
Abstract
Similar to epigenetic DNA and histone modifications, epitranscriptomic modifications (RNA modifications) have emerged as crucial regulators in temporal and spatial gene expression during eukaryotic development. To date, over 170 diverse types of chemical modifications have been identified upon RNA nucleobases. Some of these post-synthesized modifications can be reversibly installed, removed, and decoded by their specific cellular components and play critical roles in different biological processes. Accordingly, dysregulation of RNA modification effectors is tightly orchestrated with developmental processes. Here, we particularly focus on three well-studied RNA modifications, including N6-methyladenosine (m6A), 5-methylcytosine (m5C), and N1-methyladenosine (m1A), and summarize recent knowledge of underlying mechanisms and critical roles of these RNA modifications in stem cell fate determination, embryonic development, and cancer progression, providing a better understanding of the whole association between epitranscriptomic regulation and mammalian development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Overall, gene expression is tightly orchestrated with mammalian developmental processes such as stem cell fate determination, embryonic development, and cancer susceptibility; and gene expression programs are precisely modulated with mRNA metabolism and protein synthesis during development. Recently, RNA chemical modifications upon diverse RNA subtypes have emerged as a regulatory mechanism to coordinate cellular transcriptomes and proteomes in different physiological processes. To date, in addition to the canonical four RNA residues, over 170 different types of post-synthesized modifications have been identified in cellular RNA (Boccaletto et al. 2018). Ribosomal RNA (rRNA) and transfer RNA (tRNA), the highly abundant RNA species, are heavily modified. With the development of RNA modification detection methods, internal RNA modifications have been characterized increasingly in message RNA (mRNA) and non-coding RNA (ncRNA) (Dominissini 2014; Li et al. 2016b). Of these, N6-methyladenosine (m6A) serves as the best characterized RNA modification, and occurs in mRNA (Dominissini et al. 2012), rRNA (Maden 1986), and ncRNA (Patil et al. 2016). Other internal RNA modifications such as 5-methylcytosine (m5C) and N1-methyladenosine (m1A) are embedded into coding and non-coding RNAs (Squires et al. 2012; Delatte et al. 2016; Dominissini et al. 2016; Li et al. 2016a). Similar with DNA epigenetics and histone modifications, RNA modifications can be dynamically deposited, removed, and recognized by their specific cellular components (known as "writers", "erasers", and "readers", respectively), which play crucial roles in modulating RNA metabolism during mammalian development and cancer (Frye et al. 2018).
In this review, we will focus on the well-studied epitranscriptomic modifications including m6A, m5C, and m1A as notable examples (Fig. 1) and describe its functional mechanisms in the regulation of gene expression. Additionally, we summarize recent studies regarding the critical roles of these RNA modifications in stem cell differentiation, embryonic development, and cancer progression.

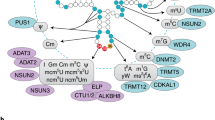
Internal RNA methylation modifications. Schematic representation of three internal RNA methylation modifications (m1A, m6A, and m5C) in mRNA (left panel) and tRNA (right panel). Methylated nucleotides are highly enriched in the regions where they are most frequent in mRNA and tRNA. The chemical structures and the highlighted methyl groups of these methylated nucleotides are represented in the figure
The regulatory networks of the three well-studied RNA methylation
N 6-Methyladenosine
As the best characterized and the most prevalent mRNA modification (Jia et al. 2013), m6A occurs 0.1–0.4% in total adenosine residues (Wei et al. 1975), and mediates almost every aspect in mRNA metabolism. m6A greatly enriches near stop codons, 3′UTR, and long internal exons, and appears to be confined within a conserved motif DRACH (where D = A, G or U; R = A or G; H = A, C or U) (Dominissini et al. 2012; Meyer et al. 2012). m6A deposition within RNA polymerase II–transcribed transcripts is co-transcriptionally formed by the m6A writer complex comprising the asymmetric METTL3–METTL14 heterodimer, where METTL14 functions as a scaffold along with the catalytic component METTL3 to perform the methylation reaction (Wang et al. 2016a, b). The subunit of the m6A writer complex, WTAP, physically interacts with METTL3/14, and functions in the nuclear speckle localization of METTL3 and METTL14 (Ping et al. 2014). WTAP is required for the methylation capability, and its depletion paradoxically shows larger effects on m6A than does depletion of METTL3 or METTL14 (Schwartz et al. 2014; Liu et al. 2014). Furthermore, VIRMA, ZC3H13, RBM15 and/or RBM15B, and CBLL1 proteins have been characterized as the subunits of the m6A-methylation complex and function critical roles in m6A deposition (Fig. 2a). Removal of VIRMA results in a substantial loss of m6A and induces longer 3′ UTR selection (Schwartz et al. 2014; Yue et al. 2018); ZC3H13 is required for nuclear localization of the writer proteins, and its depletion leads to a reduction of m6A (Wen et al. 2018); RBM15 and RBM15B are RNA-binding proteins and can associate with METTL3 and promote particular methylation in mRNAs (Patil et al. 2016) or long non-coding RNA such as XIST (Patil et al. 2016); CBLL1 is also known as HAKAI, and its deletion causes a partial loss of m6A level (Fujita et al. 2002; Ruzicka et al. 2017). Besides, the formation of m6A is also catalyzed by METTL16, which could alone deposit m6A in U6 small nuclear RNA (snRNA) and U6-m6A-consensus sequence in the MAT2A transcript (Pendleton et al. 2017).

Regulation of RNA methylation in gene expression. a m6A is co-transcriptionally installed by an m6A methyltransferase complex and removed by demethylases FTO or ALKBH5. m6A binding proteins specifically recognize m6A-modified transcripts and function diverse roles in RNA metabolism. b The deposition, removal, recognition, and downstream gene expression regulation of m5C and m1A upon diverse RNA subtypes are shown
The m6A-conserved motif DRACH appears heavily frequent in mRNA, suggesting that there are many putative m6A sites in each transcript. However, only very limited DRACH sequences have been methylated. Thus, the transcript- and site-specific m6A deposition requires more precise regulatory mechanism. Co-transcriptional m6A deposition confers specificity in transcripts through the interplay of the methyltransferase complex with histone modification, H3K36me3, or transcription factors that guide it to specific promoters (Huang et al. 2019; Bertero et al. 2018). Alternatively, the selectivity of m6A particular locations within transcripts depends on the interaction of its writer complex with RNA polymerase II and RNA-binding proteins, such as RBM15 and RBM15B (Slobodin et al. 2017; Patil et al. 2016). Until now, details of achieving the intracellular transcript- and site-specific methylation are still not clear, and there are apparently other regulatory pathways awaiting exploration.
Unlike m6A methyltransferase that needs multiple complex proteins to achieve the activity, m6A demethylase alone removes m6A modification. Until now, only two Fe(II)/α-KG-dependent m6A demethylases, FTO and ALKBH5, have been characterized in mammals (Fig. 2a) (Jia et al. 2011; Zheng et al. 2013). FTO is the first determined m6A demethylase and can dynamically remove m6A from nuclear mRNA in vitro and in vivo, implying the reversible regulation of post-transcriptional modification upon mRNA (Jia et al. 2011). ALKBH5 is the second endogenous m6A demethylase and influences mRNA export in nuclear speckles (Zheng et al. 2013). Distinct from the substrate specificity of ALKBH5, FTO has been found to demethylate multiple RNA modifications. Mauer and colleagues showed that FTO has extensively higher catalytic capability for demethylating N6,2′-O-dimethyladenosine (m6Am), adjacent to the m7G cap on mRNA (termed cap m6Am), than for m6A (Mauer et al. 2017). Cap m6Am was suggested to impair DCP2-mediated mRNA decapping for stabilization of mRNA (Mauer et al. 2017). Controversially, the identification of cap m6Am writer PCIF1 reveals that cap m6Am functions in protein translation, not in mRNA stability (Sendinc et al. 2019). In vitro biochemistry and cell-based assays showed that FTO not only can demethylate both internal m6A and cap m6Am in mRNA, but also can demethylate internal m6Am in snRNA and m1A in tRNA (Mauer et al. 2019; Wei et al. 2018). The crystal complex structure of human FTO bound to N6-methyldeoxyadenosine-modified ssDNA is solved and explains the catalytic mechanism how FTO recognizes and demethylates these diverse substrates (Zhang et al. 2019), revealing that m6A is the most favorable nucleobase substrate of FTO. Moreover, it is also found that FTO exhibits the same demethylation activity toward m6A and m6Am of the same RNA sequence. Due to the total amount of m6A was substantially tenfold more than that of cap m6Am in mRNAs, internal m6A modification is still the most favorable substrate of FTO (Wei et al. 2018; Zhang et al. 2019).
The precise regulation of m6A on RNA metabolism is achieved by the recognition of its binding proteins directly and indirectly. The YT521-B homology (YTH) domain was discovered by the in vitro pull-down assay with the m6A-modified probe (Dominissini et al. 2012). Subsequent crystal structure studies revealed that the m6A-dependent binding capability of YTH domain is achieved chiefly through an "aromatic pocket", in which two or three tryptophans enclose m6A spatially (Xu et al. 2014). The YTH domain, around 150 amino acids, is highly conserved in a wide range of eukaryotes, including human, fruit fly, yeast, and plant (Stoilov et al. 2002). In mammals, there are five YTH proteins (YTHDF1-3, YTHDC1-2) tightly bound to m6A-modified transcripts in the cytoplasm (YTHDF1-3, YTHDC2) and nucleus (YTHDC1-2) (Wang et al. 2014a, 2015; Shi et al. 2017; Xiao et al. 2016; Roundtree et al. 2017; Wojtas et al. 2017). Additionally, other putative m6A readers (IGF2BP1-3, eIF3) are also identified and directly interact with the methylated transcripts through its RNA binding domain (Huang et al. 2018; Meyer et al. 2015). HNRNP family members HNRNPA2B1, HNRNPC, and HNRNPG are indirect m6A binding proteins and recognize the m6A switch, of which m6A unfolds the RNA–protein structures and renders transcripts more accessible, instead of m6A itself (Liu et al. 2015, 2017; Alarcón et al. 2015).
As the subcellular localization is known to affect the types of regulatory pathways, the separate subcellular location of m6A readers confer distinctive functions in gene expression, ranged from epigenetic silencing, alternative splicing, nuclear export, transcription, and chromatin state in the nucleus to RNA stability and translation efficiency in the cytoplasm (Fig. 2a). In the nucleus, YTHDC1, together with splicing factor SRSF3 but antagonize with SRSF10, recognizes exon m6A and promotes exon inclusion type pre-mRNA alternative splicing (Xiao et al. 2016). YTHDC1 also tunes the nuclear export of m6A-modified transcripts via the interaction with SRSF3 and NXF1 (Roundtree et al. 2017). Ribonucleoproteins, HNRNPC and HNRNPG, depend on the recognition of m6A switch to affect the occurrence of splicing events (Liu et al. 2015; Zhou et al. 2019). In addition to the mRNA regulation, HNRNPA2B1 binds to m6A-modified primary microRNA (pri-miRNA) transcripts and associates with the microprocessor protein DGCR8, thereby facilitating the pri-miRNA processing (Alarcón et al. 2015). The long noncoding RNA (lncRNA) XIST is highly methylated, and m6A promotes XIST-mediated gene repression by recruiting YTHDC1 (Patil et al. 2016). Markedly, recent works in mouse embryonic stem cells identified that the co-transcriptional m6A methylation is tightly orchestrated with chromatin state: removal of either METTL3 or YTHDC1 activates transcription and enhances chromatin accessibility via deposition and recognition of methylated chromosome-associated regulatory RNAs (carRNAs) (Liu et al. 2020); Li and colleagues exhibited a genome-wide correlation between METTL3-mediated methylation and chromatin modification H3K9me2 (Li et al. 2020); dysregulated m6A methylation by METTL3 depletion impairs the deposition of multiple heterochromatin modifications onto intracisternal A particle (IAP) transcripts and upregulates its transcription (Xu et al. 2021); one more recent study also reported that knockout of YTHDC1 activates these repressed retrotransposons transcripts (such as IAPs, ERVK, and LINE1) in a SETDB1-mediated H3K9me3-dependent manner. These results reveal critical roles for m6A in the regulation of chromatin modification, heterochromatin integrity, and retrotransposon repression (Liu et al. 2021). In the cytoplasm, YTHDF family proteins function in m6A-containing mature RNA metabolism. YTHDF1 mostly interacts with methylated sites near the 3′ end of transcripts and promotes the cap-dependent translation initiation and efficiency, probably via a looping association with eIF3 (Wang et al. 2015). YTHDF2 destabilizes the m6A-modified transcripts by recruiting CCR4-NOT deadenylation complex, which deadenylase the poly(A) tail and accelerate the degradation of targeted transcripts (Wang et al. 2014a; Du et al. 2016). YTHDF3 binds either YTHDF1 or YTHDF2 and facilitates translation and decay of m6A-containing transcripts, separately (Shi et al. 2017). Apart from canonical cap-dependent translation initiation, elF3 could bind 5′UTR m6A and initiate translation with recruitment of the 43S ribosomal complex in a cap-independent translation manner (Meyer et al. 2015). Contrary to YTHDF2 functions, IGF2BPs could enhance the stability of its targeted transcripts in an m6A-dependent manner, suggesting that YTHDF2 and IGF2BPs may confer specific recognition toward m6A-methylated sites or transcripts (Huang et al. 2018). These distinctive regulatory mechanisms of m6A binding proteins could basically tune every aspect of RNA processing and metabolism, reflecting the accuracy and complexity of m6A regulation.
5-Methylcytosine
Methylated cytosine residues at the position 5 are very common in DNA. Despite being discovered for decades (Dubin and Taylor 1975), m5C in RNA did not gain enough attention due to the lesser abundance. Until recently, with the development of high-throughput sequencing approaches, m5C is identified to distribute widely in various RNA subtypes, including tRNA, rRNA, mRNA, and ncRNA as well as enhance RNA (eRNA), and performs diverse functions (Bohnsack et al. 2019). In mRNA, m5C appears 0.4% in total cytosine residues (Squires et al. 2012), and greatly distributes either in locations near to the Argonaute-binding regions within the 3′ UTR (Squires et al. 2012) or in the vicinity of the translational start site of mRNA (Yang et al. 2017a).
Two different types of enzyme proteins have been identified as m5C writers, including NSUN1-7 and DNMT2 (Bohnsack et al. 2019). Members in NSUN family have respective RNA targets and catalytic mechanisms (Fig. 2b). As for rRNAs and tRNAs, NSUN1/Nop2 and NSUN5/Rcm1 are in charge of m5C installation in human eukaryotic 28S rRNA/yeast 25S rRNA (Sharma et al. 2013b; Schosserer et al. 2015). And these methylated rRNAs are considered to possibly function in stabilizing rRNA folding in ribosome by promoting base stack and enhancing hydrogen bond with guanine (Hayrapetyan et al. 2009; Motorin and Helm 2010). Meanwhile, Rcm1-deleted yeast leads to a structural change in rRNA and facilitates read-through of the premature stop codon under oxidative stress (Schosserer et al. 2015). NSUN2, NSUN6, and DNMT2 are responsible for the methylation of tRNA (Tuorto et al. 2012; Haag et al. 2015; Goll et al. 2006). NSUN2 mediates the m5C methylation within the anticodon (C34) of pre-tRNALeu(CAA) (Brzezicha et al. 2006), likely contributing to the translation process by affecting codon-anticodon interaction. However, those m5C located out of anticodon are thought to function in regulating secondary structure of tRNA, such as DNMT2-mediated methylation at position 38 (Tuorto et al. 2012). Besides, it is spotted that the dysregulated NSUN2- and DNMT2-mediated m5C is related to the formation of tRNA fragments (tRFs) from tRNA cleavage, further causing a mistranslation process upon cellular and environmental cues (Blanco et al. 2016). Moreover, NSUN3 and NSUN4 are synthesized on cytoplasmic ribosomes and localized in mitochondria, enabling m5C deposition in mitochondrial tRNA and 12S rRNA, respectively (Nakano et al. 2016; Metodiev et al. 2014). NUSN7 is thought to be related to eRNA methylation and functions in metabolic stress response (Aguilo et al. 2016). In addition to rRNAs and tRNAs, NSUN2 also acts as the m5C writer in mRNAs and promotes m5C-mediated mRNA export (Yang et al. 2017a). As the traceable hm5C is found in RNAs from those species lacking of TET genes (Fu et al. 2014), m5C can be oxidized by the TET-family enzymes. TET2 functions in promoting the conversion of m5C to hm5C on tRNA and regulating the processing or stability of different classes of tRFs (He et al. 2021). Meanwhile, TET2-mediated oxidation of m5C in tRNA leads to a significant increase in translation in vitro, but not obvious in vivo (Shen et al. 2020). Additionally, TET proteins in Drosophila have been revealed to catalyze the oxidative hydroxylation of m5C to hm5C in mRNA, where hm5C is notably present in coding sequences and can favor mRNA translation (Delatte et al. 2016). In mammalian mRNA, TET2 mediates oxidation of m5C and its disruption causes the transcriptome-wide upregulation of m5C, such as the ones in the 3′UTR of SOCS3, further influencing the pathogen infection–induced myelopoiesis (Shen et al. 2018).
Functionally, ALYREF is identified as m5C reader and promotes nuclear export of m5C-containing transcripts (Yang et al. 2017a). Using pull-down along with LC–MS assay, YBX1 is captured as m5C binding protein, and coordinately maintains mRNA stability in zebrafish with Pabpc1a, thereby enhancing the stabilization of m5C-containing transcripts (Yang et al. 2019) (Fig. 2b).
N 1-Methyladenosine
As another important RNA modification, m1A is methylated at the N1 position of adenosine and appears in tRNA, rRNA, mRNA, and mitochondrial (mt) transcripts (El Yacoubi et al. 2012; Sharma et al. 2013a; Li et al. 2017a; Safra et al. 2017). The m1A methylation endows various RNA types with a positive charge that may affect its structures and interactions with potential partner proteins. m1A is first detected in yeast tRNAPhe (RajBhandary et al. 1966) and presents at the position 9, 14, 58 of tRNA (Anderson and Droogmans 2005). Among them, m1A 58 upon tRNA is highly conserved in bacteria, archaea, and eukaryote, and functions in tRNA stabilization, especially that of eukaryotic initiator tRNAiMet (Anderson et al. 1998; Liu et al. 2016). Contrary to the high abundance of m1A in tRNA, m1A occurs in 0.015–0.054% of total cytosine residues in mammalian mRNA (Dominissini et al. 2016; Li et al. 2016a). The development of m1A sequencing and m1A single-base resolution methods unravels specific m1A sites in mRNA and mt-encoded transcripts (Dominissini et al. 2016; Li et al. 2016a, 2017a; Safra et al. 2017). Li and colleagues identified over 400 m1A sites in mRNA and lncRNA, most of which are highly located within 5′ UTR (Li et al. 2017a). Additionally, m1A modification is prevalent in mt-encoded transcripts and interferes with mitochondrial translation (Li et al. 2017a; Safra et al. 2017).
TRMT6 and TRMT61 are tRNA m1A writers (Vilardo et al. 2020; Guy and Phizicky 2014), while RRP8 installs m1A in 28S rRNA (Peifer et al. 2013). Human mt-tRNAs have been reported to contain m1A sites at position 9 and 58, which could be methylated by TRMT10C and TRMT61B, respectively (Vilardo et al. 2012; Chujo and Suzuki 2012). As for mRNAs, TRMT6 and TMRT61 complex also functions in the regulation of m1A methylation in very few mRNAs (Safra et al. 2017; Li et al. 2017a). TRMT10C deposits m1A in the mitochondrial ND5 mRNA (Safra et al. 2017). TRMT61B also acts as a mitochondrial m1A methyltransferase, and TRMT61B-mediated m1A deposition in mt-mRNAs interferes with translation (Li et al. 2017a). ALKBH1 and ALKBH3 mediate the m1A demethylation process, in which ALKBH1 demethylates m1A 58 in tRNA and plays critical roles in attenuating translation elongation (Liu et al. 2016; Chen et al. 2019b); and ALKBH3 is established as the only known m1A eraser in mRNA (Woo and Chambers 2019; Li et al. 2016a) (Fig. 2b). Still now, there are no specific m1A binding proteins identified. Therefore, more knowledge toward m1A readers and biological functions needs further study.
RNA methylation functions in stem cell fate
Stem cells have been widely utilized to decipher the regulatory elements underlying cell fate decisions. The determination of cell fate is tightly orchestrated with global gene expression alterations, some of which are regulated by the epigenetic mechanism. Apart from epigenetic marks (DNA methylations and histone modifications), epitranscriptomic modifications (m6A and m5C) are also required for stem cell state maintenance, differentiation, and development.
m6A functions in embryonic stem cell pluripotency
Embryonic stem cells (ESCs) reside in the early embryo blastocyst and have the pluripotent capability to form all tissues of the embryo. ESCs are notably separated into two states: naïve-state ESCs and primed epiblast stem cells (EpiSCs). Naïve-state ESCs are originated from the inner cell mass of developing pre-implantation blastocysts and represented as a naïve state, whereas EpiSCs are isolated from the post-implantation epiblasts and primed to differentiation.
The transcriptome-wide m6A profiling in mouse and human ESCs revealed that most transcripts encoding the core pluripotency factors were methylated, implicating a possible role in affecting ESC fate determination (Batista et al. 2014). Two individual studies showed that the removal of METTL3 in ESCs delays turnover of self-renewal state and impedes cell specification in vitro (Batista et al. 2014; Geula et al. 2015). The homozygous METTL3-deficient mice are embryonic lethal. Lacking METTL3 embryos appear normal before implantation, but initiate to die at the post-implantation stage, in which METTL3-depleted ESCs constantly transcribe pluripotency factor NANOG, but fail to undertake differentiation into downstream lineages (Geula et al. 2015). Consistently, depletion of METTL14 in embryos blocks differentiation and displays embryonic growth retardation from embryonic d 6.5, leading to embryonic lethality (Meng et al. 2019). However, other studies reported that deficiency of METTL3 and METTL4 in mESCs lost its self-renewal capability and stabilizes the methylated developmental regulators via the HuR/microRNA pathway (Wang et al. 2014b). These conflicting results could be explained by the association between the current stem cell state and m6A-mediated regulation. At the naïve pluripotent state, METTL3-deleted ESCs delay turnover of dominating pluripotency factors, and are locked in a hyper-naїve state. In the primed pluripotent state, deficiency of METTL3 upregulates already-abundant lineage-commitment genes in a YTHDF2-dependent manner, diminishing self-renewal and triggering cell specification (Fig. 3a). Notably, upon Activin–NODAL signaling pathway, transcription factors Smad2/3 recruit the METTL3-METTL14-WTAP complex to facilitate m6A deposition and destabilize its transcriptional targets, particularly pluripotency gene NANOG, thereby enabling timely exit from pluripotency and differentiation into downstream lineages (Bertero et al. 2018).

m6A regulation of stem cell fate determination. a m6A balances pluripotency and lineage commitment in ESCs. As m6A enhances the degradation of methylated transcripts, deletion of METTL3-mediated m6A deposition in naïve ESCs upregulates dominating pluripotency factors to maintain a hyper-naïve state. However, deficiency of METTL3 in EpiSCs boosts the already-abundant lineage-priming genes to trigger cell specification. b In normal hematopoiesis, m6A promotes the early hematopoiesis and exerts divergent effects (activation or inhibition) to myeloid differentiation in different study systems. c In neurogenesis, m6A plays conflicting roles (activation or inhibition) in NPC differentiation. d In spermatogenesis, m6A promotes the SSCs specification. ESC, embryonic stem cell; EpiSC, primed epiblast stem cell; EHT, endothelial-to-hematopoietic transition; HSPC, hematopoietic stem/progenitor cell; NPC, neural progenitor cell; SSC, spermatogonial stem cell
Recently, increasing studies highlight the critical function of METTL3-mediated methylation in the regulation of chromatin state upon mammalian ESCs. It is reported that dysregulated m6A methylation in METTL3-knockout mESCs enhances carRNA levels and activates chromatin state and downstream transcription (Liu et al. 2020). The carRNAs include promoter-associated RNAs, eRNAs, and repeat RNAs. The METTL3-mediated m6A modifications on carRNAs (such as repeat RNA LINE1, elements of the long interspersed element-1 family) are recognized by YTHDC1 and processed to decay through the nuclear exosome targeting-mediated nuclear degradation pathway (Liu et al. 2020). Li and colleagues established a genome-wide correlation between m6A and chromatin modification H3K9me2; they found that YTHDC1 directly recruits the KDM3B, a H3K9me2 demethylase, to m6A-associated chromatin regions, facilitating H3K9me2 demethylation and transcription (Li et al. 2020). A more recent study showed that METTL13-deficient mESCs impair the deposition of multiple heterochromatin marks upon the IAP-type family of endogenous retroviruses. These methylated transcripts are bound by YTHDC1, which has an interaction with METTL3 and in turn contributes to the association of METTL3 with heterochromatin through the physical interplay of METTL3 and H3K9me3 methyltransferase SETDB1 and its cofactor TRIM28 (Xu et al. 2021). Consistently, Liu and colleagues showed that the retrotransposon repression and maintenance of mESCs identity is achieved by the m6A reader YTHDC1 (Liu et al. 2021). Mechanistically, YTHDC1 targets these m6A-modified retrotransposon transcripts (such as IAPs, ERVK, and LINE1), and that its depletion activates these silenced retrotransposons in a SETDB1-mediated H3K9me3-dependent manner. Furthermore, YTHDC1 and its targets act upstream of SETDB1 to repress the expression of Dux, the major inducer of the two-cell stage (2C)-like state transition, thereby guarding mESCs’ identity (Liu et al. 2021). Collectively, these surprising findings that m6A functions in chromatin states may provide a new regulatory mechanism for gene expression in ESC development.
m6A functions in other pluripotent stem cell fate determination
As a conserved mRNA modification, m6A has been described as a modulator or balancer, playing critical roles in stem cell fate determination during hematopoiesis, neurogenesis, and gametogenesis.
In the hematopoietic system, m6A was demonstrated to promote hematopoietic stem and progenitor cell (HSPC) specification during zebrafish and mouse embryogenesis (Zhang et al. 2017a; Lv et al. 2018) (Fig. 3b). In zebrafish, the lacking of METTL3 stabilizes the arterial endothelial transcripts, notch1a and rhoca, with YTHDF2-dependent degradation of m6A-modified transcripts, resulting in the continuous activation of the Notch signaling, block of the endothelial-to-hematopoietic transition (EHT), and repression of the earliest HSPC generation (Zhang et al. 2017a). Two key subunits of m6A writer complex, METTL3 and METTL14, transcripts are highly expressed in normal HSPCs, suggesting that METTL3 and METTL14 might act as crucial regulators in normal hematopoiesis differentiation (Martin and Park 2018). Removal of METTL3 in mouse cord blood–derived CD34 + HSPCs results in the inhibition of cell proliferation and facilitates myeloid differentiation in an in vitro myeloid differentiation culture condition. Overexpression of wild-type METTL3, instead of the catalytic mutant of METTL3, could inhibit cell differentiation and increase cell growth accordingly (Vu et al. 2017). Consistently, acceleration of myeloid differentiation and the upregulation of differentiation marker genes are also observed upon METTL14 silencing in normal HSPCs. Mechanistically, METTL14 knockdown downregulates the stability and translation of its targets such as MYB and MYC in an m6A-dependent manner, thereby promoting terminal myeloid differentiation (Weng et al. 2018). These two results suggested that METTL3/14-mediated m6A methylation inhibits the differentiation of HSPCs. In contrast, two studies reported that METTL3-depleted mice adult hematopoietic system blocks hematopoietic stem cell (HSC) differentiation and accumulates HSCs in adult bone marrow (Lee et al. 2019; Yao et al. 2018). METTL3-mediated m6A methylation in MYC transcript promotes protein translation in HSCs; depletion of METTL3 in HSCs downregulates MYC protein level and inhibits HSC differentiation (Lee et al. 2019). Conditional depletion of METTL13 from myeloid cells in mice does not affect myeloid cell number or function (Lee et al. 2019). Additionally, knockout of YTHDF2 results in functional HSPC expansion (Li et al. 2018b). As such, it needs more researches to reconcile the distinct m6A roles in HSPC differentiation.
The abundance of m6A in nervous system is higher than other organs, and raises in overall abundance ranged from the embryonic brain to the adult brain, implicating its essential roles in neuro-development. In the embryonic mouse cortex, neural progenitor cells (NPCs) generate neurons locating different cortical layers and sequentially convert to glial production before their depletion during early postnatal stages (Taverna et al. 2014). It is shown that the decreasing METTL3-mediated m6A levels leads to the protracted cell-cycle progression of cortical NPCs and reduced differentiation during mouse cortical neurogenesis, in which m6A facilitates the decay of neurogenesis-related transcripts (Yoon et al. 2017). Wang and colleagues found that conditional lacking METTL14 in mouse model displays markedly decreased NPC proliferation and premature differentiation (Fig. 3c). Mechanistically, a genome-wide increase in specific histone modifications corresponds with gene expression alteration and cellular phenotypes, providing an m6A-mediated histone modification upon regulating gene expression pathway (Wang et al. 2018). In particular, two specific m6A binding proteins in neural cells, FMRP and PRCC2A, are characterized and function in neural differentiation and oligodendrocyte specification, respectively (Edens et al. 2019; Wu et al. 2019). FMRP tunes neural differentiation via promoting nuclear export of methylated transcripts (Edens et al. 2019). PRRC2A controls oligodendrocyte progenitor cell (OPC) proliferation and specification by stabilizing a crucial oligodendroglial lineage determination transcription factor, Olig2, in an m6A-dependent manner (Wu et al. 2019).
In the process of gametogenesis, including spermatogenesis and oogenesis, the diploid primordial germ cell undertakes mitosis, meiosis, and cellular differentiation into haploid mature gametes: oocytes in females or spermatozoa in males. This heavily specialized process is accurately regulated at the transcriptional, post-transcriptional, and translational levels. The epitranscriptomic mark m6A has shown to play important roles in spermatogenesis and oogenesis. An early clue that m6A functions in spermatogenesis was the observation that removal of the m6A eraser ALKBH5 leads to impaired fertility (Zheng et al. 2013). In mammals, spermatogonial stem cells (SSCs) either self-renew or undergo twice mitotic divisions to generate spermatocytes. One study revealed that conditional depletion of either METTL3 or METTL14 causes translation dysregulation of m6A-modified transcripts, which is required for SSC proliferation and differentiation (Lin et al. 2017). Likewise, another independent study showed that the germ cell-specific METTL3-knockout mice impedes spermatogonial specification and blocks meiosis initiation by changing the alternative splicing of spermatogenesis-related genes and the global transcript expression pattern in testes (Xu et al. 2017). As an m6A cytoplasmic reader, YTHDC2 is indispensable in cell fate transition during oocyte and spermatogonial development. In spermatogenesis, YTHDC2 interacts with an essential meiosis process factor MEIOC (Abby et al. 2016) and facilitates a clean switch from mitosis to meiosis (Bailey et al. 2017). Furthermore, transcriptome analysis of YTHDC2-deficient male germ cells showed that meiosis-related transcripts are downregulated and mitotic cell–related genes are upregulated, ensuring that YTHDC2 recognizes m6A-modified germline transcripts to modulate the mitosis closure and transition to meiosis process (Wojtas et al. 2017). YTHDC2 also regulates oogenesis, in which YTHDC2-deficient mouse oocytes are arrested in the prophase of meiosis I and cause female fertility (Bailey et al. 2017; Wojtas et al. 2017). However, the specific molecular mechanism underlying what YTHDC2’s m6A binding function is and how it regulates oocyte and spermatogonial development needs to be further investigated. Additionally, YTHDC1, as another m6A nuclear reader, mediates the polyadenylation process in oocytes by recruitment of 3' end processing factors CPSF6, SRSF3, and SRSF7. Lacking of YTHDC1 blocks oocyte at the primary follicle stage and fails to achieve transition into secondary follicles (Kasowitz et al. 2018).
m5C functions in stem cell fate
Similar to m6A, m5C is also a remarkable epitranscriptomic modification and is involved in balancing stem cell self-renewal and specification and maintaining cellular homeostasis of most tissues. The functional studies of either DNMT2 or NSUN2 demonstrate that m5C on tRNAs is required for stem cell fate decision. Although DNMT2 is a homolog of DNA methyltransferase, it only has tRNA m5C methylation activity and no DNA m5C methylation activity (Raddatz et al. 2013). DNMT2 specifically installs m5C at the C38 position of tRNAs (Tuorto et al. 2015), and NSUN2 methylates tRNAs at C34, C48, C49, and C50 positions (Blanco et al. 2011; Blanco et al. 2014; Tuorto et al. 2012), which prevent tRNA cleavage. DNMT2-mediated tRNA m5C methylation is essential to ensure codon fidelity for accurate protein synthesis, and loss of DNMT2 disrupts cell-autonomous differentiation during hematopoiesis (Tuorto et al. 2015). NSUN2-mediated m5C on tRNAs is required for balancing self-renewal and differentiation, and knockout of NSUN2 in epidermal stem cells fails to initiate anagen and causes a substantial delay in accurate hair lineage differentiation (Blanco et al. 2011). In male gonads, NSUN2-mediated m5C on tRNAs is dispensable in spermatogonial stem cells, and specifically required for the meiotic progression of germ cells into the pachytene stage (Hussain et al. 2013). Likewise, NSUN2 is expressed in early neuroepithelial progenitors during human brain development and required for differentiation capacity. Loss of NSUN2 increases tRNA fragmentation and impairs migration and differentiation in neuroepithelial stem cells (Flores et al. 2017). A recent finding shows that NSUN2 is also an mRNA m5C methyltransferase (Yang et al. 2017a); however, whether the mRNA methylation activity NSUN2 is involved in the aforementioned functions remains unclear. NSUN7 installs m5C in eRNA, and loss-of-function mutation of NSUN7 has been associated with male infertility, implicating that the NSUN7-regulated m5C methylation pathway may serve as an essential balancer in germ cell differentiation (Harris et al. 2007). Notably, recent studies discovered that when cells respond to differentiation signals or other cellular and environmental incentives, m5C upon tRNA could coordinate translation rates of transcripts encoding functional proteins via the biogenesis of tRFs (Blanco et al. 2016). Removal of NSUN2- and DNMT2-mediated tRNA m5C methylation modulates the tRF formation and dysregulated intracellular translation (Tuorto et al. 2015; Blanco et al. 2016). Accordingly, the altered tRNA modification pattern, in particular m5C, could shape tRF biogenesis and their intracellular abundances. These generated tRFs influence global and/or gene-specific protein translation by occupying distinctive RNA-binding proteins, and therefore play critical roles in diverse biological developmental processes (Blanco et al. 2016; Zhang et al. 2018; Goodarzi et al. 2015).
RNA methylation functions in embryonic development
After gametogenesis, these two terminally specialized gametes are fused into a fertilized egg, followed by a programmed transition into a totipotent and pluripotent embryonic state, and then cell-fate determination and lineage-specific differentiation. In mammalian embryonic development, epigenetic remodeling of histone modifications, DNA methylation, chromatin accessibility, and 3D chromatin organization play indispensable roles in the regulation of gene expression. Additionally, the newly emerging epitranscriptomic marks (m6A and m5C) are found to function in embryonic development (Fig. 4).

Regulation of RNA methylation in embryonic development. The steps of mouse embryonic development along the timeline are represented, and the key processes implicated by the epitranscriptomic effectors are noted
m6A functions in embryo development
Following fertilization, the newly generated zygote sustains a transcriptionally quiescent state and initiates the early maternally programmed embryogenesis because it entirely depends on RNA and proteins from the mature oocyte. However, maternal stores (such as RNA and proteins) are insufficient to support later development, therefore followed by a gradual switch from clearance of maternal stores to zygote genome activation (ZGA). This original program is termed as the maternal-to-zygotic transition (MZT). Recently, increasing studies have illustrated that epitranscriptomic mark m6A and its effectors are critical to ensure suitable gene expression in both pre-implantation and post-implantation embryonic development (Zhang et al. 2020). In the pre-implantation embryonic development, m6A promotes clearance of maternal mRNA and activation of MZT process. As the essential component of the methyltransferase complex, METTL3-mediated m6A installation has a critical role in pre-implantation embryonic development. Knockdown of METTL3 severely impedes murine oocyte maturation by downregulating mRNA translation efficiency and causes defects in the MZT and ZGA processes (Sui et al. 2020). An mRNA interactome capture study identified m6A reader proteins YTHDF1-3 as maternal mRNA-binding partners during zebrafish MZT (Despic et al. 2017), implicating that YTHDF protein–mediated m6A regulation may engage in the MZT process. Indeed, over one-third of transcripts in zebrafish are methylated, and the rapid clearance of these maternal mRNAs is accelerated by undertaking an m6A-mediated YTHDF2-dependent mRNA decay pathway (Zhao et al. 2017). Lacking of zebrafish YTHDF2 embryos impedes the degradation of m6A-methyalted maternal mRNAs, thereby failing to initiate timely MZT and remaining developmental interruption during larval period (Zhao et al. 2017). Recently, another study reported that m6A promotes maternal mRNA deadenylation and degradation, but these effects are not simply dependent on YTHDF2 (Kontur et al. 2020). m6A binding proteins YTHDF1-3 function redundantly in ovary development and zebrafish viability, confirming the indispensable of m6A in MZT and early embryogenesis (Kontur et al. 2020). Another type of m6A readers IGF2BPs can also exert distinctive functions during early embryonic development. Removal of IGF2BP1 induces cell apoptosis in early mouse parthenogenetic embryogenesis (Hao et al. 2020). Mouse embryos with maternal deletion of IGF2BP2 are arrested at the 2‐cell‐stage with and ZGA-related genes such as CCAR1 and RPS14 are downregulated (Liu et al. 2019). IGF2BP3 is identified as a crucial regulator for maintaining maternal mRNA stability in zebrafish. Lacking of maternal IGF2BP3 accelerates maternal mRNAs decay prior to MZT and leads to severely impaired development, including abnormal cytoskeleton organization and cell division (Ren et al. 2020). However, whether IGF2BPs serve as m6A readers to regulate mRNA stability in ZGA and MZT processes and whether YTHDF2 and IGF2BPs recognize distinct targeted sites in early embryogenesis remain to be deciphered.
In addition to pre-implantation embryogenesis, m6A is also indispensable in post-implantation embryonic development. For example, METTL3-deficient mouse embryos retain expression of pluripotency factors and display a state of hyperpluripotency, thereby failing to undertake lineage differentiation and leading to lethality during the post-implantation embryonic development (Batista et al. 2014; Geula et al. 2015). Other m6A effectors are also crucial in embryonic development. METTL16 is identified as a U6 snRNA m6A writer and specifically deposits m6A on 3′ UTR hairpin of SAM synthetase MAT2A pre-mRNA for intron retention, which promotes MAT2A nuclear degradation and affects SAM homeostasis (Pendleton et al. 2017). Further mechanism study revealed that YTHDC1 recognizes the m6A modifications on MAT2A hairpin for mRNA degradation (Shima et al. 2017). The function of METTL16 in SAM homeostasis control is required for mouse early embryonic development. Mettl16-depleted embryos appear normal before implantation but exhibit developmental arrest around the time of implantation, which is due to the consequence that the SAM limitation leads to massive transcriptome dysregulation at the embryonic day 3.5 blastocysts stage (Mendel et al. 2018). Notably, given that mice with knockout of cytoplasmic m6A reader YTHDF2 can survive to late embryonic developmental stages but removal of either METTL3 or the nuclear m6A reader YTHDC1 causes embryonic lethality (Kasowitz et al. 2018; Geula et al. 2015; Batista et al. 2014; Ivanova et al. 2017) implies that m6A could play critical roles in the nucleus during embryogenesis.
m5C functions in embryo development
During zebrafish embryogenesis, m5C was demonstrated to maintain maternal mRNA stability in MZT process, in which transcripts with more m5C-targeted sites and higher m5C-methylated levels exhibited more stable expression levels during both 2 to 4-h and 4 to 6-h post-fertilization (hpf) stages, and its binding protein YBX1 functions in impeding early gastrulation arrest. The mechanism study showed that YBX1 directly interacts with m5C-containing mRNAs via a π-π interaction and enhances m5C-modified maternal genes by recruitment of PABPC1A (Yang et al. 2019). Furthermore, Rai and colleagues indicated that knockdown of DNMT2 in zebrafish embryos confers deficiency of terminal cellular differentiation toward the retina, liver, and brain. Considering that DNMT2 functions primarily as a genomic DNA methyltransferase and a cytoplasmic tRNA methyltransferase, the results showed that the organ differentiation requires cytoplasmic DNMT2 activity, not in the nucleus, therefore establishing a potential contact between cellular differentiation and tRNA m5C modifications (Rai et al. 2007).
RNA methylation functions in cancer
Cancer stem cells (CSCs) are termed as intratumor heterogeneity and hitherto-unappreciated subclass of neoplastic cells within tumors, and have stem cell-like capacities to self-renew, differentiate, and generate tissue to propagate the tumor (Hanahan and Weinberg 2011). Currently, amount lines of evidence illustrate that the dysregulation of epitranscriptomic modifications is engaged in the pathogenesis of CSCs, including tumorigenesis, cell proliferation, and cell fate decision. Epitranscriptomic modifications are also involved in various cancer subtypes, such as acute myeloid leukemia, glioblastoma, liver cancer, breast cancer, cervical cancer, and bladder cancer (Fig. 5), further implying its potential roles in cancer program.

RNA methylation in cancer progression. Epitranscriptomic effectors in diverse types of human cancers play oncogenic (red) or tumor-suppressive roles (blue) in cancer progression. AML, acute myeloid leukemia; GBM, glioblastoma; HCC, hepatocellular carcinoma; ESCC, esophageal squamous cell carcinoma
m6A functions in acute myeloid leukemia
AML is a prevalent hematopoietic malignancy, caused by the advance of stem cell-like renewal capability and the impediment of normal differentiation. Epigenetic modifications, including DNA methylations and histone modifications, are crucial to AML pathogenesis (Chen et al. 2010). Accordingly, many studies have pointed to m6A as an important factor for hematopoietic homeostasis and leukemogenesis.
The aberrant m6A methylation of mRNA facilitates myeloid differentiation and cell proliferation. The expression levels of METTL3 and METTL14 are more abundant in AML (Vu et al. 2017; Weng et al. 2018), and METTL3 is captured by a genome-wide CRISPR dropout screening, and identified as a necessary gene for AML cell growth (Barbieri et al. 2017). Two independent studies illustrated the oncogenic function of METTL3 in AML. Vu and colleagues found that METTL3 depletion in AML cells induces cell differentiation and apoptosis, coupled with reduced cell proliferation, by downregulating translation of its RNA targets such as C-MYC, BCL-2, and PTEN, and stimulating the activated AKT level (Vu et al. 2017). Barbieri and colleagues found that METTL3 binds chromatin and localizes to the transcriptional start sites (TSS) of active genes in an independent manner of METTL14 (Barbieri et al. 2017). METTL3 is stably recruited to promoters of transcription factors (SP1 and SP2) by CEBPZ, resulting in m6A methylation and enhanced translation. Given that SP1 promotes MYC transcription, the resulting increased translation of SP1 in turn leads to upregulated MYC signaling in leukemogenesis (Barbieri et al. 2017). Likewise, METTL14 also functions as an oncogene in the self-renewal of leukemia stem cells (LSCs) and AML maintenance via an m6A-meditated MYB/MYC-dependent regulatory pathway (Weng et al. 2018). Additionally, other methyltransferase components such as WTAP and RBM15 are also implicated in leukemogenesis, while its association with m6A remains to be elucidated (Bansal et al. 2014; Ma et al. 2001). FTO is significantly upregulated in AMLs with t(11q23)/MLL rearrangements, t(15;17)/PML-RARA, FLT3-ITD, and/or NPM1 mutations, and exerts an oncogenic role in AML by post-transcriptionally downregulating expression of critical transcripts such as ASB2 and RARA through m6A demethylation activity (Li et al. 2017b). This regulatory mechanism seems to specifically appear in isocitrate dehydrogenase 1 (IDH1) wild-type AML cells, while IDH mutants induce the conversion of α-KG to R enantiomer of 2-hydroxyglutarate (R-2HG). The demethylation activity of FTO is suppressed by R-2HG and plays critical oncogenic roles in leukemia through an FTO/m6A/MYC-CEBPA axis (Su et al. 2018).
m6A functions in glioblastoma
Glioblastoma (GBM) is one of the most prevailing primary malignant brain tumors, with a median survival of around 10 months (Rick et al. 2018). GBM is characterized by notable heterogeneity between intra- and inter-tumor and contains apical cells of cellular hierarchies with stem-like properties. Glioblastoma stem cells (GSCs) belong to a subclass of CSCs with capacities to facilitate self-renewal, tumor growth, and invasion (Kalkan 2015). Recent studies uncovered that m6A is correlated with GSC self-renewal, tumorigenesis, and therapy resistance in GBM.
Recently, although there are notable exceptions, several studies indicate that m6A generally impairs self-renewal and tumorigenesis in GBM. m6A methylation levels are significantly elevated upon induced GSC differentiation. Knockdown of either METTL3 or METTL14 enhances GSC growth and self-renewal and upregulates expression level of oncogenic genes with downregulated m6A methylation, including ADAM19, EPHA3, and KLF4 (Cui et al. 2017). By contrast, it is reported that METTL3 expression level is upregulated in GSCs and attenuated upon differentiation. METTL3-silenced CSCs inhibit GSC maintenance and enhance radiation sensitivity by stabilizing the downstream target SOX2 with recruitment of the HuR, suggesting that METTL3 functions as an oncogene in GBM (Visvanathan et al. 2018). These conflicting results are possibly caused by the variability of GBM cell types. Additionally, ALKBH5 is highly expressed in GSCs and positively functions in cell proliferation, self-renewal, and tumorigenicity. Removal of ALKBH5 increases m6A methylation on FOXM1 pre-mRNA and downregulates FOXM1 transcript expression level, leading to impaired proliferation and tumorigenesis of GSCs. The ALKBH5-FOXM1 regulatory pathway can be further strengthened by a lncRNA antisense to FOXM1 (FOXM1-AS) (Zhang et al. 2017b). Likewise, FTO undertakes oncogenic functions in GBM program (Cui et al. 2017; Su et al. 2018), and MA2, the FTO chemical inhibitor, can coordinately prolong the life span in GSC-transplanted mice (Cui et al. 2017), suggesting that FTO-regulated m6A awaits to be a promising therapeutic target for GBM treatment.
m6A functions in hepatocellular carcinoma
Hepatocellular carcinoma (HCC) is one of the most commonly diagnosed primary liver cancer and rates as the fifth malignant tumor worldwide. Aberrant expression of m6A effectors is associated with HCC progression. For example, METTL3 negatively predicts prognostic survival, and its overexpression remarkably enhances HCC growth. Knockdown of METTL3 significantly reduces HCC tumorigenicity by increasing SOCS2 expression via an m6A-mediated YTHDF2-dependent mRNA decay pathway (Chen et al. 2018). However, Ma and colleagues showed that METTL14 exhibits a converse role in HCC development. Decreased METTL14 is observed in HCC and closely associated with tumor metastasis and prognosis. In particular, METTL14 interacts with DGCR8 and positively processes pri-miR126 into mature miR126, a typical tumor repressor in HCC metastasis, in an m6A-dependent manner (Ma et al. 2017). Moreover, two studies exhibited controversial functions of YTHDF2 in the HCC program. One study showed that miR-145 is suppressed in HCC patients and downregulates the expression level of oncogenic gene, YTHDF2 (Yang et al. 2017b). Zhong and colleagues found that YTHDF2 represses cell proliferation and growth in HCC and functions as a HCC tumor suppressor via destabilizing EGFR transcript (Zhong et al. 2019).
m6A functions in breast cancer
Breast cancer is the highest incidence of malignant tumor among women. Aberrant expression of m6A effectors are involved in CSC maintenance under tumor microenvironment in breast cancer. As the typical feature of tumor microenvironment, hypoxia enhances ALKBH5 expression in breast cancer stem cells (BCSCs) in a HIF-dependent manner. The upregulated ALKBH5 decreases m6A methylation in NANOG transcript for mRNA stabilization, thereby enhancing BCSC percentage and phenotype (Zhang et al. 2016a). Meanwhile, zinc finger protein 217 (ZNF217) expression is also upregulated in an HIF-dependent manner under hypoxia. The increased ZNF217 leads to m6A hypomethylation by disrupting METTL3-mediated m6A methylation and cooperating with ALKBH5, therefore resulting in NANOG upregulation and promoting BCSC maintenance (Zhang et al. 2016b). Another study showed that METTL3 is highly expressed in clinical breast cancer samples and engages in a positive feedback loop, cooperated with HBXIP and let-7g miRNA (HBXIP/let-7g/METTL3/HBXIP), to accelerate proliferation in breast cancer. Mechanistically, the oncoprotein HBXIP upregulates METTL3 by suppressing miRNA let-7g, and the increased METTL3, in turn, enhances HBXIP expression through the promotion of m6A modification (Cai et al. 2018).
m5C functions in cancer
NSUN2 has been identified as an MYC target correlated to the MYC-dependent proliferation in tumors (Frye and Watt 2006), and DNMT2 is highly expressed in various tumor tissue cells (Forbes et al. 2015), implying that DNMT2 functions an oncogenic role in tumorigenesis. Usage of azacytidine could inhibit DNMT2-mediated RNA methylation and reduce metabolism of cancer cells (Schaefer et al. 2009). A homozygous splice mutation in NSUN2 brings about a loss of m5C on known NSUN2 targets in tRNA, and likely acts as a cause of a Dubowitz-like syndrome (Martinez et al. 2012). These potential lines of evidence demonstrate that tRNA m5C and its effectors may function in cancer development.
Recently, two independent studies revealed that m5C methylation in mRNA and lncRNA enhances the cancer susceptibility. One study showed that NSUN2 enhances tumor metastasis by m5C methylation of NMR lncRNA in esophageal squamous cell carcinoma (ESCC). The m5C methylated NMR along with BPTF play oncogenic roles in promoting expression of key drivers of esophageal cancer such as MMP3 and MMP10 (Li et al. 2018a). The other research showed that NSUN2-mediated m5C methylation highly occurred on oncogenic mRNAs of human urothelial carcinoma of the bladder (UCB). Among these, the heparin-binding growth factor (HDGF) transcript is methylated by NSUN2 and stabilized by m5C reader YBX1, which in turn facilitates the pathogenesis of bladder cancer (Chen et al. 2019a). Together, m5C on tRNA may function in modulating cancer metabolism, and m5C on mRNA potentially plays roles in enhancing the stability of cancer-related transcripts. Nevertheless, the complete prospect of functions of m5C in cancers awaits to be discovered.
m1A functions in cancer
Compared with normal tissues, ALKBH3 is highly expressed in pancreatic (Konishi et al. 2005), lung (Tasaki et al. 2011), and urothelial cancers (Shimada et al. 2012), and is evenly identified as the high-grade prostate cancer marker, implying that ALKBH3 may function in the tumorigenesis process and would be helpful for the early diagnosis.
In diverse cancer subtypes, ALKBH3 functions as an m1A demethylase and plays an oncogenic role in cancer progression via various regulatory mechanisms. In breast and ovarian cancer, ALKBH3 functions as an oncogene for cancer invasiveness via stabilizing colony-stimulating factor 1 (CSF-1) transcript in an ALKBH3-mediated m1A demethylation manner (Woo and Chambers 2019). As a tRNA m1A demethylase, the ALKBH3-mediated demethylation responds sensitively to angiogenin (ANG)-dependent tRNA cleavage, followed by generating tRNA-derived sRNAs (tDRs). The produced tDRs are engaged in ALKBH3-mediated cancer progression via modulating ribosome assembly and preventing apoptosis pathway triggered by cytochrome c (Cyt c) (Chen et al. 2019b). Additionally, ALKBH3 is also responsible for DNA damage caused by alkylating agents (Duncan et al. 2002; Dango et al. 2011), which means that ALKBH3 may play potential roles in tumorigenesis by an RNA m1A-independent manner.
Conclusions
Recently, increasing studies showed that epitranscriptomic marks are established as critical regulators in gene expression, and are of underlying importance for diverse biological developments. In the process of stem cell differentiation, the dynamic deposition of RNA modifications maintains the balance between pluripotency and differentiation into downstream lineages in an ordered manner to enable appropriate cellular development. As for early embryogenesis, RNA modifications, in particular m6A, are critical to eliminate maternal mRNA in an m6A-mediated YTHDF2-dependent mRNA decay pathway and to promote the ZGA process, ensuring suitable gene expression. Furthermore, RNA modifications along with their effectors also have oncogenic or tumor-suppressor functions in the cancer pathogenesis, including tumorigenesis, cancer cell proliferation, and cancer cell fate decision of numerous cancer types. In some cases, the same enzyme can play conflicting functions in diverse cancer types, which can be explained by tumor specificity as well as the diversity of RNA modifications. Peculiarly, both m6A writers (METTL3 and METTL14) and m6A eraser (FTO) function as an oncogene in AML development and progression. Considering that there are numerous m6A-modified tumor-related mRNAs and opposite roles of m6A binding proteins (YTHDF2 and IGF2BPs), the dysregulation of m6A installation could disrupt the homeostasis of post-transcriptional gene expression, thereby leading to oncogenic function in cancerogenesis. Notably, increasing studies showed that METTL3-mediated m6A methylation modulates chromatin state in mESCs, providing a new regulatory mechanism in gene expression of development.
Code availability
Not applicable.
References
Abby E, Tourpin S, Ribeiro J, Daniel K, Messiaen S, Moison D, et al. Implementation of meiosis prophase I programme requires a conserved retinoid-independent stabilizer of meiotic transcripts. Nat Commun. 2016;7:10324.
Aguilo F, Li S, Balasubramaniyan N, Sancho A, Benko S, Zhang F, et al. Deposition of 5-methylcytosine on enhancer RNAs enables the coactivator function of PGC-1α. Cell Rep. 2016;14(3):479–92.
Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 is a mediator of m6A-dependent nuclear RNA processing events. Cell. 2015;162(6):1299–308.
Anderson JT, Droogmans L. Biosynthesis and function of 1-methyladenosine in transfer RNA. In: Grosjean H, editor. Fine-tuning of RNA functions by modification and editing. Berlin, Heidelberg: Springer Berlin Heidelberg; 2005. p. 121–39.
Anderson J, Phan L, Cuesta R, Carlson BA, Pak M, Asano K, et al. The essential Gcd10p-Gcd14p nuclear complex is required for 1-methyladenosine modification and maturation of initiator methionyl-tRNA. Genes Dev. 1998;12(23):3650–62.
Bailey AS, Batista PJ, Gold RS, Chen YG, de Rooij DG, Chang HY, et al. The conserved RNA helicase YTHDC2 regulates the transition from proliferation to differentiation in the germline. eLife. 2017;6:e26116.
Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Penalva LO, et al. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia. 2014;28(5):1171–4.
Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millán-Zambrano G, Robson SC, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature. 2017;552(7683):126–31.
Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, et al. m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15(6):707–19.
Bertero A, Brown S, Madrigal P, Osnato A, Ortmann D, Yiangou L, et al. The SMAD2/3 interactome reveals that TGFβ controls m6A mRNA methylation in pluripotency. Nature. 2018;555(7695):256–9.
Blanco S, Bandiera R, Popis M, Hussain S, Lombard P, Aleksic J, et al. Stem cell function and stress response are controlled by protein synthesis. Nature. 2016;534(7607):335–40.
Blanco S, Dietmann S, Flores JV, Hussain S, Kutter C, Humphreys P, et al. Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J. 2014;33(18):2020–39.
Blanco S, Kurowski A, Nichols J, Watt FM, Benitah SA, Frye M. The RNA-methyltransferase Misu (NSun2) poises epidermal stem cells to differentiate. PLoS Genet. 2011;7(12):e1002403.
Boccaletto P, Machnicka MA, Purta E, Piątkowski P, Bagiński B, Wirecki TK, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018;46(D1):D303-d7.
Bohnsack KE, Höbartner C, Bohnsack MT. Eukaryotic 5-methylcytosine (m5C) RNA methyltransferases: mechanisms, cellular functions, and links to disease. Genes. 2019;10(2):102.
Brzezicha B, Schmidt M, Makalowska I, Jarmolowski A, Pienkowska J, Szweykowska-Kulinska Z. Identification of human tRNA:m5C methyltransferase catalysing intron-dependent m5C formation in the first position of the anticodon of the pre-tRNA Leu (CAA). Nucleic Acids Res. 2006;34(20):6034–43.
Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang Z, et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018;415:11–9.
Chen J, Odenike O, Rowley JD. Leukaemogenesis: more than mutant genes. Nat Rev Cancer. 2010;10(1):23–36.
Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67(6):2254–70.
Chen X, Li A, Sun BF, Yang Y, Han YN, Yuan X, et al. 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat Cell Biol. 2019a;21(8):978–90.
Chen Z, Qi M, Shen B, Luo G, Wu Y, Li J, et al. Transfer RNA demethylase ALKBH3 promotes cancer progression via induction of tRNA-derived small RNAs. Nucleic Acids Res. 2019b;47(5):2533–45.
Chujo T, Suzuki T. Trmt61B is a methyltransferase responsible for 1-methyladenosine at position 58 of human mitochondrial tRNAs. RNA. 2012;18(12):2269–76.
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18(11):2622–34.
Dango S, Mosammaparast N, Sowa ME, Xiong LJ, Wu F, Park K, et al. DNA unwinding by ASCC3 helicase is coupled to ALKBH3-dependent DNA alkylation repair and cancer cell proliferation. Mol Cell. 2011;44(3):373–84.
Delatte B, Wang F, Ngoc LV, Collignon E, Bonvin E, Deplus R, et al. Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science. 2016;351(6270):282–5.
Despic V, Dejung M, Gu M, Krishnan J, Zhang J, Herzel L, et al. Dynamic RNA-protein interactions underlie the zebrafish maternal-to-zygotic transition. Genome Res. 2017;27(7):1184–94.
Dominissini D. Roadmap to the epitranscriptome. Science. 2014;346(6214):1192.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–6.
Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS, et al. The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA. Nature. 2016;530(7591):441–6.
Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, et al. YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. 2016;7:12626.
Dubin DT, Taylor RH. The methylation state of poly A-containing messenger RNA from cultured hamster cells. Nucleic Acids Res. 1975;2(10):1653–68.
Duncan T, Trewick SC, Koivisto P, Bates PA, Lindahl T, Sedgwick B. Reversal of DNA alkylation damage by two human dioxygenases. Proc Natl Acad Sci. 2002;99(26):16660–5.
Edens BM, Vissers C, Su J, Arumugam S, Xu Z, Shi H, et al. FMRP modulates neural differentiation through m6A-dependent mRNA nuclear export. Cell Rep. 2019;28(4):845-54.e5.
El Yacoubi B, Bailly M, de Crécy-Lagard V. Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu Rev Genet. 2012;46:69–95.
Flores JV, Cordero-Espinoza L, Oeztuerk-Winder F, Andersson-Rolf A, Selmi T, Blanco S, et al. Cytosine-5 RNA methylation regulates neural stem cell differentiation and motility. Stem Cell Rep. 2017;8(1):112–24.
Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(D1):D805–11.
Frye M, Harada BT, Behm M, He C. RNA modifications modulate gene expression during development. Science. 2018;361(6409):1346–9.
Frye M, Watt FM. The RNA methyltransferase Misu (NSun2) mediates Myc-induced proliferation and is upregulated in tumors. Curr Biol. 2006;16(10):971–81.
Fu L, Guerrero CR, Zhong N, Amato NJ, Liu Y, Liu S, et al. Tet-mediated formation of 5-hydroxymethylcytosine in RNA. J Am Chem Soc. 2014;136(33):11582–5.
Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, et al. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol. 2002;4(3):222–31.
Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, et al. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science. 2015;347(6225):1002–6.
Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh CL, Zhang X, et al. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science. 2006;311(5759):395–8.
Goodarzi H, Liu X, Nguyen HC, Zhang S, Fish L, Tavazoie SF. Endogenous tRNA-derived fragments suppress breast cancer progression via YBX1 displacement. Cell. 2015;161(4):790–802.
Guy MP, Phizicky EM. Two-subunit enzymes involved in eukaryotic post-transcriptional tRNA modification. RNA Biol. 2014;11(12):1608–18.
Haag S, Warda AS, Kretschmer J, Günnigmann MA, Höbartner C, Bohnsack MT. NSUN6 is a human RNA methyltransferase that catalyzes formation of m5C72 in specific tRNAs. RNA. 2015;21(9):1532–43.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Hao J, Hu H, Jiang Z, Yu X, Li C, Chen L, et al. microRNA-670 modulates Igf2bp1 expression to regulate RNA methylation in parthenogenetic mouse embryonic development. Sci Rep. 2020;10(1):4782.
Harris T, Marquez B, Suarez S, Schimenti J. Sperm motility defects and infertility in male mice with a mutation in Nsun7, a member of the Sun domain-containing family of putative RNA methyltransferases. Biol Reprod. 2007;77(2):376–82.
Hayrapetyan A, Grosjean H, Helm M. Effect of a quaternary pentamine on RNA stabilization and enzymatic methylation. Biol Chem. 2009;390(9):851–61.
He C, Bozler J, Janssen KA, Wilusz JE, Garcia BA, Schorn AJ, et al. TET2 chemically modifies tRNAs and regulates tRNA fragment levels. Nat Struct Mol Biol. 2021;28(1):62–70.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20(3):285–95.
Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M, et al. Histone H3 trimethylation at lysine 36 guides m6A RNA modification co-transcriptionally. Nature. 2019;567(7748):414–9.
Hussain S, Tuorto F, Menon S, Blanco S, Cox C, Flores JV, et al. The mouse cytosine-5 RNA methyltransferase NSun2 is a component of the chromatoid body and required for testis differentiation. Mol Cell Biol. 2013;33(8):1561–70.
Ivanova I, Much C, Di Giacomo M, Azzi C, Morgan M, Moreira PN, et al. The RNA m6A reader YTHDF2 is essential for the post-transcriptional regulation of the maternal transcriptome and oocyte competence. Mol Cell. 2017;67(6):1059-67.e4.
Jia G, Fu Y, He C. Reversible RNA adenosine methylation in biological regulation. Trends Genet. 2013;29(2):108–15.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–7.
Kalkan R. Glioblastoma stem cells as a new therapeutic target for glioblastoma. Clin Med Insights Oncol. 2015;9:95–103.
Kasowitz SD, Ma J, Anderson SJ, Leu NA, Xu Y, Gregory BD, et al. Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte development. PLoS Genet. 2018;14(5):e1007412.
Konishi N, Nakamura M, Ishida E, Shimada K, Mitsui E, Yoshikawa R, et al. High expression of a new marker PCA-1 in human prostate carcinoma. Clin Cancer Res. 2005;11(14):5090–7.
Kontur C, Jeong M, Cifuentes D, Giraldez AJ. Ythdf m6A readers function redundantly during zebrafish development. Cell Rep. 2020;33(13):108598.
Lee H, Bao S, Qian Y, Geula S, Leslie J, Zhang C, et al. Stage-specific requirement for Mettl3-dependent m6A mRNA methylation during haematopoietic stem cell differentiation. Nat Cell Biol. 2019;21(6):700–9.
Li X, Xiong X, Wang K, Wang L, Shu X, Ma S, et al. Transcriptome-wide mapping reveals reversible and dynamic N1-methyladenosine methylome. Nat Chem Biol. 2016a;12(5):311–6.
Li X, Xiong X, Yi C. Epitranscriptome sequencing technologies: decoding RNA modifications. Nat Methods. 2016b;14(1):23–31.
Li X, Xiong X, Zhang M, Wang K, Chen Y, Zhou J, et al. Base-resolution mapping reveals distinct m1A methylome in nuclear- and mitochondrial-encoded transcripts. Mol Cell. 2017a;68(5):993-1005.e9.
Li Y, Li J, Luo M, Zhou C, Shi X, Yang W, et al. Novel long noncoding RNA NMR promotes tumor progression via NSUN2 and BPTF in esophageal squamous cell carcinoma. Cancer Lett. 2018a;430:57–66.
Li Y, Xia L, Tan K, Ye X, Zuo Z, Li M, et al. N6-Methyladenosine co-transcriptionally directs the demethylation of histone H3K9me2. Nat Genet. 2020;52(9):870–7.
Li Z, Qian P, Shao W, Shi H, He XC, Gogol M, et al. Suppression of m6A reader Ythdf2 promotes hematopoietic stem cell expansion. Cell Res. 2018b;28(9):904–17.
Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N6-methyladenosine RNA demethylase. Cancer Cell. 2017b;31(1):127–41.
Lin Z, Hsu PJ, Xing X, Fang J, Lu Z, Zou Q, et al. Mettl3-/Mettl14-mediated mRNA N6-methyladenosine modulates murine spermatogenesis. Cell Res. 2017;27(10):1216–30.
Liu F, Clark W, Luo G, Wang X, Fu Y, Wei J, et al. ALKBH1-mediated tRNA demethylation regulates translation. Cell. 2016;167(7):1897.
Liu HB, Muhammad T, Guo Y, Li MJ, Sha QQ, Zhang CX, et al. RNA-binding protein IGF2BP2/IMP2 is a critical maternal activator in early zygotic genome activation. Adv Sci. 2019;6(15):1900295.
Liu J, Dou X, Chen C, Chen C, Liu C, Xu MM, et al. N6-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science. 2020;367(6477):580–6.
Liu J, Gao M, He J, Wu K, Lin S, Jin L, et al. The RNA m6A reader YTHDC1 silences retrotransposons and guards ES cell identity. Nature. 2021;591(7849):322–6.
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–5.
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518(7540):560–4.
Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017;45(10):6051–63.
Lv J, Zhang Y, Gao S, Zhang C, Chen Y, Li W, et al. Endothelial-specific m6A modulates mouse hematopoietic stem and progenitor cell development via Notch signaling. Cell Res. 2018;28(2):249–52.
Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH, Wang F, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N6-methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65(2):529–43.
Ma Z, Morris SW, Valentine V, Li M, Herbrick JA, Cui X, et al. Fusion of two novel genes, RBM15 and MKL1, in the t(1;22)(p13;q13) of acute megakaryoblastic leukemia. Nat Genet. 2001;28(3):220–1.
Maden BE. Identification of the locations of the methyl groups in 18 S ribosomal RNA from Xenopus laevis and man. J Mol Biol. 1986;189(4):681–99.
Martin GH, Park CY. Meddling with METTLs in normal and leukemia stem cells. Cell Stem Cell. 2018;22(2):139–41.
Martinez FJ, Lee JH, Lee JE, Blanco S, Nickerson E, Gabriel S, et al. Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome. J Med Genet. 2012;49(6):380–5.
Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, et al. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature. 2017;541(7637):371–5.
Mauer J, Sindelar M, Despic V, Guez T, Hawley BR, Vasseur JJ, et al. FTO controls reversible m6Am RNA methylation during snRNA biogenesis. Nat Chem Biol. 2019;15(4):340–7.
Mendel M, Chen KM, Homolka D, Gos P, Pandey RR, McCarthy AA, et al. Methylation of structured RNA by the m6A writer METTL16 is essential for mouse embryonic development. Mol Cell. 2018;71(6):986-1000.e11.
Meng TG, Lu X, Guo L, Hou GM, Ma XS, Li QN, et al. Mettl14 is required for mouse postimplantation development by facilitating epiblast maturation. FASEB J. 2019;33(1):1179–87.
Metodiev MD, Spåhr H, LoguercioPolosa P, Meharg C, Becker C, Altmueller J, et al. NSUN4 is a dual function mitochondrial protein required for both methylation of 12S rRNA and coordination of mitoribosomal assembly. PLoS Genet. 2014;10(2):e1004110.
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5′ UTR m6A promotes cap-independent translation. Cell. 2015;163(4):999–1010.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149(7):1635–46.
Motorin Y, Helm M. tRNA stabilization by modified nucleotides. Biochemistry. 2010;49(24):4934–44.
Nakano S, Suzuki T, Kawarada L, Iwata H, Asano K, Suzuki T. NSUN3 methylase initiates 5-formylcytidine biogenesis in human mitochondrial tRNAMet. Nat Chem Biol. 2016;12(7):546–51.
Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, et al. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537(7620):369–73.
Peifer C, Sharma S, Watzinger P, Lamberth S, Kötter P, Entian KD. Yeast Rrp8p, a novel methyltransferase responsible for m1A 645 base modification of 25S rRNA. Nucleic Acids Res. 2013;41(2):1151–63.
Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, et al. The U6 snRNA m6A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell. 2017;169(5):824-35.e14.
Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24(2):177–89.
Raddatz G, Guzzardo PM, Olova N, Fantappié MR, Rampp M, Schaefer M, et al. Dnmt2-dependent methylomes lack defined DNA methylation patterns. Proc Natl Acad Sci U S A. 2013;110(21):8627–31.
Rai K, Chidester S, Zavala CV, Manos EJ, James SR, Karpf AR, et al. Dnmt2 functions in the cytoplasm to promote liver, brain, and retina development in zebrafish. Genes Dev. 2007;21(3):261–6.
RajBhandary UL, Stuart A, Faulkner RD, Chang SH, Khorana HG. Nucleotide sequence studies on yeast phenylalanine sRNA. Cold Spring Harb Symp Quant Biol. 1966;31:425–34.
Ren F, Lin Q, Gong G, Du X, Dan H, Qin W, et al. Igf2bp3 maintains maternal RNA stability and ensures early embryo development in zebrafish. Commun Biol. 2020;3(1):94.
Rick J, Chandra A, Aghi MK. Tumor treating fields: a new approach to glioblastoma therapy. J Neurooncol. 2018;137(3):447–53.
Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, et al. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. eLife. 2017;6:e31311.
Ruzicka K, Zhang M, Campilho A, Bodi Z, Kashif M, Saleh M, et al. Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol. 2017;215(1):157–72.
Safra M, Sas-Chen A, Nir R, Winkler R, Nachshon A, Bar-Yaacov D, et al. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature. 2017;551(7679):251–5.
Schaefer M, Hagemann S, Hanna K, Lyko F. Azacytidine inhibits RNA methylation at DNMT2 target sites in human cancer cell lines. Cancer Res. 2009;69(20):8127–32.
Schosserer M, Minois N, Angerer TB, Amring M, Dellago H, Harreither E, et al. Methylation of ribosomal RNA by NSUN5 is a conserved mechanism modulating organismal lifespan. Nat Commun. 2015;6:6158.
Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell Rep. 2014;8(1):284–96.
Sendinc E, Valle-Garcia D, Dhall A, Chen H, Henriques T, Navarrete-Perea J, et al. PCIF1 catalyzes m6Am mRNA methylation to regulate gene expression. Mol Cell. 2019;75(3):620-30.e9.
Sharma S, Watzinger P, Kötter P, Entian KD. Identification of a novel methyltransferase, Bmt2, responsible for the N-1-methyl-adenosine base modification of 25S rRNA in Saccharomyces cerevisiae. Nucleic Acids Res. 2013a;41(10):5428–43.
Sharma S, Yang J, Watzinger P, Kötter P, Entian KD. Yeast Nop2 and Rcm1 methylate C2870 and C2278 of the 25S rRNA, respectively. Nucleic Acids Res. 2013b;41(19):9062–76.
Shen H, Ontiveros RJ, Owens MC, Liu MY, Ghanty U, Kohli RM, et al. TET-mediated 5-methylcytosine oxidation in tRNA promotes translation. J Biol Chem. 2020;296:100087.
Shen Q, Zhang Q, Shi Y, Shi Q, Jiang Y, Gu Y, et al. Tet2 promotes pathogen infection-induced myelopoiesis through mRNA oxidation. Nature. 2018;554(7690):123–7.
Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 2017;27(3):315–28.
Shima H, Matsumoto M, Ishigami Y, Ebina M, Muto A, Sato Y, et al. S-Adenosylmethionine synthesis is regulated by selective N6-adenosine methylation and mRNA degradation involving METTL16 and YTHDC1. Cell Rep. 2017;21(12):3354–63.
Shimada K, Fujii T, Tsujikawa K, Anai S, Fujimoto K, Konishi N. ALKBH3 contributes to survival and angiogenesis of human urothelial carcinoma cells through NADPH oxidase and tweak/Fn14/VEGF signals. Clin Cancer Res. 2012;18(19):5247–55.
Slobodin B, Han R, Calderone V, Vrielink J, Loayza-Puch F, Elkon R, et al. Transcription impacts the efficiency of mRNA translation via co-transcriptional N6-adenosine methylation. Cell. 2017;169(2):326-37.e12.
Squires JE, Patel HR, Nousch M, Sibbritt T, Humphreys DT, Parker BJ, et al. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012;40(11):5023–33.
Stoilov P, Rafalska I, Stamm S. YTH: a new domain in nuclear proteins. Trends Biochem Sci. 2002;27(10):495–7.
Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m6A/MYC/CEBPA signaling. Cell. 2018;172(1–2):90-105.e23.
Sui X, Hu Y, Ren C, Cao Q, Zhou S, Cao Y, et al. METTL3-mediated m6A is required for murine oocyte maturation and maternal-to-zygotic transition. Cell Cycle. 2020;19(4):391–404.
Tasaki M, Shimada K, Kimura H, Tsujikawa K, Konishi N. ALKBH3, a human AlkB homologue, contributes to cell survival in human non-small-cell lung cancer. Br J Cancer. 2011;104(4):700–6.
Taverna E, Götz M, Huttner WB. The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu Rev Cell Dev Biol. 2014;30:465–502.
Tuorto F, Herbst F, Alerasool N, Bender S, Popp O, Federico G, et al. The tRNA methyltransferase Dnmt2 is required for accurate polypeptide synthesis during haematopoiesis. EMBO J. 2015;34(18):2350–62.
Tuorto F, Liebers R, Musch T, Schaefer M, Hofmann S, Kellner S, et al. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat Struct Mol Biol. 2012;19(9):900–5.
Vilardo E, Amman F, Toth U, Kotter A, Helm M, Rossmanith W. Functional characterization of the human tRNA methyltransferases TRMT10A and TRMT10B. Nucleic Acids Res. 2020;48(11):6157–69.
Vilardo E, Nachbagauer C, Buzet A, Taschner A, Holzmann J, Rossmanith W. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase–extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res. 2012;40(22):11583–93.
Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V, et al. Essential role of METTL3-mediated m6A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37(4):522–33.
Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23(11):1369–76.
Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell. 2016a;63(2):306–17.
Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, et al. Structural basis of N6-adenosine methylation by the METTL3-METTL14 complex. Nature. 2016b;534(7608):575–8.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014a;505(7481):117–20.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N6-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–99.
Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014b;16(2):191–8.
Wang Y, Li Y, Yue M, Wang J, Kumar S, Wechsler-Reya RJ, et al. N6-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat Neurosci. 2018;21(2):195–206.
Wei CM, Gershowitz A, Moss B. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell. 1975;4(4):379–86.
Wei J, Liu F, Lu Z, Fei Q, Ai Y, He PC, et al. Differential m6A, m6Am, and m1A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol Cell. 2018;71(6):973-85.e5.
Wen J, Lv R, Ma H, Shen H, He C, Wang J, et al. Zc3h13 regulates nuclear RNA m6A methylation and mouse embryonic stem cell self-renewal. Mol Cell. 2018;69(6):1028-38.e6.
Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m6A modification. Cell Stem Cell. 2018;22(2):191-205.e9.
Wojtas MN, Pandey RR, Mendel M, Homolka D, Sachidanandam R, Pillai RS. Regulation of m6A transcripts by the 3′→5′ RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol Cell. 2017;68(2):374-87.e12.
Woo HH, Chambers SK. Human ALKBH3-induced m1A demethylation increases the CSF-1 mRNA stability in breast and ovarian cancer cells. Biochim Biophys Acta Gene Regul Mech. 2019;1862(1):35–46.
Wu R, Li A, Sun B, Sun JG, Zhang J, Zhang T, et al. A novel m6A reader Prrc2a controls oligodendroglial specification and myelination. Cell Res. 2019;29(1):23–41.
Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61(4):507–19.
Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y, et al. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol. 2014;10(11):927–9.
Xu K, Yang Y, Feng GH, Sun BF, Chen JQ, Li YF, et al. Mettl3-mediated m6A regulates spermatogonial differentiation and meiosis initiation. Cell Res. 2017;27(9):1100–14.
Xu W, Li J, He C, Wen J, Ma H, Rong B, et al. METTL3 regulates heterochromatin in mouse embryonic stem cells. Nature. 2021;591(7849):317–21.
Yang X, Yang Y, Sun BF, Chen YS, Xu JW, Lai WY, et al. 5-Methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Res. 2017a;27(5):606–25.
Yang Y, Wang L, Han X, Yang WL, Zhang M, Ma HL, et al. RNA 5-methylcytosine facilitates the maternal-to-zygotic transition by preventing maternal mRNA decay. Mol Cell. 2019;75(6):1188-202.e11.
Yang Z, Li J, Feng G, Gao S, Wang Y, Zhang S, et al. MicroRNA-145 modulates N6-methyladenosine levels by targeting the 3′-untranslated mRNA region of the N6-methyladenosine binding YTH domain family 2 protein. J Biol Chem. 2017b;292(9):3614–23.
Yao QJ, Sang L, Lin M, Yin X, Dong W, Gong Y, et al. Mettl3-Mettl14 methyltransferase complex regulates the quiescence of adult hematopoietic stem cells. Cell Res. 2018;28(9):952–4.
Yoon KJ, Ringeling FR, Vissers C, Jacob F, Pokrass M, Jimenez-Cyrus D, et al. Temporal control of mammalian cortical neurogenesis by m6A methylation. Cell. 2017;171(4):877-89.e17.
Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, et al. VIRMA mediates preferential m6A mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell Discovery. 2018;4:10.
Zhang C, Chen Y, Sun B, Wang L, Yang Y, Ma D, et al. m6A modulates haematopoietic stem and progenitor cell specification. Nature. 2017a;549(7671):273–6.
Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci. 2016a;113(14):E2047–56.
Zhang C, Zhi WI, Lu H, Samanta D, Chen I, Gabrielson E, et al. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget. 2016b;7(40):64527–42.
Zhang M, Zhai Y, Zhang S, Dai X, Li Z. Roles of N6-methyladenosine (m6A) in stem cell fate decisions and early embryonic development in mammals. Front Cell Dev Biol. 2020;8:782.
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. m6A Demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017b;31(4):591-606.e6.
Zhang X, Wei LH, Wang Y, Xiao Y, Liu J, Zhang W, et al. Structural insights into FTO’s catalytic mechanism for the demethylation of multiple RNA substrates. Proc Natl Acad Sci. 2019;116(8):2919–24.
Zhang Y, Zhang X, Shi J, Tuorto F, Li X, Liu Y, et al. Dnmt2 mediates intergenerational transmission of paternally acquired metabolic disorders through sperm small non-coding RNAs. Nat Cell Biol. 2018;20(5):535–40.
Zhao BS, Wang X, Beadell AV, Lu Z, Shi H, Kuuspalu A, et al. m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature. 2017;542(7642):475–8.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29.
Zhong L, Liao D, Zhang M, Zeng C, Li X, Zhang R, et al. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. 2019;442:252–61.
Zhou KI, Shi H, Lyu R, Wylder AC, Matuszek Ż, Pan JN, et al. Regulation of co-transcriptional pre-mRNA splicing by m6A through the low-complexity protein hnRNPG. Mol Cell. 2019;76(1):70-81.e9.
Funding
This work was supported by the National Natural Science Foundation of China (nos. 21822702, 21820102008, 92053109, and 21432002) and the National Basic Research Program of China (2017YFA0505201 and 2019YFA0802201).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Written informed consents were collected from all the participants before enrollment.
Consent for publication
All contributing authors agree to the publication of this article.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
Three well-studied epitranscriptomic marks (m6A, m5C, and m1A) are characterized as critical regulators in gene expression.
RNA methylations maintain the stem cell balance between pluripotency and differentiation into downstream lineages.
RNA methylations play regulatory roles in embryonic development.
RNA methylations affect tumorigenesis, CSCs self-renewal and proliferation in cancer program
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, P., Tayier, S., Cai, Z. et al. RNA methylation in mammalian development and cancer. Cell Biol Toxicol 37, 811–831 (2021). https://doi.org/10.1007/s10565-021-09627-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10565-021-09627-8