
Abstract
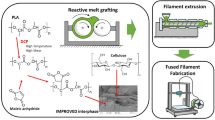
This paper aimed to produce a bio-based filament, suitable for 3D printing (fused deposition modeling), made of surface modified cellulose fiber and high density polyethylene. The cellulose fibers (CF) were first surface modified and transformed into a CF-based macroinitiator through an esterification reaction with the 2-bromoisobutyric acid. We finally studied the ability of this CF-based macroinitiator to initiate a single electron transfer-living radical polymerization (SET-LRP) with an hydrophobic monomer: the stearyl acrylate. The grafting of poly(stearly acrylate) onto the cellulose fibers did strongly increased the adhesion, compatibility of the modified fibers with the hydrophobic host matrix (HDPE). Finally, the resulting hydrophobic fibers were extruded with the high density polyethylene (HDPE) through a counter-rotating twin-screw extruder, yielding a bio-based filament suitable for FDM 3d printing. The successful surface modification, such as the correct incorporation of the modified fibers into the thermoplastic matrix, were characterized through ATR-FTIR, 13C CP-MAS NMR, FE-SEM, and mechanical testing. Throughout those characterization techniques, it was concluded that the fiber surface modification significantly improved the compatibility of the fibers with HDPE. Finally, the 3D printing properties of the composite were tested and compared to those of pure HDPE through the 3d printing of simple objects. It was concluded that the printability of the composite made with poly(stearyl acrylate)-grafted cellulose overcomes the problem (shrinkage, warpage, print fidelity) encountered with the printing of pure HDPE.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The 3D printing technology, also known as additive manufacturing or rapid prototyping, has been initially developed in the early 1980s and had since then attracted a large interest worldwide. 3D printing technology can nowadays be used in numerous and various domains, such as for biomedical applications (Hutmacher 2000; Mosadegh et al. 2015; Ng et al. 2016; Sing et al. 2017; Mi et al. 2018; Mogali et al. 2018; Li et al. 2018; Yan et al. 2018; Guo et al. 2021), aeronautics (Airbus 2016, 2017, 2018), automotive (Volkswagen 2021), or the recycling of industrial plastics waste via a re-extrusion and printing process (Mikula et al. 2021). Nowadays, 3D printing is also used domestically, allowing people to print various object such as decorative items, or simply repairing devices or components for hobbies. Fused Deposition Modeling (FDM) is one of the main method for printing plastics. The FDM process is based on the extrusion of a polymer filament through a heated nozzle, in order to print a layer-by-layer structure. Through this layer-by-layer deposition, a complex three-dimensional structure can be 3D printed. The choice of the polymer used for the 3D printing is crucial for determining its printability and the final properties of the object.
The design of Wood Plastic Composite (WPC) has a great interest in material science over the last 40 years and had been already used in molded process to produce indoor or exterior building-construction part (Peltola et al. 2014; Venkatarajan and Athijayamani 2021). WPCs are manufacture products made of wood particle and a host matrix, typically thermoplastic or thermoset. In term of wood particle, cellulose flour/fiber were already reported as potential green reinforcing agent for polymer, yielding bio-based composite with good mechanical properties (Suryanto et al. 2014; Chand and Fahim 2021). Cellulose fibers have the advantages to be easily accessible, biodegradable, renewable, and non-toxic; making them excellent candidate to address the environmental challenge and thus limiting the amount of plastic waste through the production of bio-based composite. However, due to its hydrophilic nature, cellulose fibers have a poor compatibility with the common hydrophobic thermoplastic matrix. To solve this fiber-host matrix adhesion problem, several chemical pathways have been developed to modify the surface fiber and thus decrease the hydrophilic nature of the fiber. Typical reactions are esterification, silylation, acetylation or the use of a coupling agent such as maleic anhydride derivative or methylenediphenyl 4,4′-diisocyanate (Dominkovics et al. 2007; Dányádi et al. 2010; Gregorova et al. 2011; Kaßel et al. 2018; Seo et al. 2020).
Single Electron Transfer Live Radical Polymerization (SET-LRP) is a powerful, robust and fast reaction tool to growth-graft polymeric chains with a full control of the molecular weight (Fleischmann et al. 2010; Soeriyadi et al. 2011; Levere et al. 2013; Waldron et al. 2014; Navarro et al. 2016; Navarro and Edlund 2017). Moreover, SET-LRP prove to be viable with a large variety of monomers (Nguyen et al. 2010, 2013; Liu et al. 2010; Whitfield et al. 2017). In addition to this, SET-LRP uses a Cu(0) wire as a catalyst, which greatly increases the ease of purification (Lligadas et al. 2017).
Regarding the choice of polymer matrix used in this paper, High-Density Polyethylene (HDPE) was selected. HDPE is not commonly used in 3D printing because of its technical challenge caused by warpage or shrinkage during printing (All3DP 2019). However, nowadays HDPE is one of the most commonly use thermoplastics, and exhibit interesting properties, such as food compatibility and good specific strength. Despite the numerous advantageous of using this thermoplastic matrix, HDPE is a hydrophobic polymer, making the adhesion, compatibility of the wood filler and the host matrix a sensible case.
The aim of this paper is to describe the effect of grafting a hydrophobic polymer onto the cellulose fiber surface and therefore increase its compatibility with the HDPE host matrix. The produce bio-based composite, a FDM filament, was used to produce object through 3D printing.
Firstly, a bromine initiator was grafted on the surface of the cellulose fibers to enable the growth of poly-SA by SET-LRP. Once surface was modified, the modified cellulose fibers were extruded with HDPE and a filament suitable for 3D printing was produced. The 3D-printing capability and the mechanical properties of the composite were both investigated and compared to the results obtained with the neat HDPE.
Material and methods
Materials
The solvents used were DMSO at 99% from Alfa Aesar and 2-Propanol at 99.5% from J.T.Baker. The cellulose fibers were CW 630 PU/ARBOCEL from JRS Rettenmaier & Söhne GmbH. Imidazol (99%), 2-Bromoisobutyric acid (98%), and the copper wire (1.0 mm (0.04 in) diameter, annealed, 99.9% (metal basis), 25 m) were purchased from Alfa Aesar. CDI (reagent grade) and Stearyl acrylate (contains 200 ppm monomethyl etherhydroquinone as inhibitor, 97%) were purchased from Aldrich. The polymer used was SABIC HDPE CC3054.
CF purification
Cellulose fibers were washed in DMSO then separated by centrifugation (6000 rpm, 20 min). This process was repeated until the supernatant was clear.
Synthesis of the CF-based macroinitiator
0.1 g of cellulose fibers were dispersed in 7 mL of DMSO. The solution was then degassed with N2, heated at 55 °C and 3 g of imidazole (0.044 mol) were added to it.
Simultaneously, 4 g (0.024 mol) of 2-bromo-isobutyric acid were dissolved in 20 mL of DMSO. This solution (1.2 mol.L− 1) was also degassed with N2, then 4 g of CDI (0.024 mol) were slowly added to it. It was then left for 30 min to 1 h, until the gas emission stops.
The 2-bromo-isobutyric acid solution was then slowly poured in the cellulose solution. The resulting solution was degassed with N2, then left to react overnight under stirring.
Once the reaction was finished, it was centrifuged at 6000 rpm for 20 min, then washed by 8 cycles of dispersion in DMSO followed by precipitation by centrifugation (20 min, 6000 rpm) and supernatant removal.
SET-LRP grafting of SA onto CF
15 mL of a stearyl acrylate (1 mol L− 1) solution in toluene was passed through a column of aluminum oxide to remove the stabilizing agent. The column was then washed with 15 mL of solvents to recover as much monomer as possible. A 6 cm copper wire was coiled in a spring-like shape. It was immersed in conc. HCl for 10 min, washed with water, then immersed in acetone. Finally, it was dried by air-blowing and used as such. 5 mL of DMSO were degassed using N2 and 100 µL of pure Me6-TREN (0.26 mol L− 1) were added to it with a syringe under Schlenk conditions to reach a final concentration of 5 mmol L− 1. 0.1 g of cellulose macro-initiator were dispersed in 15 mL of DMSO. The copper wire and the monomer were added to the solution, which was then heated to 40 °C and degassed with N2. Once oxygen had been removed from the solution, 0.2 mL of ligand solution (5 mmol L− 1) were added and the solution was left to react overnight under stirring. Once the reaction was over, the solid product was recovered by removal of the liquid phase. It was dissolved in toluene and precipitated with isopropanol. It was recovered by centrifugation (20 min; 6000 rpm) then centrifuged (20 min; 6000 rpm) twice in isopropanol. After this, it was washed though cycles of dissolution in toluene, precipitation in isopropanol and centrifugation (20 min; 6000 rpm) until the supernatant was clear and transparent. Finally, it was dissolved in toluene and centrifuged at 6000 rpm until obtaining a gel.
CF-based composite preparation and processing
Extruding
The extruder used was a Thermo Scientific™ HAAKE™ MiniLab 3 Mikro-Compounder. It was first pre-heated at 150 °C. Then, the screw rotation speed was set at 100 rpm, the polymer matrix and the cellulose were added into the extruder and they were let to cycle for 5 min.
Injection molding
The injector molder used was a Thermo Scientific™ HAAKE™ MiniJet II. It was set at the following parameters: cylinder temperature: 160 °C; mold temperature: 80 °C ; Pressure: 400 bar ; molding time: 4 s.
3D printing
The printer used was an Original PRUSA i3 MK3. A cube with a 1 cm dimension was printed, using the default parameters for ABS, with the following modifications: nozzle temperature: 215 °C, nozzle diameter: 0.4 mm, bed temperature: 60 °C, layer height: 0.2 mm, print speed: 20 mm/min, cooling fan (from the 4th layer): 50%, infill: 15% with a gyroid pattern.
Characterization
All samples13C NMR spectra were acquired on a Bruker 500 Avance III HD spectrometer at Larmor frequencies of 125 MHz and 500 MHz for 13C and 1H respectively. These samples were put into 4 mm zirconia rotors for magic angle spinning (MAS) at a rotation speed of 8 kHz. Creeping 13C MAS NMR spectra were recorded with a 13C mutation frequency of 50 kHz and a contact time of 1.5 ms. These spectra were acquired by Fourier transform of the FIDs, and chemical shifts were referenced to pure tetramethylsilane (TMS).
ATR-FTIR (attenuated total reflection Fourier transform infrared spectroscopy) was performed using Bruker Vector 33 FTIR Fourier-transform infrared Spectrometer I18500 PS15. Spectrums were recorded with 64 scans in the spectral region on 3800−450 cm-1. The spectral resolution is 4 cm−1.
Tensile testing were done according to the EN ISO 527-1 Norm, using a Zwick-Roell universal testing machine AllroundLine Z050 (with a load cell of 500 N). The size of the samples was 30 × 5 × 2 mm. The elongation speed was of 20 mm.min− 1 for pure HDPE, and 1 mm/minute for other samples. Each time, at least 5 samples were measured.
The morphology of modified CF in HDPE was observed via ultra-high-resolution field emission scanning electron microscopy (FE-SEM) of the Hitachi S-800. The dried samples were mounted on sample supports using carbon tape and coated with a 5 nm layer of Pd/Pt with a Cressington 208 h under an inert atmosphere.
Result and discussion
In this paper, a bromide initiator was firstly grafted on the surface of the cellulose fiber (CF) through esterification. Then, the cellulose fiber-based macroinitiator (CF-MI) was used to initiate the SET-LRP of the stearyl acrylate monomer as exposed in Scheme 1. The SET-LRP was performed in the presence of a copper wire and a tetradendate tertiary amine ligand (Me6TREN). Once the polymerization was completed, the modified cellulose fibers (CF-PSA) were extruded with the HDPE and the resulting filament was process through 3D printing.

General chemical strategy for the surface modification of the cellulose fibers
The initial and unmodified cellulose fiber, the cellulose fiber-based macroinitiator and the poly(stearyl acrylate) grafted CNF (CF-PSA) were analyzed by solid state 13C CP/MAS NMR and ATR-FTIR spectroscopies. The results are exposed in Figs. 1 and 2.

13C solid state NMR spectra of CF, CF-MI and CF-PSA
The main characteristic peaks from the cellulose can be identified on the NMR spectra (Fig. 1), for all the samples CF, CF-MI, and CF-PSA. Moreover, some additional peaks (20 ppm, 56 ppm and few weaker signals from 100 to 160 ppm) can also be observed, and were attributed to some residual lignin present in the fibers. It is to be noted that the peak of the carbons 4 and 6 are split in two, due to a difference in chemical shift between crystalline and amorphous cellulose (Park et al. 2010). In addition to those characteristic peaks, the CF-MI spectrum showed the appearance of a new peak at 171 ppm, respectively attributed to the carbonyl of the ester group. Those carbonyl groups can also be observed on the FTIR spectra (Fig. 2) with the apparition of a new band localized at 1733 cm− 1. The solid state 13C CP/MAS NMR and ATR-FTIR characterization confirmed that the cellulose fiber-based macroinitiator was successfully synthesized.

ATR-FTIR spectra of the unmodified cellulose fibers (CF), the cellulose fiber-based macroinitiator (CF-MI) and the Poly(stearyl acrylate) grafted CNF (CF-PSA).
After the synthesis of the cellulose fiber-based macroinitiator (CFMI), stearyl acrylate (SA) was grafted onto the surface through SETLRP (CF-PSA). The CF-PSA NMR spectrum showed a peak localized at 177 ppm attributed to the C12 sites (carbonyl bond, ester group) and strong peaks localized between 40−20 ppm, which are attributed to the carbon chain of the SA unit (C13C29). The FTIR spectra of the CF-PSA (Fig. 2) also showed a large increase of the C= O band localized at 1730 cm− 1, as each SA monomer grafted on the macro-initiator add an ester function. In addition to this, the C–H bands around 2900 cm− 1 were much stronger and more distinct, due to the addition of the long carbon chains from SA. From the FTIR and NMR spectra, it was concluded that the grafting of PSA on the surface of the macro-initiator was successful.
The next step of the composite preparation was the compounding of the obtained CFPSA with the thermoplastic matrix HDPE. The compounding was done with either 10 or 2% (w/w) of CFPSA (respectively labelled CF-PSA10%@HDPE and CF-PSA20%@HDPE). The composites were produced with a counter rotating twin-screw extruder, and the obtained bio-based filament was either (a) directly 3D print, or (b) processed into tensile test samples specimen using injection molding for their mechanical characterization. The mechanical properties were characterized (tensile strength and Young’s modulus) and the cross-section of the broken specimen was further analyzed with a field emission scanning electron microscope. For better comparison, several references samples were also produced: neat HPDE, HDPE with 10% of unmodified fibers (CF10%@HDPE), and finally, HDPE with 7% of unmodified fibers and 3% of maleic anhydride grafted polyethylene (MAPE). MAPE was also selected as it is one of the most widely used additives, e.g. coupling agent, for increasing the compatibility, adhesion between cellulose fibers and a thermoplastic matrix (Seo et al. 2020). The coupling reaction between the cellulose fiber and the MAPE is expose in Scheme 2. The reaction was performed directly in the counter rotating twin-screw extruder by simply mixing the cellulose fiber, the MAPE and the HDPE.

Coupling reaction between CF and the maleic anhydride grafted polyethylene MAPE
The mechanical properties of the different samples were investigated by tensile testing and the results are exposed in Fig. 3. During the characterization, the modulus of elasticity (MOE) of the bio-based composite were determined with an elongation speed of 1 mm min− 1. The pure extruded HDPE was tested with an elongation speed of 20 mm min− 1 as it could undergo strong thinning and elongation before breaking. At the same speed, CFPSA@HDPE broke without any significant deformation after only 4 mm of elongation. For this reason, the elongation speed was lowered to 1 mm min− 1.

Tensile testing curves of HDPE (20 mm/min), HDPE mixed with 10% of unmodified fibers (1 mm/min), HDPE mixed with 10% of modified fibers CF-PSA(1 mm/min), HDPE mixed with 20% of CF-PSA (1 mm/min) and HDPE mixed with 7% of unmodified fibers and 3% of MAPE (as coupling agent) (1 mm/min)
The maximal elongation before breaking, as exposed in Fig. 3 has been taken as a qualitative sign of the fiber/matrix compatibility. The pure HDPE can undergo a high strain deformation as shown in Fig. 4, while the modified cellulose fibers/HDPE composite cannot sustained such a high strain. However, the fibers increased the stiffness of the composite.

Broken tensile test samples. From top to bottom: pure HDPE, CF(7%) + MAPE(3%)@HDPE, CF-PSA(10%)@HDPE
The modulus of elasticity and the yield strength values of the different samples were listed in Table 1. The addition of the unmodified fiber to the HDPE matrix lead to a significant loss of 22% of the polymer strength while the stiffness of the material did not improve (MOE remain the same). A similar strength loss (15%) was observed for the polymer modified samples: the CF-PSA and the CF-MAPE (10% wood fiber). However the Young's modulus increased by roughly 25%, leading to a notably stiffer material. The increase of the CF-PSA amount to 20% in the composite, did not have the expected, estimated result as the stiffness of the material did not increase, if compare the same sample with 10% content.
Finally, the mechanical properties of the CF-PSA and the CF-MAPE were comparable, leading to a similar increase of the compatibility, adhesion between the cellulose fibers and HDPE. However, CF-PSA was produced through SET-LRP, meaning that the reaction condition, e.g. choice/addition of the second monomers can be optimized and therefore new functionality can be introduce onto the hydrophobic fiber.
The initial and unmodified cellulose fibers, the polymer modified fibers (CF-PSA), and the cross-section of the broken CF-PSA specimen (after tensile testing), were analyzed with a field emission scanning electron microscope. The FE-SEM pictures are exposed in Figs. 5 and 6.

FE-SEM picture of the (top) unmodified cellulose fibers and (bottom) the CF-PSA.
As seen in Fig. 5, the aspect ratio of the cellulose fibers used in this study was relatively low. This small aspect ratio could be the reason for the diminution of the yield strength values and the low gain of Young´s modulus (for both composites CFPSA & CF-MAPE). The reinforcing properties of the wood fibers also depends of their aspect ratio as reported elsewhere (Peltola et al. 2014).
The modified CF-PSA showed a different surface (Fig. 5, bottom), as for the CF surface, which one is fully embedded in the grafted SA polymer.

FE-SEM picture of a broken (up) HDPE and (bottom) CFPSA10%@HDPE test sample
Regarding the CF-PSA composite samples, it can be seen from the FE-SEM pictures exposed Fig. 6 that the cellulose fibers were evenly spread within the composite. Moreover, the modified fiber were not pull-out from the composite after reaching the fracture point during the tensile testing, since no voids in the matrix were observed. This showed a good compatibility of the modified fibers with the HDPE matrix.
Finally, the last step was to 3D print an object with our CFPSA(10%)@HDPE composite filament. The major issues with this step were the difficulties inherent to the printing of HPDE, such as shrinkage or warping, leading to the irreversible deformation of the printed object. The 3D printed object are exposed in Fig. 7. The printability of the composite could be performed without warping and with a better layer adhesion if compared to the printed object made of pure HDPE.

picture of cubes 3D-printed respectively with a composite of CFPSA(10%)@HDPE (left) and with pure HDPE (right)
Conclusion
In this paper, we reported the successful growth of polySA on the surface of cellulose fiber, which increased drastically the compatibility of cellulose to HDPE. The compatibility obtained was evaluated through mechanical testing and found to be equal to that of CF-MAPE, which is one of the main methods to do so. FE-SEM pictures showed a good dispersity of the poly-SA grafted cellulose fiber in HDPE. In addition to this, it was shown that the printability fidelity was kept for the CF-PSA@HDPE composite. However, the increase in mechanical properties was not conclusive: this could be explained by the low aspect ratio of the cellulose fibers used, which was too low to yield reinforcing effect, and therefore can only act as a filler. Finally, a great strength from this method is the versatility of the SETLRP: due to its high compatibility with many categories of monomers, it is possible to change the polymers or co-polymers grafted on the surface of cellulose, leading to many potential properties. This approach of cellulose surface modification for polymer compatibility could be used in the future as a starting point for a wide range of composites.
Data availability
All relevant data are include in the manuscript. Data can be available from the corresponding author upon request.
References
Dányádi L, Móczó J, Pukánszky B (2010) Effect of various surface modifications of wood flour on the properties of PP/wood composites. Compos Part A Appl Sci Manuf 41:199–206.https://doi.org/10.1016/j.compositesa.2009.10.008
Dominkovics Z, Dányádi L, Pukánszky B (2007) Surface modification of wood flour and its effect on the properties of PP/wood composites. Compos Part A Appl Sci Manuf 38:1893–1901. https://doi.org/10.1016/j.compositesa.2007.04.001
Fleischmann S, Rosen BM, Percec V (2010) SET-LRP of acrylates in air. J Polym Sci Part A Polym Chem 48:1190–1196. https://doi.org/10.1002/pola.23879
Gregorova A, Hrabalova M, Kovalcik R, Wimmer R (2011) Surface modification of spruce wood flour and effects on the dynamic fragility of PLA/wood composites. Polym Eng Sci 51:143–150. https://doi.org/10.1002/pen.21799
Guo Z, Dong L, Xia J et al (2021) 3D Printing unique nanoclay-incorporated double‐network hydrogels for construction of complex tissue engineering scaffolds. Adv Healthc Mater 10:2100036. https://doi.org/10.1002/adhm.202100036
Hutmacher DW (2000) Scaffolds in tissue engineering bone and cartilage. Biomaterials 21:2529–2543. https://doi.org/10.1016/S0142-9612(00)00121-6
Kaßel M, Gerke J, Ley A, Vana P (2018) Surface modification of Wood Flour via ARGET ATRP and its application as filler in thermoplastics. Polym (Basel) 10:354. https://doi.org/10.3390/polym10040354
Levere ME, Nguyen NH, Leng X, Percec V (2013) Visualization of the crucial step in SET-LRP. Polym Chem 4:1635–1647. https://doi.org/10.1039/C2PY21084C
Li Y, Jiang X, Li L et al (2018) 3D printing human induced pluripotent stem cells with novel hydroxypropyl chitin bioink: scalable expansion and uniform aggregation. Biofabrication 10:044101. https://doi.org/10.1088/1758-5090/aacfc3
Liu X-H, Zhang G-B, Li B-X et al (2010) Copper(0)-mediated living radical polymerization of acrylonitrile: SET-LRP or AGET-ATRP. J Polym Sci Part A Polym Chem 48:5439–5445. https://doi.org/10.1002/pola.24350
Lligadas G, Grama S, Percec V (2017) Single-electron transfer living radical polymerization platform to practice, develop, and invent. Biomacromolecules 18:2981–3008. https://doi.org/10.1021/acs.biomac.7b01131
Mi S, Du Z, Xu Y, Sun W (2018) The crossing and integration between microfluidic technology and 3D printing for organ-on-chips. J Mater Chem B 6:6191–6206. https://doi.org/10.1039/C8TB01661E
Mikula K, Skrzypczak D, Izydorczyk G et al (2021) 3D printing filament as a second life of waste plastics—a review. Environ Sci Pollut Res 28:12321–12333. https://doi.org/10.1007/s11356-020-10657-8
Mogali SR, Yeong WY, Tan HKJ et al (2018) Evaluation by medical students of the educational value of multi-material and multi-colored three-dimensional printed models of the upper limb for anatomical education. Anat Sci Educ 11:54–64. https://doi.org/10.1002/ase.1703
Mosadegh B, Xiong G, Dunham S, Min JK (2015) Current progress in 3D printing for cardiovascular tissue engineering. Biomed Mater 10:034002. https://doi.org/10.1088/1748-6041/10/3/034002
Navarro JRG, Edlund U (2017) Surface-initiated controlled radical polymerization approach to enhance nanocomposite integration of cellulose nanofibrils. Biomacromolecules 18:1947–1955. https://doi.org/10.1021/acs.biomac.7b00398
Navarro JRG, Wennmalm S, Godfrey J et al (2016) Luminescent nanocellulose platform: from controlled graft block copolymerization to biomarker sensing. Biomacromolecules 17:1101–1109. https://doi.org/10.1021/acs.biomac.5b01716
Ng WL, Wang S, Yeong WY, Naing MW (2016) Skin bioprinting: impending reality or fantasy? Trends Biotechnol 34:689–699. https://doi.org/10.1016/j.tibtech.2016.04.006
Nguyen NH, Rosen BM, Percec V (2010) SET-LRP of N, N ‐dimethylacrylamide and of N ‐isopropylacrylamide at 25 °C in protic and in dipolar aprotic solvents. J Polym Sci Part A Polym Chem 48:1752–1763. https://doi.org/10.1002/pola.23940
Nguyen NH, Rodriguez-Emmenegger C, Brynda E et al (2013) SET-LRP of N-(2-hydroxypropyl)methacrylamide in H2O. Polym Chem 4:2424. https://doi.org/10.1039/c3py00220a
Park S, Baker JO, Himmel ME et al (2010) Cellulose crystallinity index: measurement techniques and their impact on interpreting cellulase performance. Biotechnol Biofuels 3:10. https://doi.org/10.1186/1754-6834-3-10
Peltola H, Pääkkönen E, Jetsu P, Heinemann S (2014) Wood based PLA and PP composites: effect of fibre type and matrix polymer on fibre morphology, dispersion and composite properties. Compos Part A Appl Sci Manuf 61:13–22. https://doi.org/10.1016/j.compositesa.2014.02.002
Seo Y-R, Bae S-U, Gwon J et al (2020) Effects of methylenediphenyl 4,4′-diisocyanate and maleic anhydride as coupling agents on the properties of polylactic acid/polybutylene succinate/wood flour biocomposites by reactive extrusion. Mater (Basel) 13:1660. https://doi.org/10.3390/ma13071660
Sing SL, Wang S, Agarwala S et al (2017) Fabrication of titanium based biphasic scaffold using selective laser melting and collagen immersion. Int J Bioprinting 3:65–71. https://doi.org/10.18063/IJB.2017.01.007
Soeriyadi AH, Boyer C, Nyström F et al (2011) High-order multiblock copolymers via iterative cu(0)-Mediated radical polymerizations (SET-LRP): toward biological precision. J Am Chem Soc 133:11128–11131. https://doi.org/10.1021/ja205080u
Suryanto H, Marsyahyo E, Irawan YS, Soenoko R (2014) Morphology, structure, and mechanical properties of natural cellulose fiber from mendong grass (Fimbristylis globulosa). J Nat Fibers 11:333–351. https://doi.org/10.1080/15440478.2013.879087
Venkatarajan S, Athijayamani A (2021) An overview on natural cellulose fiber reinforced polymer composites. Mater Today Proc 37:3620–3624. https://doi.org/10.1016/j.matpr.2020.09.773
Waldron C, Zhang Q, Li Z et al (2014) Absolut “copper catalyzation perfected”; robust living polymerization of NIPAM: guinness is good for SET-LRP. Polym Chem 5:57–61. https://doi.org/10.1039/C3PY01075A
Whitfield R, Anastasaki A, Nikolaou V et al (2017) Universal conditions for the controlled polymerization of acrylates, methacrylates, and styrene via Cu(0)-RDRP. J Am Chem Soc 139:1003–1010. https://doi.org/10.1021/jacs.6b11783
Yan Q, Dong H, Su J et al (2018) A review of 3D printing technology for medical applications. Engineering 4:729–742. https://doi.org/10.1016/j.eng.2018.07.021
All3DP (2019) HDPE filament in 3D printing: the basics. https://all3dp.com/2/hdpe-3d-printing-material-all-you-need-to-know/. Accessed 25 Aug 2021
Airbus (2016) Innovative 3D printing solutions are “taking shape” within Airbus. https://www.airbus.com/en/newsroom/news/2016-04-innovative-3d-printing-solutions-are-taking-shape-within-airbus
Airbus (2017) First titanium 3D-printed part installed into serial production aircraft. https://www.airbus.com/en/newsroom/press-releases/2017-09-first-titanium-3d-printed-part-installed-into-serial-production
Airbus (2018) Bridging the gap with 3D printing. https://www.airbus.com/en/newsroom/news/2018-04-bridging-the-gap-with-3d-printing
Chand N, Fahim M (2021) Tribology of natural fiber polymer composites, 2nd edn. Woodhead Publishing, an imprint of Elsevier, Duxford Cambridge, MA, Kidlington (ISBN 978-1-84569-393-0)
Volkswagen (2021) Volkswagen plans to use new 3D printing process in vehicle production in the years ahead. https://www.volkswagen-newsroom.com:443/en/press-releases/volkswagen-plans-to-use-new-3d-printing-process-in-vehicle-production-in-the-years-ahead-7269
Acknowledgments
We thank J. Beruda (Thünen-Institut für Holzforschung) and D.A. Bielenberg (Thünen-Institut für Holzforschung) for the opportunity to perform tensile testing. We thank T. Potsch (Thünen-Institut für Holzforschung) for the chance to perform FE-SEM.
Funding
Open Access funding enabled and organized by Projekt DEAL. J. Navarro, and all authors thank Fachagentur Nachwachsende Rohstoffe e.V. (FNR/BMEL, Project HolzMat3D, Number 2220HV024X), for financial support.
Author information
Authors and Affiliations
Contributions
All authors: EB, BM, JN contributed to the study conception and the design of the experiments. The synthesis and materials preparations were performed by EB. The data collection and analysis were performed by EB, BM, JN. All authors: EB, BM, JN contributed in writing the first manuscript draft of the manuscript and the submitted version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest for the content of this article.
Ethical approval
The authors certify that this manuscript is original and has not been published elsewhere and will not be submitted for publication in another journal while being considered by Cellulose. Moreover, this study is not split up into several parts to increase the quantity of submissions and submitted to several journals. This article does not contain any studies with human participants or animals performed by any of the authors.
Consent for publication
All authors agreed with the paper content and all gave explicit consent to submit the paper to Cellulose. Moreover, we obtained consent from the responsible authorities at the institute of wood science where the study has been carried out, before the paper was submitted.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barba, E., Mietner, J.B. & Navarro, J.R.G. Grafting of poly(stearyl acrylate) on cellulose fibers as 3D-printable HDPE composites. Cellulose 30, 2267–2278 (2023). https://doi.org/10.1007/s10570-022-05021-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-022-05021-7