
Abstract
In the last decade, it has become clear that the neuropeptide “ghrelin” and its principal receptor have a large impact on anxiety and stress. Our recent studies have uncovered a link between plasma butyrylcholinesterase (BChE) and ghrelin. BChE actually turns out to be the key regulator of this peptide. This article reviews our recent work on manipulating ghrelin levels in mouse blood and brain by long term elevation of BChE, leading to sustained decrease of ghrelin. That effect in turn was found to reduce stress-induced aggression in group caged mice. Positive consequences were fewer bite wounds and longer survival times. No adverse effects were observed. Further exploration may pave the way for BChE-based treatment of anxiety in humans.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The present era is rife with anxiety and stress across the globe. In modern cultures, stress is pervasive and nearly inescapable. When paired with individual biological and/or psychological vulnerability, chronic stress can lead to serious mental and physical health issues that impact broad populations (Grippo and Johnson 2009; Puterman et al. 2016; Walker et al. 2014). These issues include heightened risk of emotional disorders and their progression across the lifespan (Puterman et al. 2016; Walker et al. 2014). Bolstering resilience to stress should be one of our best options for preventing and treating the significant health consequences of this epidemic. Richard Kvetnansky was a pioneer researcher in this arena for many years. At the NIH in the 1970s, using mice and rats, he provided solid evidence that repeated stress from prolonged restraint leads to chronically elevated levels of hormones in the adrenocortical axis. These hormones included norepinephrine, epinephrine, adrenocorticotropic releasing hormone (ACTH), and corticosterone (the rodent homologue of cortisol) (Gewirtz et al. 1971; Kvetnansky et al. 1995). Such effects are now well-established as having a substantial impact on physical and mental health, as repeatedly shown over the last four decades in Kvetnansky’s laboratory and those of increasingly many investigators worldwide.
A relatively new approach to prevention and treatment of anxiety disorders is based on the concept that stress-resilience is anchored in the promotion of biological resilience to stress at the cellular level (Puterman et al. 2016; Walker et al. 2014; Yamanaka et al. 2016). In part, this is achieved by enhancing metabolic functions essential to maintain adequate levels of usable energy in the brain (Bayliss and Andrews 2013; Packer et al. 1997). In other words, metabolic capacity and efficiency are emerging as keys to prevent stress-induced dysfunction in the brain circuitry that governs emotions and related phenomena (Walker et al. 2014). As yet, the specific factors that differentiate vulnerable from resilient human subjects are still far from understood. However, it is clear that, from mouse to man, individuals who are particularly vulnerable to the deleterious health effects of stress are likely to react by expressing persistent responses of “learned helplessness” (King et al. 1993). That is, passive, helpless, or pessimistic responses to stress have been shown to exacerbate and extend stressful states once they are triggered (Petty et al. 1997; Zhukov and Vinogradova 2002). In addition to impairing an individual’s ability to cope effectively with the stressor, such a behavioral response triggers a sequelae of biological events that further exacerbate the stress response and associated processes including inflammation and mitochondrial impairment that in turn impact neural circuit function, particularly in regions regulating mood and anxiety (Enkel et al. 2010; Petty and Sherman 1982). Over time, chronic stress exposure has been shown to accelerate cellular aging as demonstrated by reduced telomerase activity and telomere length. In contrast, resilient individuals demonstrate much more robust ability to cope actively with stressors at both the behavioral and the physiological levels (Oliveira et al. 2016). Recent works from our two laboratories and others suggest that cellular resilience to metabolic stress is crucial for buffering the behavioral and physiological impacts of emotional stress (Tye 2013; Tye et al. 2014). What is more, both of these phenomena are strongly affected by the peptide hormone “ghrelin,” originally regarded as just a “hunger hormone”.
Roles for Ghrelin in Emotional States
The intersection of basal metabolism with emotional balance is plainly evident in the manifold and widespread roles of ghrelin. This acylated octapeptide is produced primarily in gastric tissue and released into the blood stream, but it is also generated by specific neuronal populations in the hypothalamus and selected other brain regions (Delporte 2013; Kojima et al. 1999). Phasic increases of circulating ghrelin stimulate gastric muscle contractions colloquially referred to as “hunger pangs.” More or less simultaneously with gastric effects, ghrelin activates vagal afferent terminals leading directly from the stomach to stimulate brain areas involved in appetite and food seeking (Delhanty et al. 2013).
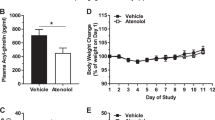
Numerous recent studies also implicate ghrelin in behaviors and states that have no obvious connection to nourishment. In particular, it has become apparent that ghrelin directly affects anxiety, stress, and fear-related behaviors by virtue of its actions on “GHSR,” the growth hormone secretagogue receptor (Harmatz et al. 2017). This fact became clear to us as we followed long-term, group-housed, sibling male mice to determine the safety of interventions for sustaining elevated plasma levels of butyrylcholinesterase (BChE) to treat cocaine abuse (Chen et al. 2015). These animals unexpectedly showed a sharp drop in spontaneous aggression. Computer searches for stress-related hormones with ester groups that BChE might hydrolyze led to a report by Chuang and Zigman (2010) focusing on the enzyme and ghrelin. We soon found that our mice with BChE overexpression had substantially lower plasma ghrelin than did the controls. Such an outcome, though focused on fighting behaviors, was plausibly related to a reduction in social anxiety that would ordinarily have promoted inter-sibling attacks (Chen et al. 2015). Continued research along those lines soon led us to conclude that BChE, long considered a nonspecific drug metabolizer, has a true physiological role in ghrelin inactivation and is, essentially, a ghrelin hydrolase. As such, BChE may serve as a useful agent for direct modulation of stress-related physiological and behavioral responses.
Given ghrelin’s multiplicity of effects it is unsurprising that there has been much confusion regarding the precise nature of the interactions with its target, the growth hormone secretagogue receptor, GHSR1a. One reason for the confusion is the unusual behavior of that receptor. Unlike many others, GHSR1a typically exist in a partially active and tonically desensitized state, even in the absence of agonist. Early on, this peculiar behavior led to confusion among researchers in the field. In particular, there was uncertainty as to whether ghrelin was a GHSR agonist or antagonist. We initially assumed that ghrelin was a stress-producer, given that untreated mice with normal plasma ghrelin were fighting much more than the BChE-treated mice with low plasma ghrelin. Abundant new evidence, from our lab and others (Chen et al. 2015; Harmatz et al. 2017; Lutter et al. 2008; Meyer et al. 2014) indicates that the opposite is true.
What appears to resolve the superficially contradictory evidence at hand is that there are two different processes involving ghrelin’s actions in the brain. Thus, ghrelin release from the stomach rises and falls in waves that coincide with increased appetite and food seeking, but the range of peptide concentrations in the blood is not great, and the rate of change is not rapid. In contrast, ghrelinergic neurons that synapse on feeding centers in brain provide rapid spikes of ghrelin to act on postsynaptic receptors. Such an arrangement means that synaptic release of ghrelin should be more effective on brain receptors in animals or humans with low levels of the circulating peptide. That is, low tonic ghrelin levels in brain should confer increased postsynaptic receptor sensitivity to transient, phasic fluxes of ghrelin in the brain. In contrast, high levels of circulating ghrelin will weaken ghrelin signaling at brain synapses, owing to reduced sensitivity or expression of the GHSR1a receptor. The implication of the impact of ghrelin tone on receptor sensitivity is of great importance given that the apparent mission of those receptors is to release growth hormone in the pituitary and other brain regions which in turn function as antianxiety, antifear, antistress, and antiaggression agents (Harmatz et al. 2017).
We serendipitously developed a model based on an AAV viral vector that reduces circulating ghrelin for the life of a mouse by driving large and sustained elevations of BChE (Schopfer et al. 2015). This approach provides an important new tool for modulating circulating ghrelin levels. Specifically, our research has shown that long-term lowering of plasma ghrelin by BChE bolsters physiologic resilience (increased health and longevity (Fig. 1) and reduces social stress-related behaviors (reduced spontaneous fighting and associated injuries (Fig. 2) and (Brimijoin et al. 2016; Chen et al. 2015). We hypothesize that altered stress reactivity is key to these observations. If so, then a stable reduction of “background ghrelin” should increase central cellular responses to ghrelin to bolster stress resilience and alleviate anxiety and depression-like behaviors. This hypothesis can easily be tested by manipulating ghrelin levels up or down with viral gene transfer or by utilizing a range of gene knockouts to reach similar outcomes. Complementing this, it also remains possible that our findings can be explained, at least in part, via an increase in the efficiency of ghrelin-mediated signaling. That is, a stable reduction of ghrelin may increase constitutive signaling through the ghrelin receptor as a direct result of an increase in membrane presence of GHSR1a, relative to when levels of ghrelin are high. Indeed, it may be that this constitutive signaling is particularly important for the antistress/antianxiety effects.

Unpublished findings from long-term daily observations of group-housed male mice (32 pooled controls and 30 mice transduced at 6 weeks with adeno-associated virus (AAV) or helper-dependent virus vector encoding mutated mouse BChE: “mBChE mut”). Plasma BChE levels in the vector-treated groups were ~100-fold above normal and were sustained for the following 2 years. Initially, mice were housed five per cage, until signs of fighting arose (4–5 months), when they moved to single-cage housing. The varied housing conditions prevent a definitive judgment of true lifespan although ANOVA showed a significant main effect of treatment (controls vs. pooled vector-treated groups): F 1.60 = 13.64, p < 0.001). No post hoc testing was performed

Bite scores in confrontations between a resident male mouse and a male intruder (Chen et al. 2015). a 3-month Balb/c with AAV-luciferase vector (n = 18) versus mBChE mutant vector-treated (n = 7); AAV-CocH-6 ΔT (human BChE E1-V529 with A199S/F227A/S287G/A328 W/Y332G/E441D)-treated mice (n = 14). b AAV-luciferase-treated 3-month C57BL/6 wild type (n = 9) versus same age C57BL/6 treated simultaneously with AAV vectors encoding cDNA for ghrelin and ghrelin octanoyl acyl transferase (GOAT) (n = 6); and same-age mice treated triply, with vectors for ghrelin, GOAT, and mBChE mutant (n = 9). *p < 0.05; ***p < 0.001; n.s. not significant
Ghrelin Signaling and Neuroprotection/Proliferation
It is already clear that ghrelin is a mediator of cellular metabolic capacity, and that it plays a key role in bolstering cellular resilience by increasing mitochondrial function and regulating cell proliferation, apoptosis and inflammation-related signaling pathways in a GHSR-1a dependent manner (Chung et al. 2007). GHSR-1a signaling may play an important role in maintaining stress resilience by protecting cellular integrity and buffering the potentially deleterious effects of chronic HPA-axis activation. We suspect that the neuroprotective effects of GHSR-1a can be enhanced by long term modulation of ghrelin tone. In our view, clarifying ghrelin’s neuroprotective mechanisms would be a good step toward developing novel prevention and treatment strategies.
Ghrelin stimulates release of growth hormone by activating GHSR1a, the growth hormone secretagogue receptor (Howard et al. 1996) and has multiple nonendocrine functions that serve to regulate neuronal behavior (Chung et al. 2013). Consequently, ghrelin signaling impacts diverse brain functions, including learning and memory (Diano et al. 2006), reward, and motivation (Naleid et al. 2005, Abizaid et al. 2006, Jiang et al. 2006), stress-responsivity and mood (Carlini et al. 2004, Lutter et al. 2008), as well as neuroprotection and neurogenesis (Jiang et al. 2006, 2008, Chung et al. 2007, 2013, Miao et al. 2007, Hwang et al. 2009, Moon et al. 2009a, b, Lee et al. 2010a, b).
Ghrelin can boost cellular resilience, increase mitochondrial function, and reduce apoptosis after ischemic insult in hippocampal, cortical or hypothalamic neurons, either in vivo or in vitro (Bali and Jaggi 2016; Bayliss and Andrews 2013, 2016; Diano et al., 2006). This occurs through direct, GHSR1a-dependent, modulation of mitogen-activated kinase (MAPK) and extracellular-signal-regulated kinase (ERK1/2) signaling (Bayliss and Andrews 2013). Ghrelin has also been shown to increase the proliferation of cultured hippocampal neural stem cells in a GHSR1a-dependent manner (Chung et al. 2013). Receptor levels rose substantially after treatment and rapid activation of MAPK, ERK1/2, and PI3 k/Akt pathways. Modulation of downstream effectors, such as glycogen synthase kinase (GSK)-3b and mammalian target of rapamycin (mTOR), were also observed. However, pretreatment with specific inhibitors of MAPK, ERK1/2, PI3 K/Akt, and mTOR attenuated ghrelin-induced cell proliferation (Chung et al. 2013).
Ghrelin Signaling and Behavioral Resilience
Chronic stress and its long-term physiological and psychological sequelae are harmful to both brain and body. The mechanism by which stress induces adaptations in the neural circuitry to impair behavioral resilience has recently been elucidated to a certain degree. The molecular pathways involved in neuroprotection and glial proliferation bolster not only resilience against damaging effects on neurons and supporting cells. They also enhance behavioral stress resilience, by facilitating ‘active coping’ responses (Duman and Voleti 2012). There is mounting evidence that ghrelin, via actions at GHSR1, plays an important role in regulating stress-responsivity—although the direction of effect remains unclear. Systemic and central administration of ghrelin has been shown to induce anxiety-like behavior in mice (Asakawa 2001; Carlini 2002) by activating the hypothalamic–pituitary–adrenal axis (Zigman et al. 2006). However, anxiolytic effects of ghrelin have also been reported. Thus, increased ghrelin levels (achieved through calorie restriction or direct peptide injection) demonstrably decrease anxiety and depressive-like behaviors in a GHSR1a-dependent fashion (Lutter 2008). The reasons for the disparate responses are unclear. In light of these conflicting data, together with development of new tools for modulating ghrelin tone, we are placing a high priority on further research into stress-related phenomena and the potential roles of BChE and ghrelin in exacerbating or reducing the impact of stress disorders. The potential to buffer stress effects at the cellular, systems, and behavioral levels now seems to have real therapeutic potential, warranting full investigation and rapid clinical translation.
References
Abizaid A, Liu Z-W, Andrews ZB, Shanabrough M, Borok E, Elsworth JD, Roth RH, Sleeman MW, Picciotto MR, Tschöp MH, Gao X-B, Horvath TL (2006) Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest 116(12):3229–3239
Asakawa A, Inui A, Kaga T, Yuzuriha H, Nagata T, Fujimiya M, Katsurra G, Makino S, Fujino MA, Kasuga M (2001) A role of ghrelin in neuroendocrine and behavioral responses to stress in mice. Neuroendocrinology 74:143–147
Bali A, Jaggi AS (2016) An integrative review on role and mechanisms of ghrelin in stress, anxiety and depression. Curr Drug Targets 17:495–507
Bayliss JA, Andrews ZB (2013) Ghrelin is neuroprotective in Parkinson’s disease: molecular mechanisms of metabolic neuroprotection. Ther Adv Endocrinol Metab 4:25–36
Bayliss JA, Andrews ZB (2016) Ghrelin is the metabolic link connecting calorie restriction to neuroprotection. Neural Regen Res 11:1228–1229
Brimijoin S, Chen VP, Pang YP, Geng L, Gao Y (2016) Physiological roles for butyrylcholinesterase: a BChE-ghrelin axis. Chem Biol Interact 259:271–275
Carlini VP, Monzón ME, Varas MM, Cragnolini AB, Schioth HB, Scimonelli TN, de Barioglio SR (2002) Ghrelin increases anxiety-like behavior and memory retention in rats. Biochem Biophys Res Commun 299:739–774
Carlini VP, Varas MM, Cragnolini AB, Schioth HB, Scimonelli TN, de Barioglio SR (2004) Differential role of the hippocampus, amygdala, and dorsal raphe nucleus in regulating feeding, memory, and anxiety-like behavioral responses to ghrelin. Biochem Biophys Res Commun 313(3):635–641
Chen VP, Gao Y, Geng L, Parks RJ, Pang YP, Brimijoin S (2015) Plasma butyrylcholinesterase regulates ghrelin to control aggression. Proc Natl Acad Sci USA 112:2251–2256
Chuang JC, Zigman JM (2010) Ghrelin’s roles in stress, mood, and anxiety regulation. Int J Pept. doi:10.1155/2010/460549
Chung H, Kim E, Lee DH, Seo S, Ju S, Lee D, Kim H, Park S (2007) Ghrelin inhibits apoptosis in hypothalamic neuronal cells during oxygen-glucose deprivation. Endocrinology 148(1):148–159
Chung H, Li E, Kim Y, Kim S, Park S (2013) Multiple signaling pathways mediate ghrelin-induced proliferation of hippocampal neural stem cells. J Endocrinol 218(1):49–59
Delhanty PJ, Huisman M, Baldeon-Rojas LY, van den Berge I, Grefhorst A, Abribat T, Leenen PJ, Themmen AP, van der Lely AJ (2013) Des-acyl ghrelin analogs prevent high-fat-diet-induced dysregulation of glucose homeostasis. Faseb J 27:1690–1700
Delporte C (2013) Structure and physiological actions of ghrelin. Scientifica 2013:518909
Diano S, Farr SA, Benoit SC, McNay EC, da Silva I, Horvath B, Gaskin FS, Nonaka N, Jaeger LB, Banks WA et al (2006) Ghrelin controls hippocampal spine synapse density and memory performance. Nat Neurosci 9:381–388
Duman RS, Voleti B (2012) Signaling pathways underlying the pathophysiology and treatment of depression: novel mechanisms for rapid-acting agents. Trends Neurosci 35:47–56
Enkel T, Spanagel R, Vollmayr B, Schneider M (2010) Stress triggers anhedonia in rats bred for learned helplessness. Behav Brain Res 209:183–186
Gewirtz GP, Kvetnansky R, Weise VK, Kopin IJ (1971) Effect of ACTH and dibutyryl cyclic AMP on catecholamine synthesizing enzymes in the adrenals of hypophysectomized rats. Nature 230:462–464
Grippo AJ, Johnson AK (2009) Stress, depression and cardiovascular dysregulation: a review of neurobiological mechanisms and the integration of research from preclinical disease models. Stress 12:1–21
Harmatz ES, Stone L, Lim SH, Lee G, McGrath A, Gisabella B, Peng X, Kosoy E, Yao J, Liu E, et al (2017) Central ghrelin resistance permits the overconsolidation of fear memory. Biol Psychiatry 81(12):1003–1013.
Howard AD, Feighner SD, Cully DF, Arena JP, Liberator PA, Rosenblum CI, Hamelin M, Hreniuk DL, Palyha OC, Anderson J et al (1996) A receptor in pituitary and hypothalamus that functions in growth hormone release. Science 273:974–977
Hwang S, Moon M, Kim S, Hwang L, Ahn KJ, Park S (2009) Neuroprotective effect of ghrelin is associated with decreased expression of prostate apoptosis response-4. Endocr J 56(4):609–617
Jiang H, Betancourt L, Smith RG (2006) Ghrelin amplifies dopamine signaling by cross talk involving formation of growth hormone secretagogue receptor/dopamine receptor subtype 1 heterodimers. Mol Endocrinol 20(8):1772–1785
Jiang H, Li L-J, Wang J, Xie J-X (2008) Ghrelin antagonizes MPTP-induced neurotoxicity to the dopaminergic neurons in mouse substantia nigra. Exp Neurol 212(2):532–537
King JA, Campbell D, Edwards E (1993) Differential development of the stress-response in congenital learned helplessness. Int J Dev Neurosci 11:435–442
Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K (1999) Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 402:656–660
Kvetnansky R, Pacak K, Fukuhara K, Viskupic E, Hiremagalur B, Nankova B, Goldstein DS, Sabban EL, Kopin IJ (1995) Sympathoadrenal system in stress. Interaction with the hypothalamic-pituitary-adrenocortical system. Ann N Y Acad Sci 771:131–158
Lee J, Lim E, Kim Y, Li E, Park S (2010a) Ghrelin attenuates kainic acid-induced neuronal cell death in the mouse hippocampus. J Endocrinol 205(3):263–270
Lee JY, Chung H, Yoo YS, Oh YJ, Oh TH, Park S, Yune TY (2010b) Inhibition of apoptotic cell death by ghrelin improves functional recovery after spinal cord injury. Endocrinology 151:3815–3826
Lutter M, Sakata I, Osborne-Lawrence S, Rovinsky SA, Anderson JG, Jung S, Birnbaum S, Yanagisawa M, Elmquist JK, Nestler EJ, Zigman JM (2008) The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat Neurosci 11:752–753
Meyer RM, Burgos-Robles A, Liu E, Correia SS, Goosens KA (2014) A ghrelin-growth hormone axis drives stress-induced vulnerability to enhanced fear. Mol Psychiatry 19:1284–1294
Miao Y, Xia Q, Hou Z, Zheng Y, Pan H, Zhu S (2007) Ghrelin protects cortical neuron against focal ischemia/reperfusion in rats. Biochem Biophys Res Commun 359(3):795–800
Moon M, Kim HG, Hwang L, Seo JH, Kim S, Hwang S, Kim S, Lee D, Chung H, Oh MS, Lee KT, Park S (2009a) Neuroprotective effect of ghrelin in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease by blocking microglial activation. Neurotox Res 15:332–347
Moon M, Kim S, Hwang L, Park S (2009b) Ghrelin regulates hippocampal neurogenesis in adult mice. Endocr J 56:525–531
Naleid AM, Grace MK, Cummings DE, Levine AS (2005) Ghrelin induces feeding in the mesolimbic reward pathway between the ventral tegmental area and the nucleus accumbens. Peptides 26(11):2274–2279
Oliveira BS, Zunzunegui V, Quinlan J, Fahmi H, Tu MT, Guerra RO (2016) Systematic review of the association between chronic social stress and telomere length: a life course perspective. Ageing Res Rev 26:37–52
Packer L, Tritschler HJ, Wessel K (1997) Neuroprotection by the metabolic antioxidant alpha-lipoic acid. Free Radic Biol Med 22:359–378
Petty F, Sherman A (1982) A neurochemical differentiation between exposure to stress and the development of learned helplessness. Drug Develop Res 2:43–45
Petty F, Kramer GL, Wu JH, Davis LL (1997) Posttraumatic stress and depressio—a neurochemical anatomy of the learned helplessness animal model. Ann NY Acad Sci 821:529–532
Puterman E, Gemmill A, Karasek D, Weir D, Adler NE, Prather AA, Epel ES (2016) Lifespan adversity and later adulthood telomere length in the nationally representative US Health and Retirement Study. Proc Natl Acad Sci USA 113:E6335–E6342
Schopfer LM, Lockridge O, Brimijoin S (2015) Pure human butyrylcholinesterase hydrolyzes octanoyl ghrelin to desacyl ghrelin. Gen Comp Endocrinol 224:61–68
Tye SJ (2013) Allostatic overload: transcriptomic insights into the molecular basis of antidepressant resistance. Bipolar Disord 15:1–163
Tye SJ, Walker AJ, Hu C, Frye MA (2014) Behavioral and neurobiological effects of ketamine in a preclinical model of antidepressant resistance. Biol Psychiatry 75:164S
Walker AJ, Kim Y, Price JB, Kale RP, McGillivray JA, Berk M, Tye SJ (2014) Stress, inflammation, and cellular vulnerability during early stages of affective disorders: biomarker strategies and opportunities for prevention and intervention. Front Psychiatry 5:34
Yamanaka R, Tabata S, Shindo Y, Hotta K, Suzuki K, Soga T, Oka K (2016) Mitochondrial Mg(2+) homeostasis decides cellular energy metabolism and vulnerability to stress. Sci Rep 6:30027
Zhukov DA, Vinogradova KP (2002) Learned helplessness or learned inactivity after inescapable stress? Interpretation depends on coping styles. Integr Physiol Behav Sci 37:35–43
Zigman JM, Jones JE, Lee CE, Saper CB, Elmquist JK (2006) Expression of ghrelin receptor mRNA in the rat and the mouse brain. J Comp Neurol 494:528–548
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Brimijoin, S., Tye, S. Favorable Impact on Stress-Related Behaviors by Modulating Plasma Butyrylcholinesterase. Cell Mol Neurobiol 38, 7–12 (2018). https://doi.org/10.1007/s10571-017-0523-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-017-0523-z