
Abstract
Nanoporous microspheres with divinylbenzene (DVB), styrene (St), and lignin were synthesized by an emulsion-suspension polymerization method. Several types of lignins were used: (1) kraft lignin before (L-unmod) and after modification with methacryloyl chloride (L-Met) and (2) low-molecular-weight kraft lignin unmodified (LWL-unmod) and modified with methacrylic anhydride (LWL-Met). LWL was prepared by ultrafiltration of industrial black liquor using a ceramic membrane with a molecular weight (Mw) cut-off of 5 kDa. The synthesis was optimized by addition of different amounts of lignins. The microsphere texture was characterized using low-temperature nitrogen adsorption and small angle X-ray scattering analyses. The microspheres were nano- and mesoporous with a specific surface area in the range of 0.1–409 m2/g. The morphology of the copolymers was studied using field emission scanning electron microscopy and atomic force microscopy. The thermal properties were studied using differential scanning calorimetry and thermogravimetric analysis methods. A significant difference in the microsphere roughness is affected by lignins due to the presence of lignin nanoparticles at the surface of the microspheres. Molecular modeling was used to predict the sorption properties of the copolymers affected by various fields around the particles. The particle size, polydispersity and zeta potential of the St + DVB, L-Met + St + DVB and L-unmod + St + DVB samples were measured by dynamic light scattering. Additionally, the point of zero charge of the samples was determined using potentiometric titration. The materials studied have a great potential for sorption processes due to their developed porosity and the presence of a number of active surface functionalities.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
There is a growing interest in the development of biomaterials originating from renewable feedstocks. The driving force behind this interest is the limited supply of fossil fuels (oil and gas) and related economic factors and environmental considerations (Razavi 2018; Bose et al. 2018; Calcea et al. 2017; Ramesha et al. 2017; Óscar et al. 2018). The challenges in using bio-based polymers are their inferior performance compared to petrol-based materials, relatively high cost of extraction and processing and the risk of competition with food production. In this context, a more efficient usage of biopolymers formed as by-products of existing processing, e.g., in the forestry and agricultural industries producing lignin, hemicelluloses, and various lignocellulosic residues, are of especially high importance.
Lignins, composed of aromatic macromolecules, have a great potential for replacing oil-based polymers. Lignin is one of the main constituents of lignocellulosic biomass. It is a complex polymer containing aromatic alcohols and a high number of polar functionalities. Industrial lignins are formed as by-products of such pulping processes as kraft, sulfite or alkaline pulping. Lignin’s intrinsic structure is usually modified during the pretreatment processes of lignocellulose that results in the formation of more condensed and less reactive products (Berlin and Balakshin 2014). Additional purification and fractionation treatments of kraft lignin thus are needed prior to its usage in polymeric applications (Duval and Lawoko 2014; Sevastyanova et al. 2014). One of such methods (LignoBoost process) is based on sequential precipitation and filtration steps of lignin from black liquor (Tomani 2010). Another way to obtain lignin materials of high purity with a suitable molecular weight and polydispersity is the ultrafiltration process using ceramic membranes to isolate a lignin fraction from black liquor (Sevastyanova et al. 2014; Aminzadeh et al. 2018). To improve the chemical reactivity, the initial activation of industrial lignin, e.g., through chemical modification, is often needed (Podkościelna et al. 2015; Podkościelna et al. 2017b). Usually, chemical modification is performed through the creation of new active sites or the functionalization of hydroxyl groups (Laurichesse and Averous 2014). It is typically aimed at the derivatization of phenolic and aliphatic hydroxyl groups to introduce more reactive functionalities in the macromolecules. Several types of chemical modification reactions involving hydroxyl groups of lignin have been proposed previously such as etherification, esterification, reaction with isocyanates, silylation, phenolation, and oxidation/reduction (Laurichesse and Averous 2014). Three types of reagents can be used in the esterification reactions: acidic compounds, acid anhydrides, and acid chlorides. The latter two are more reactive than the acidic compounds. The esterification is the easiest type of lignin modification.
A promising application of lignin is linked to its incorporation into various polymeric materials. Different methods of lignin modification and new composite creation have been described previously (Rabinovich et al. 2016; Monteil-Rivera et al. 2013; Hatakeyama and Hatakeyama 2010; Gómez-Fernández et al. 2017; Henry 1975; Zou et al. 2008; Chung et al. 2013; Langholtz et al. 2014). Lignin has been modified and used to prepare functional materials for various applications, including lignin-based composites, epoxy resins, antioxidants, biomedical materials, activated carbons, and carbon fibers.
Lignin as a naturally plentiful biopolymer with polyphenol structure shows potential antioxidant properties (Ge et al. 2014; Huang et al. 2017). Many researchers are currently investigating these properties. For example, Li et al. (2018) proposed improving the antioxidant properties of two lignins by enzymatic treatment with laccase. The obtained results showed that the enzymatically treated lignin with the highest Ph–OH content and lower Mw value possessed the strongest antioxidant activity, which is higher than that of commercial antioxidants. This study suggested that the enzymatic treatment is an effective way to obtain lignin derivatives, which in turn have a high potential for antioxidant applications.
Lignin-derived carbons can be used as an inexpensive alternative to graphite and petroleum-based carbons allowing the manufacture of functional carbon materials for different applications (Chen et al. 2017). The availability of abundant hydroxyl groups in lignin macromolecules and its low cost make it a preferred precursor for the synthesis of sustainable mesoporous carbons (Jeon et al. 2015). However, the highly branched structure of lignin makes it difficult to obtain highly controlled porosity (Carrott and Carrott 2007). The use of lignin for the synthesis of mesoporous carbons will generate new value-added products and revenue streams for the pulping industry and biorefineries (Fierro et al. 2013). It has recently been demonstrated that lignin-containing nanocomposites, such as lignin/polypyrrole (Kim et al. 2014), lignin/graphene (Milczarek and Inganas 2012), and lignin/NiO hybrid electrodes (Chen et al. 2013; Zhou et al. 2018) can be used to solve some energy storage problems.
Another application of lignin is linked to the preparation of sorbents used to remove metal or metalloid species, nanoparticles, and dissolved organic/inorganic compounds from various solutions (Tonin et al. 2016; Li et al. 2017; Ge and Li 2018). Lignin is soluble in some pore-forming solvents that allows the creation of porous composite materials in combination with styrene and divinylbenzene (Podkościelna et al. 2015, 2017a, b; Sobiesiak et al. 2017). Porous styrene–divinylbenzene (St–DVB) materials are among the most popular and first developed polymeric sorbents (Podkościelna et al. 2017a). Due to its hydrophobic character, the St–DVB co-polymer interacts with analytes through π-electron interactions of the aromatic rings and van der Waals forces. To improve the sorption of polar analytes, polar functional groups should be introduced into the copolymer; thus, the sorption capacity can be enhanced.
Sorption studies of Pb(II), Cu(II), Zn(II), and Ni(II) ions on various materials based on modified lignin have been carried out. Li et al. (2015) produced a green porous lignin-based spheres from lignosulfonate cross-linked with sodium alginate and epichlorohydrin. They demonstrated that the spheres with high porosity of 87.66% had a large amount of mesopores. Batch-wise sorption experiments indicated excellent sorption efficiency of the material for lead ions. Qin et al. (2017) synthesized an eco-friendly composite by grafting poly(ethylene imine) onto a natural polymer-based lignin matrix with dithiocarbamate groups. The obtained material was found to be an effective sorbent for uptake of heavy metal ions such as Cu(II), Zn(II), and Ni(II) from the aqueous solutions. In turn, Li et al. (2015) prepared a lignin xanthate resin by a two-step method from alkaline lignin and carbon disulfide. They used the material as a sorbent to remove Pb2+ from the aqueous solution. The low cost, ease to prepare, and effective removal of Pb2+ from wastewater makes this material very efficient in the purification of wastewater containing heavy metal ions. The sorption results obtained by many researchers show that modified lignins can be successfully used as a sorbent of heavy metal ions from the aqueous solutions.
The increasing emphasis on environmental protection means that new, more environmentally friendly sorbents are in demand. Although lignin itself is not characterized by a developed porous structure, it can become very useful in sorption processes as a functional and renewable component of porous polymeric microspheres. Porous microspheres with lignin have a great potential in sorption processes due to their well-developed porous structure, the presence of various functional groups and improved thermal resistance (Qu et al. 2010).
In our previous work devoted to the modification of lignin, new copolymers in the form of microspheres were synthesized using commercial kraft lignin. These materials were porous at the specific surface area (SBET) of 105–196 m2/g, but they had a limited sorption efficiency. The highest values of recovery in the solid phase extraction (SPE) experiments reached only 47% (Podkościelna et al. 2015). These results were caused by a limited reactivity of lignin and its partial presence as a filler in the structure of microspheres. The next studies were focused on additional modification of lignin and synthesis of microspheres with a highly developed porous structure (Podkościelna et al. 2017a, b).
The main aim of this work was to incorporate the maximum possible amount of lignin into the St–DVB microspheres without negative effects on their sorption capabilities. To improve the reactivity, lignin was activated by modification with methacryloyl chloride (or methacrylic anhydride) before copolymerization with DVB or St and DVB. The main role of lignin is to improve the functionalities of St–DVB microspheres accessible to various solutes. The optimized synthetic process and a detailed investigation of the morphology, sorption and thermal properties of the materials are analyzed here in detail.
Materials and methods
Materials
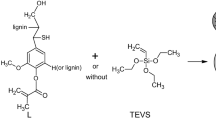
Kraft lignin (L-unmod) was obtained from Sigma-Aldrich (Sweden). This lignin sample was modified with methacryloyl chloride as described elsewhere (Podkościelna et al. 2017b) to obtain a L-Met sample. LWL kraft lignin (LWL-unmod) was obtained by ultrafiltration of industrial black liquor using a ceramic membrane with Mw cut-off of 5 kDa. Then, this lignin was modified with methacrylic anhydride (Henry 1975) to obtain an LWL-Met sample. Methacryloyl chloride and benzyl alcohol were from Fluka AG (Buchs, Switzerland). α,α’-azoiso-bis-butyronitrile (AIBN) and divinylbenzene (DVB) were obtained from Merck (Darmstadt, Germany). Acetone, methanol, chlorobenzene, chloroform, toluene, tetrahydrofuran (THF) and acetonitrile were obtained from Avantor Performance Materials (Poland).
Copolymerization
The copolymerization of divinylbenzene with styrene and different amounts of lignins (modified or unmodified) (see Tables S1 and S2 in Electronic Supplementary Material (ESM) file) was performed in an aqueous medium. The suspension-emulsion polymerization method was used. With regard to the mechanism of the polymerization, the initiator and the obtained product are specific to the suspension polymerization method, but instead of the suspension stabilizer, a surfactant (bis(2-ethylhexyl)sulfosuccinate sodium salt)) was applied that is common to the emulsion polymerization methods. A mixed method was applied to obtain the microspheres of a smaller size. In a 250 cm3 three-necked flask fitted with a water condenser, a stirrer and a thermometer, 75 ml of bidistilled water and 0.75 g of bis(2-ethylhexyl)sulfosuccinate sodium salt were stirred at 80 °C for 0.5 h. Then, the solutions containing DVB, styrene, lignin, initiator AIBN (1%), and pore-forming diluents were added while stirring. The reaction mixture was stirred at 350 rpm and 80 °C for 18 h.
After the synthesis, multistage purification procedure for the obtained microspheres are made. Firstly, the obtained materials are filtered off and washed with hot water until the filtrate is clear. In the second step of purification, the microspheres are moved to Soxhlet apparatus and again cleaned using boiling acetone (6 h). This procedure allows to eliminate unreacted monomers and additives (e.g. pore-forming diluents), if they are not chemically bonded. Then, the microspheres are fractionated with sieves. The scheme of the polymerization reaction is presented in Fig. 1. For comparison, microspheres without lignin were prepared with DVB and St–DVB (Podkościelna et al. 2015).

Scheme of copolymerization of lignin with St and DVB
Characterization methods
The molecular weight characteristics of the two lignin samples were determined by a gel-permeation chromatography (GPC) method using a Waters instrument system (Waters Sverige AB, Sollentuna, Sweden) consisting of a 515 HPLC-pump, 2707 autosampler, and 2998 photodiode array detector (operated at 254 and 280 nm). Prior to the analysis, approximately five mg of a dried lignin sample was acetobrominated using 1 ml mixture of acetyl bromide and glacial acetic acid (1/9 v/v) at room temperature for 2 h as described (Aminzadeh et al. 2018). The lignin residue dried with nitrogen gas was dissolved in HPLC-grade tetrahydrofuran (THF) used as a mobile phase at a flow rate of 0.3 ml/min. Waters Ultrastyragel HR4, HR2, and HR0.5 4.6 × 300 mm solvent-efficient columns connected in series and operated at 35 °C were used for the separation. A sample volume of 20 μL was injected into the instrument and UV signal was recorded at two wavelengths, 254 and 280 nm. Calibration was performed at 254 nm using polystyrene standards with nominal Mw’s ranging between 480 and 176,000 Da. The final analysis of the intensity of the UV signal at 280 nm was performed using Waters Empower 3 build 3471 software.
The structural characterization of lignins, including the amounts of various functionalities, was carried out using 31P NMR spectroscopy as described in detail elsewhere (Granata and Argyropolous 1995). The 31P NMR analysis with a 90° pulse angle, an inverse gated proton decoupling and a time delay of 6 s was used for the identification and quantification of hydroxyl and carboxyl groups. Prior to the analysis, 30 mg of a lyophilized sample was accurately weighed (± 0.01 mg) and mixed in 100 µl of DME and 100 of µl pyridine under rotation at 50 °C for 30 min until complete dissolution. Then, 100 µl of internal standard (endo-N-hydroxy-5-norbornene-2,3-dicarboximide (e-HNDI) at a concentration of 40 mg/ml was added and stirred for 30 min (718.3 mg, 10 ml of pyridine, 50 mg of Cr(AcAc)3, 100 µl equivalent to 40 µmol). A mixture of 100 µl of derivatization agent (2-chloro-4,4,5,5-tetrametyl-1,3,2-dioxaphospholane) and 400 µl CDCl3-d was added dropwise to the lignin solution. The reaction proceeded for 30 min before NMR analysis. The NMR analysis was performed with 256 scans (1500 scans for LCC), a time delay of 6 s and a total runtime of 34 min. Measurements were performed in duplicate. The assignments and integration areas for various signals from the aliphatic OH, various phenolic OH and carboxylic groups were performed as shown in Fig. 2. “G units” corresponds to “noncondensed” phenolic OH groups.

The assignments and integration areas of various functional groups in lignin sample by 31P NMR
Attenuated total reflectance (ATR) FTIR spectra were recorded on a Bruker TENSOR 27 FTIR spectrophotometer (resolution of 4 cm−1, 32 scans accumulated).
The appearance and morphology of the microspheres were characterized using a Hitachi S-4800 field emission scanning electron microscope (FE-SEM) operated at an accelerating voltage of 1 kV. Prior to the FE-SEM analysis, the samples were grounded with a 1 nm-thick Pt–Pd layer, sputtered with a Cressington 208HR high-resolution coater.
Atomic force microscopy (AFM) images were recorded using a MultiMode™8, NanoScope® V(BRUKER-VEECO, USA) microscope in PeakForce QNM mode (RTESPA, fo = 300 kHz, k = 40 N/m, tip radiusNom = 5 nm, tip radiusMax = 10 nm).
The pore size distribution (PSD) of the microspheres was characterized using nitrogen adsorption–desorption isotherms recorded at 77.4 K with a Micrometric ASAP 2405 or ASAP 2420 adsorption analyzer. Prior to the analysis, the copolymers were degassed at 140 °C for 2 h. The specific surface area (SBET) was calculated according to the standard BET method (Gregg and Sing 1982). The total pore volume (Vp) was evaluated from the nitrogen adsorption at p/p0 = 0.98–0.99 (p and p0 denote the equilibrium and saturation pressure of nitrogen at 77.4 K, respectively) (Adamson and Gast 1997) (see ESM file for additional details).
Thermogravimetric analysis (TGA) was carried out with a TGA/SDTA 851 METTLER TOLEDO apparatus. To determine the thermal degradation behavior of lignins and lignin-containing microspheres, 5–10 mg of a sample was tested under a nitrogen atmosphere (50 ml/min) at a heating rate of 10 °C/min from 25 °C to 800 °C.
Differential scanning calorimetry (DSC) measurements were carried out with a Netzsch DSC 204 calorimeter (Germany) operated in dynamic mode. DSC measurements were performed using aluminum pans with pierced lids and a sample mass of ~ 5–10 mg under a nitrogen atmosphere (30 ml min−1). Dynamic scans were performed at a heating rate of 10 K min−1 in the temperature range of 20–500 °C.
The swelling coefficient (B) was determined using equilibrium swelling in acetone, acetonitrile, methanol, toluene, chloroform, THF, and distilled water. B is expressed as:
where Vs is the volume of a sample after swelling and Vd is the volume of a dry sample.
Small-angle X-ray scattering (SAXS) analysis of polymeric microparticles was performed using an Empyrean (PANalytical, Netherlands) diffractometer with Cu Kα radiation (with a parallel beam X-ray mirror with W/Si crystal) using a transmission mode with scans over the 0.115–5° range with an increment of 0.01° at continuous scan mode at 293 K. Before the SAXS measurements, the samples were poured onto a mylar film (6 μm in thickness), leveled and gently kneaded by hand. The beam weakness after passing through the sample was measured, and was corrected taking into account the background observed calculating the absorption factor of each sample. SAXS patterns were analyzed using PANanalytical EasySAXS V. 2.0.0.405 program and approaches described in details elsewhere (Brumberger 1965; Dieudonné et al. 2000; Fairén-Jiménez et al. 2006; Gun’ko et al. 2011).
The pore size distribution (PSD) f(r) based on the SAXS data was calculated using an integral equation for the scattering intensity I(q) (Pujari et al. 2007)
where q = 4πsin(θ)/λ is the scattering vector value, 2θ is the scattering angle, λ is the wavelength of the incident X-ray, v(r) is the volume of a pore with radius r (proportional to r3), and f(r)dr represents the probability of having pores with a radius ranging from r to r + dr. Rmin(= π/qmax) and Rmax (= π/qmin) correspond to the lower and upper limits of the resolvable real space due to instrument resolution, respectively.
The chord size distribution, G(r), as a geometrical statistic description of a multiphase medium, was calculated from the SAXS data (Brumberger 1965; Dieudonné et al. 2000).
where K is the Porod constant (scattering intensity I (q) ~ Kq−4 in the Porod range). The G(r) distribution function characterizes the thickness distribution of pore walls.
The specific surface area from the SAXS data was calculated (in m2/g) using equation
where ϕ = ρa/ρ0 is the solid fraction of adsorbent and Q is the invariant.
The Q value is sensitive to the range used to integrate Eq. (4) (since experimental q values are measured between the qmin and qmax values instead of from 0 and ∞). Therefore, the invariant value Q was calculated using equation (Fairén-Jiménez et al. 2006)
where b is a constant determined using equation
valid in the Porod range. This SAXS-based approach was used for various materials and compared with different methods used to analyze the nitrogen adsorption data (Gun’ko et al. 2011).
The particle size (effective and mean diameter) of the representative samples (St + DVB, L-Met + St + DVB, L-unmod + St + DVB) suspensions dispersed in 10−3 M NaCl (as electrolyte base) and polydispersity were measured by dynamic light scattering (DLS) (also known as photon correlation spectroscopy, PCS or quasi-elastic light scattering, QELS) using a Zeta Pals-BiMass Zetameter (Brookhaven Instruments Corporation, USA) with a laser light of wavelength 670 nm at once after mechanical mixing. Stability of the system was verified at the same concentration 0.1 g/100 ml of electrolyte. To even the coverage and to separate loosely agglomerated particles, these samples were energetically stirred and homogenized just before measurements. The average results of five repeated runs were calculated. Smoluchowski approximation was applied for the zeta potential evaluation from the electrophoretic mobility data. All experiments were performed at room temperature 25 ± 1 °C (Wiącek 2011a, b, c; Wiącek et al. 2008; Grabowski and Morison 1983).
The point of zero charge of samples was determined using potentiometric titration by a high-end titrator with 807 Dosino dosing units and stirrer (907 Titrando, Metrohm, Switzerland). Titration was performed using as ionic strength of 0.1 M NaCl. 0.5 g of each sample was titrated with 0.1 M HCl and 0.1 M NaOH.
The microspheres were further estimated by the X-ray photoelectron spectroscopy (XPS, multi-chamber analytical system UHV, PREVAC, Poland).
Results and discussion
Characterization of unmodified lignins
The molecular weights of both initial kraft lignin samples, L-unmod and LWL-unmod were determined by the GPC method, and the values of average molecular weight (Mw), number average molecular weight (Mn), and polydispersity index (PDI) are shown in Table 1. Both lignin samples have quite low molecular weights, as with the kraft lignins that are the result of certain purification steps (Sevastyanova et al. 2014). LWL-unmod lignin obtained by the ceramic membrane filtration has only a slightly lower molecular weight, but significantly improved polydispersity (2.6) compared to the L-unmod sample (4.3). Additionally, a significant difference is observed in the contents of different functionalities for the initial samples of lignin as shown by the results of the 31P NMR spectroscopy (Granata and Argyropolous 1995). The content of the hydroxyl groups is quite similar for both lignin samples. LWL-unmod lignin has a smaller number of aliphatic, but a higher total content of phenolic groups that is quite typical for kraft lignin fractions of a low molecular weight (Sevastyanova et al. 2014). Additionally, LWL-unmod kraft lignin contains a higher proportion of non-condensed phenolic units than L-unmod lignin has (Table 1). The ratios of non-cond/cond units are 1.62 and 1.19 for LWL-unmod and L-unmod lignins, respectively. This is an indication that LWL-unmod lignin may have a higher chemical reactivity during the modification reaction and subsequent synthesis of porous microspheres (Table 1).
Spectroscopic characterization of copolymers
Chemical structures of the copolymers were confirmed by the ATR/FTIR spectral analysis. The FTIR spectra of polyDVB and St–DVB copolymers without and with modified and unmodified kraft lignin and LWL kraft lignin are presented in Fig. 3. The aromatic C=C bonds produce a band at 1509–1510 cm−1, and the carbonyl groups produce a band at 1728–1732 cm−1 (visible only for the copolymers with modified lignin). The hydroxyl groups are characterized by a broad band at 3243 cm−1 (visible for the samples with unmodified lignin). After the modification, a significant reduction in the band of the hydroxyl groups is observed. Additionally, a new band corresponding to the carbonyl groups is clearly visible. This observation demonstrates the successful modification of lignin with methacrylate groups. Different amounts of lignin, i.e., 1, 2, and 3 g (relative contents of 7, 33 and 50 wt%, respectively), were used to optimize the process and to incorporate as much lignin as possible into the microspheres. With higher amounts of lignin, a shift in the spectra toward greater absorbance is observed due to the dark color of lignin. This is similar for both types of lignin. A sample with LWL kraft unmodified lignin was not prepared because it was washed off, but kraft lignin was more easily trapped during copolymerization. Note that the content of water bound in the composites is small (Fig. 3, 3600–3200 cm−1) even for samples containing lignins. Hydrophobic samples DVB and St + DVB do not contain water because intensity of the FTIR spectra is zero at 3600–3200 cm−1 (Fig. 3a).

ATR/FTIR spectra of copolymers with kraft lignin (a) and LWL kraft lignin (b)
Thermal properties
The thermal properties of the microspheres were investigated using the DSC and TGA methods. On all DSC curves, an endothermic effect is observed in the range of 430–450 °C (Fig. 4, and Tables S3 and S4 in ESM file). This effect is caused by the maximum degradation of the samples. There is a small exothermic effect at 170 °C that is associated with the crosslinking process. For the copolymers with styrene, the incorporation of lignin into microspheres results in a higher temperature of degradation, and the temperature is higher when unmodified lignin is used. The observations are similar for the copolymers with kraft and LWL kraft lignins. Thus, unmodified lignin has higher thermal stability. For the copolymers with DVB and an increasing amount of lignin, faster decomposition occurs.

DSC curves of the copolymers with kraft lignin (a) and LWL kraft lignin (b)
For the lignin-containing microspheres, TGA curves are shown in Fig. 5 and Fig. S2; the decomposition temperatures and char (residue at 800 °C) are shown in Tables 2 and 3. In general, the decomposition of the microspheres takes place within a narrow temperature range of 400–460 °C. There were no significant differences observed between two pairs of samples (L-Met and L-unmod and LWL-Met and L-unmod). Within each pair of samples, the following tendencies could be identified. First, in the microspheres with various modified lignin samples (L-Met and LWL-Met) and DVB, the addition of the larger quantities of lignin component resulted always in decreased thermal stability: both initial and maximum degradation temperatures decreased when adding increasing amount of modified lignin (from 1L-Met + DVB to 3L-Met + DVB and from 1LWL-Met + DVB to 3LWL-Met + DVB) (Tables 2 and 3). The char content increased with the addition of higher quantity of the lignin component, from 17 to 245 for the copolymers with L-Met and from 21 to 28% for the copolymers with LWL-Met component. The degradation of the lignin-containing copolymers with both St and DVB took place in the similar temperature range of 400–460 °C. The addition of the lignin component resulted always in the increased thermal stability of the St–DVB copolymers and in the higher quantity of the formed char. When comparing the effect of the addition of the unmodified and modified lignin samples, the unmodified lignin samples (L-unmod and LWL-unmod) had a higher positive impact on the thermal stability of the copolymers with St and DVB. The analysis of the DSC/TG data leads to the conclusion that the synthesized lignin-containing composites are characterized by improved thermal resistance compared to the DVB or St–DVB materials.

TG curves of the copolymers with kraft lignin (a) and LWL kraft lignin (b)
Morphology
Taking chromatographic applications as an example, sorbent particles should possess a uniform spherical shape in order to have a decent efficiency in the sorption process that is caused by facilitating the regular flow of a mobile phase and minimal diffusion effects.
As discussed above, porous St–DVB materials are already widely used for the fabrication of polymeric sorbents (see Ref. (Podkościelna et al. 2015) for examples of the St–DVB microsphere structure and Fig. 6g for the DVB structure). Lignin could be considered as a more environmentally friendly material than styrene. In this study, we investigate a possible replacement of styrene with lignin. We also incorporate lignin in the St–DVB and DVB matrices as a follow up to previously published studies (Podkościelna et al. 2017a).

SEM images of all copolymers synthesized with L (a–f) and LWL (h–l) kraft lignin, DVB (c–f, j–l) and St–DVB (a–b, h–i), before (a, c, h) and after (b, d–f, i–l) modification, including a reference image of DVB (g). The scale bar is the same for all images
Figure 6 shows SEM images of the lignin-containing St–DVB and DVB microspheres. Two types of kraft lignin are compared: L (Fig. 6a–f) and LWL (Fig. 6h–l) kraft lignin before and after modification (Figs. 6a, c, h and 6b, d–f, i–l, respectively). As seen in Fig. 6, all samples mainly contain uniformly distributed microspheres of different sizes with diameters in the range from a few micrometers to a few tens of micrometers. It can be observed that the lignin modification process facilitates the formation of more agglomerated structures. On the other hand, a larger amount of the finer-sized microspheres is formed in the samples. The microspheres with LWL kraft lignin have more uniform spherical shapes than those with L kraft lignin. The integrity of the microspheres is better preserved in the structures containing modified LWL lignin.
The morphology data for the microspheres containing only lignin and DVB (without styrene) are presented in Fig. 6c–f for the L lignin and (Fig. 6j–l) for the LWL lignin. The effect of different weight percentages of lignin is investigated, including 17 wt%, 33 wt% and 50 wt% (Fig. 6d–f for L-Met and Fig. 6j–l for LWL-Met kraft lignin). For reference, the lignin weight in the samples shown in Fig. 6 was equal to 33 wt% (see Tables S1 and S2 in the ESM file for more details). It is observed that the application of L-Met kraft lignin in the copolymerization processes with DVB does not lead to the formation of microspheres (Fig. 6d–e). However, the utilization of the highest-tested lignin addition level (50 wt%, Fig. 6f) led to an encouraging tendency of microsphere formation. At the same time, the microsphere formation was achieved after the synthesis process of DVB with unmodified L kraft lignin at the weight of 33 wt% (Fig. 6c). In the case of LWL-Met kraft lignin, it was possible to fabricate the microspheres at any concentration of lignin. It was observed that with increasing LWL-Met lignin weight, the microspheres tend to agglomerate and form spheres of smaller sizes (down to the diameter of a few micrometers). These results are very promising and provide evidence of the high potential of styrene substitution with lignin in polymeric sorbents, making them a more sustainable class of materials.
Textural characteristics
The well-developed textural characteristics of adsorbents are very important for effective sorption processes. The characteristics of the porous structures of all the copolymers are summarized in Tables 4, S5, and S6. The materials are mainly mesoporous. The largest specific surface area and pore volume are observed for samples with unmodified lignin. With increasing lignin content, a decrease in the values of SBET and VTOT is noticeable. This is probably due to the blocking of pores by the lignin molecules, which are characterized by much greater intermolecular interaction energy (because of the presence of polar proton-donor and electron-donor functionalities) compared with the nonpolar structures of St and DVB. The main factors influencing the BET surface area and porous structure of the microspheres are (1) the structure of the applied monomers (the homopolymer of DVB has more developed specific surface area of 542 m2/g than copolymer St–DVB at 269 m2/g), (2) the application of pore-forming diluents and (3) the structure and quantity of the biocomponent (lignin is a macromolecule, which can block the pores and decrease the specific surface area).
Figure 7 shows the incremental PSD (SCV/SCR method; the sample numbers correspond to those in Table 4). Figure 8 shows the incremental PSD calculated from the nitrogen adsorption–desorption isotherms and SAXS data for samples (a) 4 and (b) 7 (numbers from Table 4). The difference in the SAXS and SCV/SCR PSD is due to features of the methods. The SAXS method provides the PSD for (1) both open and closed pores and (2) pores in a much broader range of nano/macropores of 0.2 nm < R < 300 nm than that of SCV/SCR (0.34–100 nm). Thus, the SAXS data show that a significant proportion of the pores (especially in the 1–10 nm range) in the polymeric microspheres are closed. It is difficult to estimate the PSD for pores at R > 50 nm using nitrogen adsorption data because adsorbed molecules do not sense the presence the pore walls in broad macropores. Therefore, the use of both SAXS and nitrogen adsorption methods provides a more complete picture of the texture of the materials that is useful to evaluate their potential as adsorbents and the potential ways to improve the textural/sorption characteristics.

Incremental pore size distribution (SCV/SCR method). Sample numbers correspond to those in Table 4

Incremental pore size distributions calculated from nitrogen adsorption–desorption isotherms and SAXS data for samples a 4 and b 7 (numbers from Table 2)
Swelling
Figure 9 shows the swelling effect results for the microspheres studied. The swellability coefficients for all the samples were determined using typical organic solvents. The smallest tendency to swell is observed for the composites with unmodified kraft lignin + DVB and 17 wt% modified kraft lignin + DVB (Table S7). The largest swellability coefficients are observed for the samples (for all solvents) with 50 wt% LWL kraft and kraft lignin. The samples with lignin usually reached the greatest swelling coefficient values in THF and chloroform. In water, the smallest tendency to swell is observed because of the nature of the composite components, since the microspheres have a higher affinity for nonpolar solvents than for polar ones. In contrast to a decrease in the BET surface area, the swelling properties improve with increasing lignin content. The differences can be caused by medium type and features of the interaction of the molecules of adsorbates and solvents with polymer functionalities that is much stronger for the latter.

Swelling studies
AFM
AFM images show the surface roughness of the polymeric microspheres. Figure 10 presents 3D-rendered AFM topography images of two different samples: without (St–DVB) and with lignin (L-Met + St + DVB). As seen, the top surface of the sample without lignin is rather smooth, but the one with lignin is quite rough. As derived from AFM analysis, the Rq roughness (route mean square, RMS) of the microsphere surface is equal to ≅ 10 and ≅ 50 nm for the samples without and with lignin, respectively. This nanoscale roughness may be caused by the presence of lignin particles (macromolecules) on the microsphere surface.

3D-rendered AFM topography images of the top surface of the microspheres without (a) and with (b) lignin of the St + DVB and L-Met + St + DVB samples, respectively
DLS measurements
The representative samples were analyzed using the DLS method. Polydispersity (PD) determines how many types of particles (in terms of size) are present in the system. The smaller the PD value, the more homogenous the system is. According to Table 5, the most homogenous sample is St + DVB (particles with a size less than 10 μm).
The zeta potential was calculated from the electrophoretical mobility. The most stable ζ potential versus pH is for St + DVB material demonstrating negative values. For the samples with lignin (L-Met + St + DVB and L-unmod + St + DVB), the range of ζ potential values is much broader due to lignin functionalities affecting the pH dependence of the ζ value and sign.
Determination of the point of zero charge (pHpzc)
The point of zero charge (pHpzc) of the representative samples (St–DVB, L-Met + St + DVB, L-unmod + St + DVB) was determined using the potentiometric titration method (Fig. 11). In some cases, the surface charge is not as important because the number of charged sites is relatively small and other interactions such as the electrostatic and dispersion interactions are more significant. The pHpzc value of 6.39 was obtained for St–DVB, 7.80 for L-Met + St + DVB and 6.99 for L-unmod + St + DVB. Thus, lignins shift the pHPZC toward basicity mainly due to the features of aromatic hydroxyls.

Determination of a St–DVB, b L-Met + St + DVB, and c L-unmod + St + DVB pHpzc by potentiometric titration
X-ray photoelectron spectroscopy
Samples St–DVB, L-unmod + St + DVB, and L-Met + St + DVB) were studied using X-ray photoelectron spectroscopy (Figs. 12 and 13, Table 6). The C atoms are predominant with the main contribution of sp2 species, which becomes slightly smaller after addition of lignin. The presence of a low content of oxygen atoms in St–DVB is caused by the contamination of the sample by CO2 from the air during the synthesis. The deconvolution of the C1s spectra shows the presence of several types of the C atoms with the sp2 and sp3 the hybridizations and having the C–C (284.4–0–284.9 eV), C = C (mainly aromatics, 284.0–284.4 eV, 291–292 eV (shake-up), C–O (287 eV), C=O, C(O)O (289–290 eV), and C–H (285 eV) bonds (Briggs and Seah 1983; Briggs and Beamson 1992; Fan et al. 2017; Kovač 2011). The O/C atomic ratio increased from 0.009 (St–DVB) to 0.055 (L-Met + St + DVB) due to the presence of lignin in the microspheres that results in the appearance of new bonds of the C atoms with the O and C atoms.

C1s XPS spectra of a St–DVB, b L-unmod + St + DVB, and c L-Met + St + DVB

a Survey and b O1s XPS spectra of St–DVB, L-unmod + St + DVB, and L-Met + St + DVB
Conclusions
Two types of kraft lignin with different molecular weights and several types of functional groups were successfully modified by reactions with methacryloyl chloride or methacrylic anhydride. Using the suspension-emulsion polymerization method, the porous microspheres were prepared with both modified and non-modified lignins. To compare the properties of the polymeric microspheres with and without the addition of lignin, DVB and DVB + St samples were synthesized. The chemical structures of all the microspheres were studied by ATR/FTIR spectroscopy, which showed that higher lignin content enhances the absorbance of all characteristic bands.
The results of thermal analysis demonstrate that the lignin-containing microspheres have good thermal resistance. On the DSC curves, one endothermic peak (observed in the range of 430–450 °C) is associated with the maximum degradation of the samples. TGA analysis illustrated the improved thermal stability of the microspheres on the addition of lignin component. The morphology of the composites was studied using SEM and AFM techniques. The microspheres have a spherical shape, which is important for chromatographic purposes. Their roughness increases with increasing content of lignin.
The porosity of the microspheres was investigated using nitrogen adsorption–desorption isotherms analyzed by the SCV/SCR and SAXS methods. The largest surface area (409 and 542 m2/g) and pore volume (0.9 and 1.3 cm3/g) are observed for the samples with unmodified lignin and polyDVB, respectively. Addition of lignins results in certain compaction of the particles characterized by reduced porosity and specific surface area. The microspheres are nano- and mesoporous, and their developed porosity is important for the sorption applications of the studied composites.
Swelling of the polymeric microspheres in typical organic solvents shows the maximal effects for lignin-containing samples, especially in THF and chloroform, and the smallest effects were when water was used as a solvent due to the hydrophobicity of some microsphere components.
Thus, the materials produced using our approach have great potential to be utilized in various sorption processes due to their developed porous structure, the presence of nano- and mesopores, and their numerous polar and nonpolar functionalities. According to preliminary studies by DLS method, the obtained materials demonstrate a stronger dependence on pH values with respect to the ζ potential and surface charges in comparison to DVB and DVB + St samples due to the presence of lignin functionalities at the surface of composite particles. Therefore, the composite materials can be used as sorbents of cations and anions. These materials could also be used as components of biodegradable composites.
Abbreviations
- AFM:
-
Atomic force microscopy
- AIBN:
-
α,α′-azoiso-bis-butyronitrile
- ATR/FT-IR:
-
Attenuated total reflectance spectroscopy
- DVB:
-
Divinylbenzene
- DSC:
-
Differential scanning calorimetry
- FE-SEM:
-
Field emission scanning electron microscopy
- L-unmod:
-
Unmodified kraft lignin
- L-Met:
-
Modified kraft lignin
- LWL-unmod:
-
Unmodified low molecular weight kraft lignin
- LWL-Met:
-
Modified low molecular weight kraft lignin
- TGA:
-
Thermogravimetric analysis
- St:
-
Styrene
- THF:
-
Tetrahydrofuran
References
Adamson AW, Gast AP (1997) Physical chemistry of surface, 6th edn. Wiley, New York
Aminzadeh S, Lauberts M, Dobele G, Ponomarenko J, Mattsson T, Lindström ME, Sevastyanova O (2018) Membrane filtration of kraft lignin: structural characteristics and antioxidant activity of the low-molecular-weight fraction. Ind Crops Prod 112:200–209
Berlin A, Balakshin M (2014) Industrial lignins: analysis, properties, and applications. In: Vijai G, Maria Tuohy G, Kubicek CP, Saddler J, Xu F (eds) Bioenergy research: advances and applications. Elsevier, Amsterdam, pp 315–336
Bose S, Himanshu DK, Bandyopadhyay SA (2018) Additive manufacturing of biomaterials. Prog Mater Sci 93:45–111
Briggs D, Beamson G (1992) Primary and secondary oxygen-induced C1 s binding energy shifts in X-ray photoelectron spectroscopy of polymers. Anal Chem 64:1729–1736
Briggs D, Seah MP (eds) (1983) Practical surface analysis by Auger and X-ray photoelectron spectroscopy. Wiley, Chichester
Brumberger H (1965) Small angle X-ray scattering. Gordon and Breach, New York
Calcea E, Petriccib E, Savianoc M, De Luca S (2017) Green microwave-assisted procedure to generate bio-based pectin materials. Sustain Chem Pharm 5:127–130
Carrott P, Carrott MR (2007) Lignine from natural adsorbent to activated carbon: a review. Bioresour Technol 98(12):2301–2312
Chen F, Zhou W, Yao H, Fan P, Yang J, Fei Z, Zhong M (2013) Self-assembly of NiO nanoparticles in lignin-derived mesoporous carbons for supercapacitor applications. Green Chem 15:3057–3063
Chen F, Zhou Z, Chang L, Kuang T, Zhao Z, Fan P, Yang J, Zhong M (2017) Synthesis and characterization of lignosulfonate-derived hierarchical porous graphitic carbons for electrochemical performances. Microporous Mesoporous Mater 247:184–189
Chung Y, Olsson JV, Li RJ, Frank CW, Waymouth RM, Billington SL, Sattely ES (2013) A renewable lignin-lactide copolymer and application in biobased composites. ACS Sustainable Chem Eng 1:1231–1238
Dieudonné P, Alaoui AH, Delord P, Phalippou J (2000) Transformation of nanostructure of silica gels during drying. J Non Cryst Solid 262:155–161
Duval A, Lawoko M (2014) A review on lignin-based polymeric, micro- and nanostructured materials. React Funct Polym 85:78–96
Fairén-Jiménez D, Carrasco-Marín F, Djurado D, Bley F, Ehrburger-Dolle F, Moreno-Castilla C (2006) Surface area and microporosity of carbon aerogels from gas adsorption and small- and wide-angle X-ray scattering measurements. J Phys Chem B 110:8681–8688
Fan W, Li Q, Hu L, Yan S, Wen W, Chai Z, Liu H (2017) Polystyrene-based hollow microsphere synthesized by 3-ray irradiation-assisted polymerization and self-assembly and its application in detection of ionizing radiation. Sci Rep 7:1–7
Fierro CM, Gorka J, Zazo JA, Rodriguez JJ, Ludwinowicz J, Jaroniec M (2013) Colloidal templating synthesis and adsorption characteristics of microporous—mesoporous carbons from Kraft lignin. Carbon 62:233–239
Ge Y, Li Z (2018) Application of lignin and its derivatives in adsorption of heavy metal ions in water: a review. ACS Sustainable Chem Eng 6:7181–7192
Ge Y, Xiao D, Li Z, Cui X (2014) Dithiocarbamate functionalized lignin for efficient removal of metallic ions and the usage of the metalloaded bio-sorbents as potential free radical scavengers. J Mater Chem A 2(7):2136–2145
Gómez-Fernández S, Ugarte L, Calvo-Correas T, Peña-Rodríguez C, Corcuera MA, Eceiza A (2017) Properties of flexible polyurethane foams containing isocyanate functionalized kraft lignin. Ind Crops Prod 100:51–64
Grabowski EF, Morison JD (1983) In: Dabneke B (ed) Particle size distributions from analysis of quasi-elastic light scattering data. Wiley, New York
Granata A, Argyropolous DSJ (1995) 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane, a reagent for the accurate determination of the uncondensed and condensed phenolic moieties in lignins. J Agric Food Chem 43:1538–1544
Gregg SJ, Sing KSW (1982) Adsorption, surface area and porosity, 2nd edn. Academic Press, London
Gun’ko VM, Meikle ST, Kozynchenko OP, Tennison SR, Ehrburger-Dolle F, Morfin I, Mikhalovsky SV (2011) Comparative characterization of carbon and polymer adsorbents by SAXS and nitrogen adsorption methods. J Phys Chem C 115:10727–10735
Hatakeyama H, Hatakeyama T (2010) Lignin structure, properties, and applications. Adv Polym Sci 232:1–63
Henry P (1975) Naveau, Methacrylic derivatives of lignin. Cellulose Chem. Technol. 9:71–77
Huang WX, Zhang YH, Ge YY, Qin L, Li ZL (2017) Soft nitrogen and sulfur incorporated into enzymatic hydrolysis lignin as an environmentally friendly antioxidant and mercury adsorbent. BioResources 12(4):7341–7348
Jeon JW, Zhang L, Lutkenhaus JL, Laskar DD, Lemmon JP, Choi D, Nandasiri MI, Hashmi A, Xu J, Motkuri RK (2015) Controlling porosity in lignin derived nanoporous carbon for supercapacitor applications. Chemsuschem 8(3):428–432
Kim SK, Kim YK, Lee H, Lee SB, Park HS (2014) Superior pseudocapacitive behavior of confined lignin nanocrystals for renewable energy-storage materials. Chemsuschem 7(4):1094–1101
Kovač J (2011) Surface characterization of polymers by XPS and SIMS techniques. Mater Tehnol 45:191–197
Langholtz M, Downing M, Graham R, Baker F (2014) Lignin-Derived Carbon Fiber as a Co-Product of Refining Cellulosic Biomass. SAE Int J Mater Manuf 7(1):115–121
Laurichesse S, Averous L (2014) Chemical modification of lignins: towards biobased polymers. Prog Polym Sci 39:1266–1290
Li Z, Ge Y, Wan L (2015a) Fabrication of a green porous lignin-based sphere for the removal of lead ions from aqueous media. J Hazard Mater 285:77–83
Li Z, Kong Y, Ge Y (2015b) Synthesis of porous lignin xanthate resin for Pb2+ removal from aqueous solution. Chem Eng J 270:229–234
Li Z, Chen J, Ge Y (2017) Removal of lead ion and oil droplet from aqueous solution by lignin-grafted carbon nanotubes. Chem Eng J 308:809–817
Li Z, Zhang J, Qin L, Ge Y (2018) Enhancing antioxidant performance of lignin by enzymatic treatment with laccase. ACS Sustain Chem Eng 6:2591–2595
Milczarek G, Inganas O (2012) Renewable cathode materials from biopolymer/conjugated polymer interpenetrating networks. Science 335(6075):1468–1471
Monteil-Rivera F, Phuong M, Ye M, Halasz A, Hawari J (2013) Isolation and characterization of herbaceous lignins for applications in biomaterials. Ind Crops Prod 41:356–364
Óscar E, Ramos L, Pereira RN, Cerqueira MA, Martins JR, Teixeira JA, Malcata FX, Vicente AA (2018) Bio-based nanocomposites for food packaging and their effect in food quality and safety food packaging and preservation. Handb Food Bioeng 8:271–306
Podkościelna B, Sobiesiak M, Gawdzik B, Zhao Y, Sevastyanova O (2015) Preparation of lignin-containing porous microspheres through the copolymerization of lignin acrylate derivatives with St and DVB. Holzforschung 69:769–776
Podkościelna B, Gordobil O, Riazanova AV, Dobele G, Labidi J, Lindstrom ME, Gun’ko VM, Sevastyanova O (2017a) Novel porous materials obtained from technical lignins and their methacrylate derivatives copolymerized with styrene and divinylbenzene. Chem Sel 2:2257–2264
Podkościelna B, Goliszek M, Sevastyanova O (2017b) New approach in the application of lignin for the synthesis of hybrid microspheres. Pure Appl Chem 89:161–171
Pujari PK, Sen D, Amarendra G, Abhaya S, Pandey AK, Dutta D, Mazubder S (2007) Study of pore structure in grafted polymer membranes using slow positron beam and small-angle X-ray scattering techniques. Nuclear Instr Method Phys Res B 254:278–282
Qin L, Ge Y, Deng B, Li Z (2017) Poly(ethylene imine) anchored lignin composite for heavy metals capturing in water. J Taiwan Inst Chem Eng 71:84–90
Qu Y, Tian Y, Zou B, Zhang J, Zheng Y, Wang L, Li Y, Rong C, Wang Z (2010) A novel mesoporous lignin/silica hybrid from rice husk produced by a sol–gel method. Bioresour Technol 101:8402–8405
Rabinovich ML, Fedoryak O, Dobele G, Andersone A, Gawdzik B, Lindström ME, Sevastyanova O (2016) Carbon adsorbents from industrial hydrolysis lignin: the USSR/Eastern European experience and its importance for modern biorefineries. Renew Sust Energy Rev 57:1008–1024
Ramesha M, Palanikumarb K, Reddy KH (2017) Plant fibre based bio-composites: sustainable and renewable green materials. Renew Sust Energy Rev 79:558–584
Razavi M (2018) Bio-based nanostructured materials. Nanobiomater Nanostruct Mater Biomed Appl 2:17–39
Sevastyanova O, Helander M, Chowdhury S, Lange H (2014) Tailoring the molecular and thermo-mechanical properties of kraft lignin by ultrafiltration. J Appl Polym Sci 131:505–9515
Sobiesiak M, Podkościelna B, Sevastyanova O (2017) Thermal degradation behavior of lignin-modified porous styrene-divinylbenzene and styrene-bisphenol a glycerolate diacrylate copolymer microspheres. J Anal Appl Pyrol 123:364–375
Tomani P (2010) The lignoboost process. Cell Chem Technol 44:53–58
Tonin F, Vignali E, Pollegioni E, D’Arrigo P, Rosini E (2016) A novel, simple screening method for investigating the properties of lignin oxidative activity. Enzym Microb Tech 96:143–150
Wiącek AE (2011a) Investigations of DPPC effect on Al2O3 particles in the presence of (phospho)lipases by the zeta potential and effective diameter measurements. Appl Surf Sci 257:4495–4504
Wiącek AE (2011b) Investigation of DPPC effect on SiO2 particles and in the presence of phospho(lipases) by zeta potential and effective diameter measurements. Powder Technol 213:141–147
Wiącek AE (2011c) Changes in wetting properties of alumina surface treated with DPPC in the presence of phospholipase A2 enzyme. Colloids Surf B 87:54–60
Wiącek AE, Hołysz L, Chibowski E (2008) Effect of temperature on n-tetradecane emulsion in the presence of phospholipid DPPC and enzyme lipase or phospholipase A2. Langmuir 24(14):7413–7420
Zhou Z, Chen F, Kuang T, Chang L, Yang J, Fan P, Zhao Z, Zhong M (2018) Lignin-derived hierarchical mesoporous carbon and NiO hybrid nanospheres with exceptional Li-ion battery and pseudocapacitive properties. Electrochim Acta 274:288–297
Zou H, Wu S, Shen J (2008) Polymer/silica nanocomposites: preparation, characterization, properties, and applications. Chem Rev 108:3893–3957
Acknowledgments
The research leading to these results received funding from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7/2007-2013 under REA Grant Agreement No PIRSES-GA-2013-612484 and from the Knut and Alice Wallenberg Foundation in connection with the Wallenberg Wood Science Centre (WWSC) Program. The authors would like to thank Prof. A. Wiącek (UMCS, Lublin) for her help with DLS measurements and M.Sc. D. Fila (UMCS, Lublin) for her help with determination of pHpzc by potentiometric titration.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Goliszek, M., Podkościelna, B., Fila, K. et al. Synthesis and structure characterization of polymeric nanoporous microspheres with lignin. Cellulose 25, 5843–5862 (2018). https://doi.org/10.1007/s10570-018-2009-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-018-2009-7