
Abstract
Cellulose being the most widely available biopolymer on Earth is attracting significant interest from the industry and research communities. While molecular simulations can be used to understand fundamental aspects of cellulose nanocrystal self-assembly, a model that can perform on the experimental scale is currently missing. In our study we develop a supra coarse-grained (sCG) model of cellulose nanocrystal which aims to bridge the gap between molecular simulations and experiments. The sCG model is based on atomistic molecular dynamics simulations and it is developed with the force-matching coarse-graining procedure. The validity of the model is shown through comparison with experimental and simulation results of the elastic modulus, self-diffusion coefficients and cellulose fiber twisting angle. We also present two representative case studies, self-assembly of nanocrystal during solvent evaporation and simulation of a chiral nematic phase ordering. Finally, we discuss possible future applications for our model.
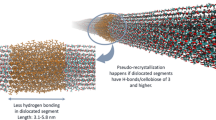
Graphic abstract

Similar content being viewed by others


Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Introduction
Cellulose is the most abundant biopolymer on Earth, and has been gaining growing interest from the research community and industry in the recent years. The main sources of cellulose are wood, plants and bacteria. These sources produce cellulose fibers with different cross sections and lengths (Habibi et al. 2010; Moon et al. 2011; Trache et al. 2017. The cellulose fibers are broken down into cellulose nanofibril (CNF) and cellulose nanocrystal (CNC), where the latter are often referred to “whiskers”. CNC are the product of acid hydrolysis of CNF, which removes the amorphous (disordered) regions of CNF (Kontturi et al. 2018). The combination of wide availability, bio-compatibility, mechanical strength and possibility for surface chemical modifications make cellulose (both CNC and CNF) promising material for numerous applications, e.g. electrically conducting materials, hydrogels and aerogels, nanocellulose films and membranes and biomedical applications, to mention a few (Kontturi et al. 2018; Berglund and Burgert 2018; Dufresne 2013; Tang et al. 2017; Eyley and Thielemans 2014; Chen et al. 2018; Sharma et al. 2019; Bayer et al. 2016). Cellulose often serves as a template scaffold in the aforementioned applications and is usually combined with other materials to achieve the desired functionality and thus forms functional composite materials. These materials often possess complex morphologies and therefore understanding and tuning the interactions between the different components can modulate and improve their properties.
The interactions and self-assembly of CNC or CNF have been subject of numerous experimental studies (Kontturi et al. 2018; Lagerwall et al. 2014; Ding et al. 2014; Orts et al. 1998; Benselfelt et al. 2019; Lombardo et al. 2019; Honorato-Rios et al. 2018). Depending on the experimental conditions, surface modifications, additives, etc. CNC and CNF can assemble into variety of different configurations (Kontturi et al. 2018; Lagerwall et al. 2014; Ding et al. 2014; Orts et al. 1998; Benselfelt et al. 2019; Lombardo et al. 2019; Honorato-Rios et al. 2018). CNC can also self-assemble into chiral nematic phases which possess interesting photonic properties and may have applications as optical material (Schütz et al. 2015; Surov et al. 2017; Liu et al. 2019, 2018; Pospisil et al. 2018; Bertsch et al. 2019; Parker et al. 2016; Yao et al. 2017). The reflected wavelengths of the chiral nematic phases are depended of the pitch (p) of the self-assembled CNC. The pitch can be controlled in many ways either chemically or physically (Park et al. 2014; Frka-Petesic et al. 2017; Nguyen et al. 2013; Parker et al. 2018a). Although, there are plentiful experiments studying this phenomenon, to the best of our knowledge there are no molecular simulations that can provide valuable insight for the self-organization and properties of CNC into chiral nematic phases. This is mainly due to the large time and spatial scales encountered in these systems compared to the systems usually studied by molecular simulations.
Cellulose has been the subject of many molecular simulation studies. Some of these studies focus only at the surface phenomena and modifications of CNC, (Chen et al. 2017; Zhou et al. 2015; Bergenstråhle et al. 2008; Paajanen et al. 2016; Chundawat et al. 2011) while many others simulate whole CNC, both at atomistic (Chen et al. 2017; Zhou et al. 2015; Bergenstråhle et al. 2008; Paajanen et al. 2016; Chundawat et al. 2011; Matthews et al. 2006; Conley et al. 2016; Oehme et al. 2015; Djahedi et al. 2015; Paajanen et al. 2019; Ciesielski et al. 2019) and coarse-grained representations (López et al. 2009; Wohlert and Berglund 2011; López et al. 2015; Mehandzhiyski and Zozoulenko 2019; Glass et al. 2012; Fan and Maranas 2015; Markutsya et al. 2013; Shishehbor et al. 2018; Shishehbor and Zavattieri 2019; Ramezani and Golchinfar 2019; Srinivas et al. 2011; Poma et al. 2015, 2016, 2017). All-atom molecular dynamics (AA-MD) studies are able to simulate the crystal structure of cellulose in a perfect agreement with the experimental crystal structure of different cellulose allomorphs and provide a detailed atomistic picture for the structure and dynamics of CNC (Chundawat et al. 2011). However, AA-MD simulations are way too expensive to study the interactions and aggregation of large CNC in the order of 100–200 nm, which are typical lengths of CNC observed experimentally. Coarse-grained MD (CG-MD) simulations on the other hand are able to significantly reduce the degrees of freedom in molecular simulations and therefore to explore larger systems at a much longer time scale compared to AA-MD. Several CG models for CNC have been developed through the years. They have different degree of mapping (grouping the atoms from the atomistic model in one interaction bead) varying from 4 heavy atoms mapped into one CG bead, in the case of the most popular CG force field, MARTINI, (López et al. 2009; Wohlert and Berglund 2011; López et al. 2015; Mehandzhiyski and Zozoulenko 2019) to 1 or even 2 glucose units represented by 1 CG bead (Glass et al. 2012; Fan and Maranas 2015; Markutsya et al. 2013; Shishehbor et al. 2018; Shishehbor and Zavattieri 2019; Ramezani and Golchinfar 2019). These approaches help to decrease the computational cost of the simulations and enable one to explore much larger systems, but sacrifices the fine atomistic details presented in the AA models. Therefore, it is of the crucial importance for the CG models to be carefully parameterized and to reproduce key aspects of CNC structure and interactions. Most commonly the parameterization of CG models is based of AA-MD, which is shown to reproduce experiments very well (Zhou et al. 2015; Chundawat et al. 2011; Oehme et al. 2015; Paajanen et al. 2019).
In the present study, we introduce a new supra coarse-grained model for the simulation of large CNC systems, spanning into the micrometer and microsecond scales. The paper is organized as follows: first, the supra coarse-grained representation and the parametrization based on the force matching procedure are presented in the next section; the validation of the model and application of the model to the self-assembly of CNC are demonstrated in section three; finally some possible applications are discussed and conclusions are presented in the last section.
Methods and models
Supra coarse-grained representation
Constructing a coarse-grained model begins with choosing the mapping scheme for a specific molecule. We already mentioned in the introduction that CNC were modeled with a different degree of mapping, varying from four heavy atoms in one CG bead to as much as 1 or 2 glucose units in one CG bead, in the most spatially reduced models. We would like to be able to simulate CNC, their interactions and self-assembly, on a mesoscopic scale (\(\approx 100\,\hbox {nm}\)–\(1\,\upmu \hbox {m}\)) and also on a longer time scale than a typical time scale of AA-MD which is on the order of hundreds of ns. These length- and time-scales are beyond the reach of already existing CG cellulose models.

Supra coarse-grained (sCG) model of cellulose showed in a schematic representation and b simulation snapshots. The red beads (O) represent the 200 surface, the green (I)—the 110 surface and the blue (C)—the interior region of 6 chains mapped into one bead
Therefore, we have chosen to build a CG model with even more reduced resolution than the existing ones. The new CNC supra coarse-grained (sCG) model is based on the 36 chains model of cellulose I\(\beta \) arranged in hexagonal shape, which has two surfaces (110 and 200) and is presented in Fig. 1. It should be here noted that we chose to model the I\(\beta \) allomorph, because this is the dominant allomorphic structure of cellulose in higher plants. Our model can be easily adapted to the I\(\alpha \) allomorph, because all the beads and form of the potential functions remain the same. A new parametrization has to be done using e.g. the force-matching coarse graining procedure as described in this paper. The 36 chain model of cellulose I\(\beta \) CNC has dimensions of 3\(\times \)5 nm and represents a wood derived CNC, which has a typical width of 3–5 nm (Habibi et al. 2010; Lagerwall et al. 2014). In our model, we have mapped a part of the CNC cross-section in one CG bead, as shown in Fig. 1. The cross-section of the CNC is divided into surface and inner region, which are represented by the O/I and C bead in Fig. 1, respectively. The I bead represents 110 surface and the O bead—the 200 surface, which are often referred to hydrophilic and hydrophobic, respectively. Both of these two type of beads (I, O) group 20 glucose units,—5 glucose chains in the cross-section and 4 glucose units in the axial direction of the crystal. The third bead type, C, groups the interior of the CNC (which corresponds to the remaining 24 glucose units—6 and 4 glucose chains in the cross-sectional and axial directions, respectively). Thus, the overall shape and the specific surfaces of the CNC are preserved by the sCG model and are represented with the minimal amount of CG beads. It should be mentioned that a similar philosophy of coarse-graining, called ultra-coarse-graining, has been applied earlier, where a region of a protein structure or large amphiphilic molecules has been represented by one CG bead (Dama et al. 2013; Mehandzhiyski and Grimes 2016). Figure 1, depicts the cellulose chains mapped into the respective CG beads and shows both schematically and with simulation screenshots the sCG CNC model. The next two sections present in details the parameterization procedure of the model.
Force matching coarse-graining - bonded interactions
Different coarse-graining techniques exist which can be utilized to derive and parameterize a CG model (Noid 2013; Rühle et al. 2009). We parameterize our model with the force matching coarse-graining method, (Izvekov and Voth 2005b, a) as implemented in the VOTCA package (Rühle et al. 2009). In general, the force-matching method (often referred to multiscale coarse-graining) is trying to match the forces acting on the respective CG bead to some reference forces obtained from AA-MD or ab initio simulations. This matching is achieved in the least-squared manner by minimizing the objective function equivalent to:
where \(F^{ref}_{il}\) and \(F^{p}_{il}\) are the reference and predicted force acting on the ith atom in the lth configuration, respectively, and depends on the set of M parameters; N is the number of atoms in the AA configuration and \(L=5000\) is the total total number of atomic configurations used in the fit (Izvekov and Voth 2005b, a).

Bonded distributions for a CO bond; b CI bond; c OCO bending; d CCC bending; e OCCO dihedral angle and f ICCI dihedral angle, where the CG distribution is given in blue and the respective atomistic distribution in violet
In order to properly capture the physical properties CNC structure at such reduced resolution, we have fitted 3 bonds, 6 bending angles, 2 dihedral angles and 1 improper dihedral. It should be noted here that one bond (CI—Fig. 2b) and one angle (ICO) possess two distinctive distributions and therefore they can not be fitted with a single harmonic potential. Therefore, in practice these interactions are represented with two terms each (CI-1, CI-2) and (ICO-1, ICO-2), see Table S1 in the Supporting Information, such that the total number of bonds and angles are 4 and 7, respectively. More detailed information of the different bonds and angles in the model can be found in the Supporting Information. The full potential function can be expressed as a sum of the bonded and non-bonded terms:
where k’s are the respective force constants, r is distance between two atoms, \(\varTheta \) is the angle between three beads, \(\phi \) is the dihedral angle, n is the multiplicity of the dihedral angle, \(\xi \) is the improper dihedral, \(\epsilon \) and \(\sigma \) are the Lennard–Jones (LJ) potential parameters. The non-bonded interactions and parameters are discussed in more details in the next section. The multiplicity of the dihedral angles in our model equals \(n=1\) and therefore produces one population in the probability distribution presented in Fig. 4e, f). The force matching procedure in VOTCA produces tables with the respective potentials which can be directly used in GROMACS (Abraham et al. 2015). Alternatively, one can fit the force field parameters to reproduce the VOTCA obtained tables and to use the standard GROMACS files. We have chosen to perform the latter and all the force field parameters can be found in the Supporting Information. We have presented in Fig. 2 the bonded probability distributions obtained from the AA-MD and with the sCG model for the (a) CO bond; (b) CI bond; (c) OCO bending; (d) CCC bending; (e) OCCO dihedral angle and (f) ICCI dihedral angle. It can be seen from Fig. 2 that the sCG model is in a perfect agreement with the atomistic model.
Umbrella sampling: non-bonded interactions
The non-bonded interactions in our model are fitted to reproduce the potential of mean forces (PMF) obtained from the AA-MD simulations. The atomistic PMF are obtained from umbrella sampling simulations which will be reported elsewhere and are similar to those reported by Paajanen et al. (2016), where two fibrils were simulated in water. It should be noted that the atomistic PMF are obtained for CNC with cross-section of 18 chains and they are normalized per unit surface area (projected surface area) of CNC. This normalization allows us to fit the non-bonded interactions of our sCG model, even though we build the model with 36 chains. The procedure for the umbrella sampling simulations is as follows: first the two CNC are brought together, then a pulling force is applied to the center of mass of one of the CNC while the other CNC is kept restrained at its original place. By pulling the second CNC we generate a series of configurations which are separated by 0.1 nm and then each configuration was run for 5 ns. Finally, to obtain the PMF we use the Weighted Histogram Analysis Method (WHAM) implemented in GROMACS (Hub et al. 2010). For a more detailed discussion of the umbrella sampling simulations please see (Paajanen et al. 2016). The results from the AA and CG-MD are presented in Fig. 3, together with representative simulation snapshots of the two models. In our model we use the Lennard–Jones 12–6 potential for the non-bonded interactions and therefore we fitted the \(\sigma \) and \(\epsilon \) parameters in the LJ potential, which are given in the Supporting Information. The CNC possesses two different types of surfaces and therefore we have calculated the interactions between the 200–200 (Fig. 3a), 110–110 (Fig. 3b) and 200–110 (Fig. 3c) surfaces. The non-bonded interactions between the central bead (C) and the surface beads (I/O) in the same fibril are excluded, since they are connected by bonds and the non-bonded interactions between different fibrils with these beads are beyond the cut-off distance used in the simulations. The PMF obtained from the sCG model are in a very good agreement with those obtained with the atomistic model. Both the magnitude of the interactions and the equilibrium distance are well represented by the sCG model, as seen from Fig. 3. However, from Fig. 3c, it can be seen that the strength of 200–110 interaction is overestimated in the sCG model. This is related to a particular problem arising during parameterization of this interaction, where the unconstrained CNC always rotate out of the 200–110 configuration, such that two CNC adopt either the 200–200 or 110–110 configuration. To overcome this problem we also constrained the second CNC during the CG umbrella sampling, thus not allowing this rotation to happen. This most likely resulted in a stronger interaction compared to the AA-MD. Nevertheless, the 200–110 interactions are still weaker than the other two types of interactions for both the sCG and the AA models; note that the equilibrium distance between the fibers is well represented in the sCG model.

Simulation snapshots from the AA and CG-MD umbrella sampling simulation and the respective potential of mean forces of the a 200–200 surfaces, b 110–110 surfaces and c 200–110 surfaces
Simulation details
The simulations were carried out with the GROMACS 2019 simulation package (Abraham et al. 2015). The sCG model is parameterized against AA-MD in water. Note that a similar procedure was used by Izvekov and Voth (2009) for a different polymer, and this approach is shown to reproduce energetics and dynamics of the system and an obvious strength of the CG model is that the solvent is not considered and it is possible to simulate large systems going from nm scale to sub-micrometer/micrometer scales. All the CG-MD simulations are performed in an implicit solvent, and the equations of motion are integrated with the stochastic dynamic integrator with the friction in the Langevin equation set with a time constant of \(\tau _{t}=4.0\hbox { ps}\). The time step \(\varDelta \hbox {t}\) was set to 40 fs and the temperature to 300 K, unless specified otherwise. The VdW cutoff and the neighbor list were set to 3.2 nm. The simulation snapshots were prepared with VMD and pymol (Humphrey et al. 1996; Schrödinger 2015).
Results and discussion
In this section we validate our model against both experimental data and atomistic simulations. Then we discuss possible applications for the model and present several examples where our model can be used.
Validation
Twisting of a single fibril
The right-hand twist of cellulose was observed and discussed in many experimental and simulation studies (Matthews et al. 2006; Conley et al. 2016; Paajanen et al. 2019; Paavilainen et al. 2011; Zhao et al. 2013; Hadden et al. 2013; Hanley et al. 1997; Arcari et al. 2019). Although the twist can be clearly seen in TEM (Conley et al. 2016) and AFM (Hanley et al. 1997; Arcari et al. 2019) pictures, it is difficult to deduce exactly how much is the twist per length of fibril. In this respect molecular simulations can be a valuable tool to determine the twist of a CNC. Generally, the twist of the CNC depends on the diameter i.e. the number of constituent chains forming the CNC (Matthews et al. 2006). Zhao et al. (2013) showed that the twisting angle varies between 9.9 and \(1.3\,^{\circ }/\hbox {nm}\) for CNC with cross-sections between \(2 \times 2\) chains and \(6 \times 6\) chains, respectively. Two earlier MD studies found a twist around \(1.4\,^{\circ }/\hbox {nm}\) (Matthews et al. 2006; Zhao et al. 2013). We calculated the twisting angle for the sCG model as a function of the simulation time and presented it together with a simulation snapshot in Fig. 4a. As Fig. 4a shows, the CNC quickly twists in the right-hand direction in the first 10 ns and remains stable afterwards with a twisting angle around \(1.5\,^{\circ }/\hbox {nm}\). The twist of the fibril in our model is described by the dihedral angles used in it. This value is in a very good agreement with the AA-MD results discussed above.

Diffusion coefficients
We calculated the translational diffusion coefficients as a function of the CNC length and presented the results in Fig. 4b and Table S3 in the Supporting Information. The CNC length is varied between 50 and 1200 nm. 20 CNC were placed randomly in a simulation box where the initial separation distance between CNC was at least 100 nm, thus the CNC did not interact with each other. Note that typical experimental conditions correspond to a solution at an infinite dilution. Hence, our simulation box was sufficiently large to prevent the interactions and aggregation between CNC. The concentration of CNC in these simulations is very low (\(\ll \) 1 %) and therefore corresponds to a very diluted solution. The simulations were run for \(1\,\upmu \hbox {s}\) each and the diffusion coefficients D is calculated in a standard way from the mean-square displacement \(\langle \varDelta r^2(t) \rangle \) and the Einstein relation \(D = \langle \varDelta r^2(t) \rangle /6t\).
The time-dependence of the mean-square displacements for all the systems is shown in the Supporting Information. The calculated diffusion coefficients of CNC show an exponential decay with the increase of the length and vary between 17 and \(\approx 1 \times 10^{-12}\hbox { m}^{2}/\hbox {s}\) for lengths between 50 and 1200 nm, respectively. Together with the calculated diffusion coefficients, we present several experimental values for CNC in Fig. 4b (Khouri et al. 2014; Lima et al. 2003; Mao et al. 2017; Boluk and Danumah 2014). It can be seen that the calculated values reproduce the experimental results very well for a wide range of CNC lengths. Although the different experiments often have CNC from different sources that might have different cross-section, the agreement of our model with the experimental values is remarkably good. It should be also noted that the diffusion coefficients for the same CNC length obtained from different experiments differ from each other as well.
Elastic modulus of a single fibril
We have calculated the axial elastic modulus (E) for the sCG model by pulling one end of the CNC while keeping the other end fixed (Glass et al. 2012; Shishehbor et al. 2018; Ramezani and Golchinfar 2019). Hereby we obtained the stress–strain curve (Fig. 4c) and by a linear fit to it, we obtain \(E=110\) GPa. Values in the literature, both experimental and theoretical estimates, vary in the range of 93–270 GPa, (Glass et al. 2012; Tanaka and Iwata 2006; Nishino et al. 1995; Wu et al. 2013, 2014) and the calculated value for our model fells into this range. It should be noted that the elastic modulus in our model is entirely depended on the C–C bond, thus on the bonded interactions. All-atom MD simulations show that E depends around 75% on the bonded interactions and the rest 25% on the non-bonded interactions - VdW, electrostatics and hydrogen bonds (Wohlert et al. 2012). This means that our sCG bonded interactions effectively includes contribution from both bonded and non-bonded AA interactions.
Evaporation simulations
Cellulose fibrils and CNC can self-assemble into various structures to produce materials with different properties and applications (Kontturi et al. 2018). Some of these materials, which find broad applications in various fields can be easily prepared by evaporating the solvent in cellulose dispersions or by freeze drying (Bayer et al. 2016; Honorato-Rios et al. 2018; Schütz et al. 2015; Zhang et al. 2019; France et al. 2017). During the evaporation and depending on the fabrication conditions CNC can form a percolating network which manifests itself in the form of hydrogels and membranes (Kontturi et al. 2018; Bayer et al. 2016; France et al. 2017). These materials are usually self-standing, porous and contain large amount of water. In the current section, we present “water” evaporation simulations prepared with our sCG model. Since in our approach we use an implicit solvent MD simulation, the solvent evaporation can not be done in the usual way as in the explicit solvent MD, where solvent molecules are removed individually (Mehandzhiyski and Zozoulenko 2019). For our simulations the system was prepared by randomly placing 200 CNC with length 100 nm in a simulation box with the periodic boundary condition with a side of 300 nm, shown in Fig. 5a. The system is then simulated for \(1\,\upmu \hbox {s}\) in the NVT ensemble at \(T=300\) K. We calculated the volume fraction (\(\phi \)) of CNC to be 1 vol\(\%\) for the initial system. After \(1\,\upmu \hbox {s}\), we perform a simulation in the NPT ensemble for 10 ns with the isotropic pressure \(P=2\) bar at \(T=300\) K. This is the evaporation step in our simulation in which the system changes its volume and the CNC are brought closer together due to the removal of solvent and thus enhancing the interaction and increasing the number of contacts between them. Due to the implicit MD simulation, the effect of water evaporation can only be modeled by shrinking the simulation box size. We used pressure of 2 bar in order to accelerate the shrinkage of the box and to speed up the simulations. After the evaporation step, the system is simulated in NVT ensemble for \(1\,\upmu \hbox {s}\), followed again by an evaporation step. This procedure is repeated several times and the total simulation time is \(15\,\upmu \hbox {s}\). The volume fraction of cellulose increases linearly during the simulation time of \(15\,\upmu \hbox {s}\), and \(\phi =4.5\hbox { vol}\%\) at the end of the simulation. Simulation snapshots for different volume fractions are shown in Fig. 4b–d and the time evolution of \(\phi \) is presented in Fig. 4e. The snapshots reveal that the CNC form highly percolated cross-linked network throughout the box. The system is in a kinetically arrested state, which represents a macroscopic gel (Honorato-Rios et al. 2018).

Simulation snapshots from the evaporation simulations of the a initial box, b at \(5\,\upmu \hbox {s}\), c\(10\,\upmu \hbox {s}\), d\(15\,\upmu \hbox {s}\). Time evolution of the volume fraction (\(\phi \)) of CNC in the system
We further characterize the final system (\(\phi =4.5\hbox { vol}\%\)) by calculating its elastic modulus. To simulate the stress–strain dependencies, we applied uniaxial deformation of the periodic simulation box in the positive x, y and z axis (Lyulin et al. 2004; Nazarychev et al. 2016). This was achieved by applying a constant rate of deformation to the box in the respective direction with two different rates, \(2 \times 10^{-4}\hbox { nm}/\hbox {ps}\) and \(1 \times 10^{-3}\hbox { nm}/\hbox {ps}\). In the direction of the applied strain the compressibility of the box was set to zero (incompressible), and in the transverse directions it was set to \(4.5 \times 10^{-5}\hbox { bar}^{-1}\) and the pressure to \(P=1\) bar. During the simulations we calculated the pressure tensor \(P_{i}\) (i= x, y, z) and monitored the system size in the direction of the deformation \(L_{i}\). The stress–strain characteristics are then obtained from:
where \(L_{0i}\) is the initial box size before the deformation. The elastic modulus E is obtained from a linear fit to the stress–strain curve in the initial linear elastic regime. E has been averaged over three deformation directions i = x, y, z. Figure 6 depicts the box prior and after deformation in the y-direction. We have obtained 7.2 MPa and 12.7 MPa for deformation rates of \(2 \times 10^{-4}\hbox { nm}/\hbox {ps}\) and \(1 \times 10^{-3}\hbox { nm}/\hbox {ps}\), respectively. These values are in the range of the reported elastic modulus of cellulose hydrogels, 1–50 MPa (France et al. 2017; Nakayama et al. 2004; Ye et al. 2018; Gao et al. 2015). Although, it is often not straightforward to directly compare the simulation with the experimental elastic modulus results, it is a good indication that our results fell in the experimental range for E and that our model is a good representation of a physical gel.

Simulation snapshots of the system with \(\phi =4.5\,\hbox {vol}\%\) prior to deformation and after applying deformation in the y-axis with the rate of \(1 \times 10^{-3}\,\hbox {nm}/\hbox {ps}\)
Chiral nematic phase simulations
The ordering of CNC into chiral nematic phases has been a topic of numerous studies and might be used in applications such as optical encryption, chiral templates or optical sensors (Schütz et al. 2015; Surov et al. 2017; Liu et al. 2019, 2018; Pospisil et al. 2018; Bertsch et al. 2019; Parker et al. 2016; Yao et al. 2017; Park et al. 2014; Frka-Petesic et al. 2017; Nguyen et al. 2013; Parker et al. 2018a; Zhang et al. 2012; Shopsowitz et al. 2010; Kamita et al. 2016; Parker et al. 2018b). One of the main characteristics of CNC chiral nematic phases is their pitch (p), where often the half pitch (p/2) is reported. CNC chiral nematic phases can form during solvent evaporation, by applying a shear force or by a geometry confinement of CNC. The pitch can be experimentally measured and usually is of the order of two to tens of micrometers, but frequently it can be as small as hundreds of nanometers (Lagerwall et al. 2014). Parker et al. have also discussed that the optical resolution limit is \(2\,\upmu \hbox {m}\), which can make it difficult to quantify the exact pitch size if it is less than \(2\,\upmu \hbox {m}\) (Parker et al. 2016). The concentration at which the chiral nematic phases occur has been determined and discussed in the literature as an essential factor for understanding the self-assembly of CNC. However, the self-assembly of CNC into chiral nematic phases is still poorly understood (Lagerwall et al. 2014; Parker et al. 2016). Here, we apply the sCG model to simulate a chiral nematic ordering of CNC. The CNC dispersion needs hours to undergo a transition from an isotropic to anisotropic phase (Lagerwall et al. 2014) which is beyond the time scales of any MD simulations. Therefore, we had to accelerate the dynamics and formation of the chiral nematic phase. To this end we construct an initial system as depicted in Fig. 7a. First, 10 CNC are placed in a row with their 200 surfaces pointing at each other and run for 40 ns in the NVT ensemble in a large periodic box. After the initial 40 ns we observed that the CNC spontaneously form a twist. Finally, this twisted structure is duplicated 18 times, rotated and put next to each other as shown in Fig. 7a. As it can be seen from the inset in Fig. 7a that the individual subset of 10 CNC are placed at a certain distance so they do not overlap. After that this system is run for \(1\,\upmu \hbox {s}\) at 300 K. Simulation snapshots of the final structure are presented in Fig. 7b. The half-pitch (p/2) is around 500 nm and the twist angle is \(\varTheta =171^{\circ }\). It should be noted that in our simulations we use native cellulose without surface modifications while in the experiments there are always sulfonate or carboxylate groups on the CNC surface. The presence of the surface groups will therefore increase the equilibrium surface-surface distance and respectively the observed pitch will be larger. However, the obtained structure represents very well the expected chiral nematic ordering of CNC and its left-handed helix. It should be noted here that the twist of a single fiber is right-handed, but the chiral nematic phase possess left-handed helix (Parker et al. 2018b).
The twisting of the individual CNC in our simulations was also observed in the chiral nematic phase composed of a bundle of cellulose fibrils. However, the degree of twisting is lower than the twisting of the single CNC, presented in Section 3.1.1. This can be explained as a competition between a tendency to twist, and an efficient packing of the fibrils in the chiral nematic phase.

Chiral nematic phase simulations where a how the simulation has been prepared and b The final structure obtained after \(1\,\upmu \hbox {s}\). The rainbow color scheme represents the position in the simulation box
Conclusions
In the present paper, we developed a novel supra coarse-grained model for molecular dynamics simulations of cellulose nanocrystal on large time and spatial scales (microsecond- and sub-micrometer/micrometer scales respectively). The model is based on all-atom MD simulations and is parameterized with the help of the force-matching coarse-graining and umbrella sampling simulations. We demonstrated that our model reproduced sufficiently well experimental and simulation results for CNC twisting angle, self-diffusion coefficients and axial elastic modulus. Additionally, we carried out simulations that represents the self-assembly of CNC during water evaporation and the formation of a cross-linked network, which closely resembles the formation of a cellulose hydrogel. Finally, a simplified and idealistic representation of a chiral nematic phase ordering was simulated and is shown to be a stable configuration of CNC. Thus, our model is versatile and can be applied to study different CNC self-assembled structures on scales comparable with the experiments, which was not possible with the existing AA and CG CNC models. The sCG model can be re-parameterized to represent CNC with other shapes, number of constituent cellulose chains, inclusion of amorphous regions and various surface functional groups. The latter is particularly important since in most of the experiments CNC possess surface functional groups like sulfonate or carboxylate with a different degree of substitution. We plan to include these functional groups in our future studies, as well as to use this model to study in more details the formation and characteristics of chiral nematic phases and gelation observed in cellulose systems.
References
Abraham M, Murtola T, Schulz R, Páll S, Smith J, Hess B, Lindahl E (2015) Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1:19–25
Arcari M, Zuccarella E, Axelrod R, Adamcik J, Sanchez-Ferrer A, Mezzenga R, Nyström G (2019) Nanostructural properties and twist periodicity of cellulose nanofibrils with variable charge density. Biomacromolecules 20:1288–1296
Bayer T, Cunning BV, Selyanchyn R, Nishihara M, Fujikawa S, Sasaki K, Lyth SM (2016) High temperature proton conduction in nanocellulose membranes: paper fuel cells. Chem Mater 28:4805–4814
Benselfelt T, Nordenström M, Hamedi MM, Wågberg L (2019) Ion-induced assemblies of highly anisotropic nanoparticles are governed by ion-ion correlation and specific ion effects. Nanoscale 11:3514–3520
Bergenstråhle M, Mazeau K, Berglund LA (2008) Molecular modeling of interfaces between cellulose crystals and surrounding molecules: effects of caprolactone surface grafting. Eur Polym J 44:3662–3669
Berglund LA, Burgert I (2018) Bioinspired wood nanotechnology for functional materials. Adv Mater 30:1704285. https://doi.org/10.1002/adma.20170428
Bertsch P, Sanchez-Ferrer A, Bagnani M, Isabettini S, Kohlbrecher J, Mezzenga R, Fischer P (2019) Ion-induced formation of nanocrystalline cellulose colloidal glasses containing nematic domains. Langmuir 35:4117–4124
Boluk Y, Danumah C (2014) Analysis of cellulose nanocrystal rod lengths by dynamic light scattering and electron microscopy. J Nanopart Res 16:2174
Chen P, Nishiyama Y, Wohlert J, Lu A, Mazeau K, Ismail AE (2017) Translational entropy and dispersion energy jointly drive the adsorption of urea to cellulose. J Phys Chem B 121:2244–2251
Chen W, Yu H, Lee SY, Wei T, Li J, Fan Z (2018) Nanocellulose: a promising nanomaterial for advanced electrochemical energy storage. Chem Soc Rev 47:2837
Chundawat SPS, Bellesia G, Uppugundla N, da Costa Sousa L, Gao D, Cheh AM, Agarwal UP, Bianchetti CM, Phillips George NJ, Langan P, Balan V, Gnanakaran S, Dale BE (2011) Restructuring the crystalline cellulose hydrogen bond network enhances its depolymerization rate. J Am Chem Soc 133:11163–11174
Ciesielski PN, Wagner R, Bharadwaj VS, Killgore J, Mittal A, Beckham GT, Decker SR, Himmel ME, Crowley MF (2019) Nanomechanics of cellulose deformation reveal molecular defects that facilitate natural deconstruction. PNAS 116(20):9825–9830
Conley K, Godbout L, Whitehead MT, van de Ven TG (2016) Origin of the twist of cellulosic materials. Carbohydr Polym 135:285–299
Dama JF, Sinitskiy AV, McCullagh M, Weare J, Roux B, Dinner AR, Voth GA (2013) The theory of ultra-coarse-graining. 1: General principles. J Chem Theory Comput 9:2466–2480
Ding SY, Zhao S, Zeng Y (2014) Size, shape, and arrangement of native cellulose fibrils in maize cell walls. Cellulose 2:1863–1871
Djahedi C, Berglund LA, Wohlert J (2015) Molecular deformation mechanisms in cellulose allomorphs and the role of hydrogen bonds. Carbohydr Polym 130:175–182
Dufresne A (2013) Nanocellulose: a new ageless bionanomaterial. Mater Today 30(1704285):1704285
Eyley S, Thielemans W (2014) Surface modification of cellulose nanocrystals. Nanoscale 6:7764
Fan B, Maranas JK (2015) Coarse-grained simulation of cellulose i\(\beta \) with application to long fibrils. Cellulose 22:31–44
France KJD, Hoare T, Cranston ED (2017) Review of hydrogels and aerogels containing nanocellulose. Chem Mater 29:4609–4631
Frka-Petesic B, Guidetti G, Kamita G, Vignolini S (2017) Controlling the photonic properties of cholesteric cellulose nanocrystal films with magnets. Adv Mater 29:1701469
Gao X, Shi Z, Liu C, Yang G, Sevostianov I, Silberschmidt VV (2015) Inelastic behaviour of bacterial cellulose hydrogel: in aqua cyclic tests. Polym Testing 44:82–92
Glass DC, Moritsugu K, Cheng X, Smith JC (2012) Reach coarse-grained simulation of a cellulose fiber. Biomacromolecules 13:2634–2644
Habibi Y, Lucia LA, Rojas OJ (2010) Cellulose nanocrystals: chemistry, self-assembly, and applications. Chem Rev 110:3479–3500
Hadden JA, French AD, Woods RJ (2013) Unraveling cellulose microfibrils: a twisted tale. Biopolymers 99(10):746–756
Hanley SJ, Revol JF, Godbout L, Gray DG (1997) Atomic force microscopy and transmission electron microscopy of cellulose from micrasterias denticulata; evidence for a chiral helical microbril twist. Cellulose 4:209–220
Honorato-Rios C, Lehr C, Schütz C, Sanctuary R, Osipov MA, Baller J, Lagerwall JPF (2018) Fractionation of cellulose nanocrystals: enhancing liquid crystal ordering without promoting gelation. NPG Asia Mater 10:455–465
Hub JS, de Groot BL, van der Spoel D (2010) gwham—A free weighted histogram analysis implementation including robust error and autocorrelation estimates. J Chem Theo Comput 6:3713–3720
Humphrey W, Dalke A, Schulten K (1996) Vmd: visual molecular dynamics. J Mol Graph 14(1):33–38
Izvekov S, Voth GA (2005a) A multiscale coarse-graining method for biomolecular systems. J Phys Chem B 109(7):2469–2473
Izvekov S, Voth GA (2005b) Multiscale coarse graining of liquid-state systems. J Chem Phys 123:134105
Izvekov S, Voth GA (2009) Solvent-free lipid bilayer model using multiscale coarse-graining. J Phys Chem B 113(13):4443–4455
Kamita G, Frka-Petesic B, Allard A, Dargaud M, King K, Dumanli AG, Vignolini S (2016) Biocompatible and sustainable optical strain sensors for large-area applications. Adv Opt Mater 4:1950–1954
Khouri S, Shams M, Tam KC (2014) Determination and prediction of physical properties of cellulose nanocrystals from dynamic light scattering measurements. J Nanopart Res 16:2499
Kontturi E, Laaksonen P, Linder MB, Nonappa Gröschel AH, Rojas OJ, Ikkala O (2018) Advanced materials through assembly of nanocelluloses. Adv Mater 30:1703779
Lagerwall JP, Schütz C, Salajkova M, Noh JH, Park JH, Scalia G, Bergström L (2014) Cellulose nanocrystal-based materials: from liquid crystal self-assembly and glass formation to multifunctional thin films. NPG Asia Mater 6:e80
Lima MMDS, Wong JT, Paillet M, Borsali R, Pecora R (2003) Translational and rotational dynamics of rodlike cellulose whiskers. Langmuir 19:24–29
Liu Y, Stoeckel D, Gordeyeva K, Agthe M, Schütz C, Fall AB, Bergström L (2018) Nanoscale assembly of cellulose nanocrystals during drying and redispersion. ACS Macro Lett 7:172–177
Liu Y, Schütz C, Salazar-Alvarez G, Bergström L (2019) Assembly, gelation, and helicoidal consolidation of nanocellulose dispersions. Langmuir 35:3600–3606
Lombardo S, Gençer A, Schütz C, Rie JV, Eyley S, Thielemans W (2019) Thermodynamic study of ion-driven aggregation of cellulose nanocrystals. Biomacromolecules 20:3181–3190
López CA, Rzepiela AJ, de Vries AH, Dijkhuizen L, Hünenberger PH, Marrink SJ (2009) Martini coarse-grained force field: extension to carbohydrates. J Chem Theory Comput 5:3195–3210
López CA, Bellesia G, Redondo A, Langan P, Chundawat SPS, Dale BE, Marrink SJ, Gnanakaran S (2015) Martini coarse-grained model for crystalline cellulose microfibers. J Phys Chem B 119:465–473
Lyulin AV, Balabaev NK, Mazo MA, Michels MAJ (2004) Molecular dynamics simulation of uniaxial deformation of glassy amorphous atactic polystyrene. Macromolecules 37:8785–8793
Mao Y, Liu K, Zhan C, Geng L, Chu B, Hsiao BS (2017) Characterization of nanocellulose using small-angle neutron, x-ray, and dynamic light scattering techniques. J Phys Chem B 121:1340–1351
Markutsya S, Devarajan A, Baluyut JY, Windus TL, Gordon MS, Lamm MH (2013) Evaluation of coarse-grained mapping schemes for polysaccharide chains in cellulose. J Chem Phys 138:214108
Matthews JF, Skopec CE, Mason PE, Zuccato P, Torget RW, Sugiyama J, Himmel ME, Brady JW (2006) Computer simulation studies of microcrystalline cellulose i\(\beta \). Carbohydr Res 341:138–152
Mehandzhiyski AY, Grimes BA (2016) Coarse-grained interface surfactant density maps for calculation of the fractional conversion of tetrameric carboxylic acids to calcium naphthenate precipitates. Ind Eng Chem Res 55(17):5090–5099
Mehandzhiyski AY, Zozoulenko I (2019) Computational microscopy of pedot:pss/cellulose composite paper. ACS Appl Energy Mater 2:3568–3577
Moon RJ, Martini A, Nairn J, Simonsen J, Youngblood J (2011) Cellulose nanomaterials review: structure, properties and nanocomposites. Chem Soc Rev 40:3914–3994
Nakayama A, Kakugo A, Gong JP, Osada Y, Takai M, Erata T, Kawano S (2004) High mechanical strength double-network hydrogel with bacterial cellulose. Adv Funct Mater 14(11):1124–1128
Nazarychev VM, Lyulin AV, Larin SV, Gurtovenko AA, Kennyad JM, Lyulin SV (2016) Molecular dynamics simulations of uniaxial deformation of thermoplastic polyimides. Soft Matter 12:3972
Nguyen TD, Hamad WY, MacLachlan MJ (2013) Tuning the iridescence of chiral nematic cellulose nanocrystals and mesoporous silica films by substrate variation. Chem Commun 49:11296
Nishino T, Takano K, Nakamae K (1995) Elastic modulus of the crystalline regions of cellulose polymorphs. J Polym Sci Part B Polym Phys 33:1647–1651
Noid WG (2013) Perspective: coarse-grained models for biomolecular systems. J Chem Phys 139:090901
Oehme DP, Downton MT, Doblin MS, Wagner J, Gidley MJ, Bacic A (2015) Unique aspects of the structure and dynamics of elementary i\(\beta \) cellulose microfibrils revealed by computational simulations. Plant Physiol 168(3–17):3–17
Orts WJ, Godbout L, Marchessault RH, Enhanced JFR (1998) Ordering of liquid crystalline suspensions of cellulose microfibrils: a small angle neutron scattering stud. Macromolecules 31:5717–5725
Paajanen A, Sonavane Y, Ignasiak D, Ketoja JA, Maloney T, Paavilainen S (2016) Atomistic molecular dynamics simulations on the interaction of tempo-oxidized cellulose nanofibrils in water. Cellulose 23:3449–3462
Paajanen A, Ceccherini S, Maloney T, Ketoja JA (2019) Chirality and bound water in the hierarchical cellulose structure. Cellulose 26:5877–5892
Paavilainen S, Rog T, Vattulainen I (2011) Analysis of twisting of cellulose nanofibrils in atomistic molecular dynamics simulations. J Phys Chem B 115:3747–3755
Park JH, Noh J, Schütz C, Salazar-Alvarez G, Scalia G, Bergström L, Lagerwall JPF (2014) Macroscopic control of helix orientation in films dried from cholesteric liquid-crystalline cellulose nanocrystal suspensions. Chem Phys Chem 15:1477–1484
Parker RM, Frka-Petesic B, Guidetti G, Kamita G, Consani G, Abell C, Vignolini S (2016) Hierarchical self-assembly of cellulose nanocrystals in a confined geometry. ACS Nano 10:8443–8449
Parker RM, Guidetti G, Williams CA, Zhao T, Narkevicius A, Vignolini S, Frka-Petesic B (2018a) The self-assembly of cellulose nanocrystals: hierarchical design of visual appearance. Adv Mater 30:1704477
Parker RM, Guidetti G, Williams CA, Zhao T, Narkevicius A, Vignolini S, Frka-Petesic B (2018b) The self-assembly of cellulose nanocrystals: hierarchical design of visual appearance. Adv Mater 30:1704477
Poma A, Chwastyk M, Cieplak M (2017) Elastic moduli of biological fibers in a coarse-grained model: crystalline cellulose and beta-amyloids. Phys Chem Chem Phys 19:28195–28206
Poma AB, Chwastyk M, Cieplak M (2015) Polysaccharide-protein complexes in a coarse-grained model. J Phys Chem B 119:12028–12041
Poma AB, Chwastyk M, Cieplak M (2016) Coarse-grained model of the native cellulose i-alpha and the transformation pathways to the i-beta allomorph. Cellulose 23:1573–1591
Pospisil MJ, Saha P, Abdulquddos S, Noor MM, Davis VA, Green MJ (2018) Orientation relaxation dynamics in cellulose nanocrystal dispersions in the chiral liquid crystalline phase. Langmuir 34:13274–13282
Ramezani MG, Golchinfar B (2019) Mechanical properties of cellulose nanocrystal (cnc) bundles: coarse-grained molecular dynamic simulation. J Compos Sci 3:57
Rühle V, Junghans C, Lukyanov A, Kremer K, Andrienko D (2009) Versatile object-oriented toolkit for coarse-graining applications. J Chem Theory Comput 5:3211–3223
Schrödinger, LLC (2015) The PyMOL molecular graphics system, version 1.8
Schütz C, Agthe M, Fall AB, Gordeyeva K, Guccini V, Salajková M, Plivelic TS, Lagerwall JPF, Salazar-Alvarez G, Bergström L (2015) Rod packing in chiral nematic cellulose nanocrystal dispersions studied by small-angle x-ray scattering and laser diffraction. Langmuir 31:6507–6513
Sharma A, Thakur M, Bhattacharya M, Mandal T, Goswami S (2019) Commercial application of cellulose nano-composites—a review. Biotechnol Rep 21:e00316
Shishehbor M, Zavattieri PD (2019) Effects of interface properties on the mechanical properties of bio-inspired cellulose nanocrystal (CNC)-based materials. J Mech Phys Solids 124:871–896
Shishehbor M, Dri FL, Moon RJ, Zavattieri PD (2018) A continuum-based structural modeling approach for cellulose nanocrystals (CNCS). J Mech Phys Solids 111:308–332
Shopsowitz KE, Qi H, Hamad WY, MacLachlan MJ (2010) Free-standing mesoporous silica films with tunable chiral nematic structures. Nature 468:422–425
Srinivas G, Cheng X, Smith JC (2011) A solvent-free coarse grain model for crystalline and amorphous cellulose fibrils. J Chem Theory Comput 7:2539–2548
Surov OV, Voronova MI, Zakharov AG (2017) Functional materials based on nanocrystalline cellulose. Russ Chem Rev 86(10):907–933
Tanaka F, Iwata T (2006) Estimation of the elastic modulus of cellulose crystal by molecular mechanics simulation. Cellulose 13:509–517
Tang J, Sisler J, Grishkewich N, Tam KC (2017) Functionalization of cellulose nanocrystals for advanced applications. J Colloid Interface Sci 494:397–409
Trache D, Hussin MH, Haafiz MKM, Thakur VK (2017) Recent progress in cellulose nanocrystals: sources and production. Nanoscale 9:1763–1786
Wohlert J, Berglund LA (2011) A coarse-grained model for molecular dynamics simulations of native cellulose. J Chem Theory Comput 7:753–760
Wohlert J, Bergenstråhle-Wohlert M, Berglund LA (2012) Deformation of cellulose nanocrystals: entropy, internal energy and temperature dependence. Cellulose 19:1821–1836
Wu X, Moon RJ, Martini A (2013) Crystalline cellulose elastic modulus predicted by atomistic models of uniform deformation and nanoscale indentation. Cellulose 20:43–55
Wu X, Moon RJ, Martini A (2014) Tensile strength of i\(\beta \) crystalline cellulose predictedby molecular dynamics simulation. Cellulose 21:2233–2245
Yao K, Meng Q, Bulone V, Zhou Q (2017) Flexible and responsive chiral nematic cellulose nanocrystal/poly(ethylene glycol) composite films with uniform and tunable structural color. Adv Mater 29:1701323
Ye D, Yang P, Lei X, Zhang D, Li L, Chang C, Sun P, Zhang L (2018) Robust anisotropic cellulose hydrogels fabricated via strong self-aggregation forces for cardiomyocytes unidirectional growth. Chem Mater 30:5175–5183
Zhang T, Zhang Y, Jiang H, Wang X (2019) Amino silane-grafted spherical cellulose nanocrystal aerogel with high Co\(_{2}\) adsorption capacity. Environ Sci Pollut Res 26:16716–16726
Zhang YP, Chodavarapu VP, Kirk AG, Andrews MP (2012) Nanocrystalline cellulose for covert optical encryption. J Nanophoton 6(1):063516
Zhao Z, Shklyaev OE, Nili A, Mohamed MNA, Kubicki JD, Crespi VH, Zhong L (2013) Cellulose microfibril twist, mechanics, and implication for cellulose biosynthesis. J Phys Chem A 117:2580–2589
Zhou A, ho Tam L, Yu Z, Lau D (2015) Effect of moisture on the mechanical properties of CFRP-wood composite: an experimental and atomistic investigation. Compos Part B 71(73):63
Acknowledgments
Open access funding provided by Linköping University. This work was supported by the Swedish Research Council (Projects 2016-05990), the Knut and Alice Wallenberg foundation, Peter Wallenberg Foundation, Åforsk, and Troëdsson foundation. IZ thanks the Advanced Functional Material Center at Linköping University for support. ML thanks SeRC (Swedish e-Science Research Center) for funding. IZ and JW acknowledge the support of Wallenberg Wood Science Center. The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at NSC and HPC2N.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mehandzhiyski, A.Y., Rolland, N., Garg, M. et al. A novel supra coarse-grained model for cellulose. Cellulose 27, 4221–4234 (2020). https://doi.org/10.1007/s10570-020-03068-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-020-03068-y