIAID), Medical Counter Measures Initiative Grant, and a CBER Pandemic Influenza Counter Measures Grant. The molecular cytogenetics work was funded in part by the NCSU Cancer Genomics Fund (MB). The CKB1-3T7 cell line was developed and its biological properties characterized in LDNAV, CBER, FDA. Cytogenomic evaluation of the CKB1-3T7 and MDCK cell lines was performed at North Carolina State University. We thank Barry Falgout, Arifa Khan, Kari Irvine, and Melissa Savage for critical review of the manuscript. All authors contributed in the development of the manuscript for publication.
Ethical standards
Experiments described in this manuscript comply with the current laws of the country in which they were performed (USA). All institutional and national guidelines for the care and use of laboratory animals were followed.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Responsible Editor: Conly Rieder
R. Omeir and R. Thomas contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
SOM Fig. 1
Morphological evolution of CKB1-3T7 cell monolayers assessed at p15 (A), p50 (B) and p90 (C). Phase-contrast micrographs were taken at 10X using a Nikon Diaphot camera (p15) and at 4X using an Olympus 1X51 microscope with a DP72 camera (p50 and p90). (PDF 10069 kb)
SOM Fig. 2
The migratory capacity (i.e., the capacity to heal a wound created by scratching a confluent monolayer) of CKB1-3T7 cells at p32, p42, p52, p74, and p94 over 36 h is shown. (PDF 996 kb)
SOM Fig. 3
Overview of CNAs identified by genome-wide oaCGH analysis of the CKB1-3T7 cell line during extended culture. Each image summarizes the distribution and amplitude of CNAs identified at one of nine timepoints (passages), indicated at the top of the image. The DNA copy number status along the length of each chromosome, relative to the reference, is shown beside the dog DAPI-banded ideogram set (Breen et al. 1999a). Genomic imbalances, defined as regions with log2 test:reference values ≥0.2 (copy number gain) or ≤−0.2 (copy number loss), are denoted by blue and red shading of the ideogram for that chromosome. Boxes of the same color highlight key CNAs emerging during the progression of the CKB1-3T7 cell line (see text for discussion). (PDF 1568 kb)
SOM Table 1
Clones from the CHORI-82 canine BAC library (https://bacpac.chori.org/library.php?id=253) used to generate single-locus FISH probes as part of the current study. Panels 3T7 i and ii were used to detect and quantify copy number status in cells from progressive passages of the cell line 3T7. Panels MDCK i and ii were used to detect and quantify copy number status in three lots of MDCK cells obtained from ATCC. (PDF 287 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Omeir, R., Thomas, R., Teferedegne, B. et al. A novel canine kidney cell line model for the evaluation of neoplastic development: karyotype evolution associated with spontaneous immortalization and tumorigenicity. Chromosome Res 23, 663–680 (2015). https://doi.org/10.1007/s10577-015-9474-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-015-9474-8