
Abstract
Progress in targeted gene editing by programmable endonucleases has paved the way for their use in gene therapy. Particularly, Cas9 is an endonuclease with high activity and flexibility, rendering it an attractive option for therapeutic applications in clinical settings. Many disease-causing mutations could potentially be corrected by this versatile new technology. In addition, recently developed switchable Cas9 variants, whose activity can be controlled by an external stimulus, provide an extra level of spatiotemporal control on gene editing and are particularly desirable for certain applications. Here, we discuss the considerations and difficulties for implementing Cas9 to in vivo gene therapy. We put particular emphasis on how switchable Cas9 variants may resolve some of these barriers and advance gene therapy in the clinical setting.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Gene therapy is a tool to treat or cure diseases by modifying a patient’s genotype. Modifications include replacing a lack-of-function gene with its wild-type sequence, silencing a disease-causing gene that is constitutively active, or introducing a new gene of novel function to treat a disease (Dangi et al. 2018; Gupta and Shukla 2017). Therefore, gene therapy is complementary to existing therapeutic approaches and provides an obvious solution to diseases with a clear genetic origin (Anguela and High 2019). Importantly, gene therapy is perhaps the only permanent and inheritable solution for diseases caused by a lack-of-function gene (Cox et al. 2015). Due to its unique advantages, there has been significant research on its use to tackle otherwise hard-to-treat diseases, invoking a surge of gene therapies entering clinical trials (Naldini 2015). Although most clinical trials rely on ex vivo gene editing, in which cells are removed from the patient, modified, and then transferred back, in recent years, more trials are being conducted to explore in vivo gene editing in animal models (Naldini 2015). The in vivo approach is particularly beneficial for diseases of the muscles and internal organs, such as Duchenne muscular dystrophy, cystic fibrosis, and tyrosinemia (Cox et al. 2015; Wang et al. 2017; Mention et al. 2019).
This review limits the discussion to in vivo gene editing for therapeutic applications. The premise of all gene editing techniques involves breaking a double-stranded DNA under the action of an endonuclease (Fig. 1a), followed by repair via cellular machinery (Fig. 1b). The mechanism of repair can result in a few different outcomes. For example, the double-strand break can be repaired into a new, defined sequence to correct a diseased gene, or be disrupted to inactivate a disease-causing gene with autosomal dominant effects. In comparison to other gene editing systems, the clustered regularly interspaced short palindromic repeats (CRISPR) system is unique. In particular, CRISPR associated protein 9 (Cas9) is specifically appealing due to its high activity and flexibility in experimental design (Wang et al. 2017; Kim and Kim 2014). Moreover, recent advances in protein engineering have yielded switchable Cas9 variants, whose activity can be controlled with spatiotemporal resolution by an external stimulus (Gangopadhyay et al. 2019; Nihongaki et al. 2018; Richter et al. 2017; Zhou and Deiters 2016), and we envisage that such variants developed by us (Suzuki et al. 2018) and others (Zetsche et al. 2015; Davis et al. 2015; Oakes et al. 2016; Liu et al. 2016; Nguyen et al. 2016; Tang et al. 2017; Senturk et al. 2017; Rose et al. 2017; Nihongaki et al. 2015; Hemphill et al. 2015; Richter et al. 2016; Jain et al. 2016; Zhou et al. 2018) can benefit clinical development of in vivo gene editing.

Two stages of precise gene editing involving a recognition of the target DNA by the endonuclease and subsequent cleavage to generate a double-strand break and b repair of the break by cellular mechanisms
In this review, we will first illustrate the molecular mechanisms of gene editing and provide examples of Cas9-mediated gene editing in disease models. We will then compare different means to deliver Cas9 for in vivo gene therapy. Finally, we will discuss how switchable Cas9 variants may advance gene therapy in a clinical setting.
Cas9 for gene editing
Endonucleases that generate a double-strand break at the targeted DNA sequence are indispensable for gene editing (Cox et al. 2015; Cornu et al. 2017). There have been many endonucleases discovered to date that can cause site-specific double-strand breaks, and many have been developed for the purpose of gene therapy. Such endonucleases include meganucleases, zinc-finger nucleases, transcriptional activator-like effector nucleases, and CRISPR/Cas9 (Cox et al. 2015). Among them, Cas9, found in the Gram-positive bacterium Streptococcus pyogenes (SpCas9), is arguably the most versatile (Wang et al. 2017). Although its use in precise gene editing was initially questioned by its relatively high off-target activity, this issue has been progressively addressed through different approaches (Wang et al. 2017), including engineering of SpCas9 variants with negligible non-specific activity (Slaymaker et al. 2016; Kleinstiver et al. 2016; Hu et al. 2018).
The popularity of SpCas9 originates from its flexibility in creating DNA double-strand breaks at different target sequences (Cox et al. 2015). The specificity of other endonucleases relies on the amino acid sequence of the DNA-binding domain. Consequently, when a new DNA target is required, researchers must alter and engineer the DNA-binding domain of the endonuclease to achieve specificity to the new target. Clearly, this process can be labor-intensive and time-consuming, if not challenging. In stark contrast, the specificity of SpCas9 can be determined by a single-strand guide RNA (gRNA) molecule, rather than the protein domain within the enzyme (Jinek et al. 2012). Accordingly, DNA double-strand breaks at desired sites can be easily introduced by the addition of corresponding gRNA molecules, which are readily achievable using standard cloning techniques. Specifically, a gRNA molecule contains two parts: (i) a 20-nucleotide guide sequence that complements the DNA target and (ii) a structural motif essential for binding the enzyme SpCas9. At the molecular level (Jiang and Doudna 2017), a ribonucleoprotein (a complex of SpCas9 and gRNA) is first formed (Fig. 2a). In order for the complex to bind to the target DNA (Fig. 2b), the existence of a protospacer adjacent motif (PAM) upstream of the DNA target sequence in the genome is essential (Jinek et al. 2012). Guided by the PAM sequence and complementarity to the DNA target, the gRNA in the ribonucleoprotein forms a double-stranded complex with DNA. Consequently, the target DNA is cleaved by SpCas9 of the ribonucleoprotein (Fig. 2c), generating a double-strand break (Jiang and Doudna 2017). For wild-type SpCas9, 5′-NGG-3′ is the required PAM sequence, but SpCas9 variants that recognize different PAM sequences have also been generated (Wang et al. 2017; Hu et al. 2018). Therefore, it is theoretically possible to use SpCas9 and its variants to target nearly any gene. Given the simplicity and versatility, there has been an exponential use of SpCas9 in in vitro and in vivo genome editing reported in the literature (Dangi et al. 2018; Nishitani et al. 2019).

Generation of a DNA double-strand break by Cas9 involving a formation of the ribonucleoprotein, b recognition of the target DNA, and c cleavage of the double-stranded DNA
Cellular DNA repair mechanisms
When a double-strand DNA break is formed inside a cell, there are two major repair mechanisms: homology-directed repair (HDR, Fig. 3a) and non-homologous end joining (NHEJ, Fig. 3b) (Wyman and Kanaar 2006). In HDR, a DNA molecule with identical nucleotide sequence (i.e., homology) to the two sides of the break is present and used by the cell as the template to repair the lesion accordingly. This ensures the repaired DNA molecule will have identical nucleotide sequence to the template. The DNA repair template can be of endogenous or exogenous origin, and an exogenously supplied template can be either single- or double-stranded DNA (Pawelczak et al. 2018). In the case of double-stranded DNA, it can be a linear fragment or a circular plasmid (Pawelczak et al. 2018). For gene editing, it is also possible to supply a template containing a new gene of novel function to the cell. On the other hand, during repair by NHEJ, the two DNA fragments are reconnected without a template. In this mechanism, accurate repair yielding an identical sequence to that before the cleavage is the most prominent outcome (Fig. 3b), although indels (insertion or deletion of nucleotides) can also be produced at the cleavage site. However, if the double-strand break is generated by an endonuclease, products of accurate repair retain the recognition sequence and are readily re-cleaved by the endonuclease, whereas indel products are not. Therefore, in the absence of a repair template, indel products will accumulate and become the predominant consequence over time. Since indels in exon sequences often lead to a frameshift with a premature stop codon, the open reading frame of the gene is disrupted. Subsequently, mRNA resulting from such a disrupted gene is either recognized and degraded by the nonsense-mediated decay pathway, or translated into truncated, non-functional protein. The overall outcome is gene silencing, and this mechanism has been commonly used for gene knockout (Wyman and Kanaar 2006; Pawelczak et al. 2018).

Consequences of repairing a double-strand break by the two cellular mechanisms, a homology-directed repair (HDR) and b non-homologous end joining (NHEJ)
Generally, in mammalian systems, most DNA repairs undergo the NHEJ pathway which is active at all stages of the cell division cycle, whereas the HDR mechanism is less efficient and restricted to the S and G2 phases (Wyman and Kanaar 2006; Pawelczak et al. 2018). Nevertheless, HDR is desired in many scenarios of gene editing therapy (see below). HDR efficiency can be increased by controlling the activity of the endonuclease at the G2 and S phases, administration of small molecules (e.g., cell cycle arrest drugs, or inhibitors of proteins involved in the NHEJ pathway), or optimization of repair DNA template format (Pawelczak et al. 2018; Robert et al. 2015; Paulk et al. 2012). However, it is still technically challenging to achieve close to 100% HDR efficiency in mammalian models even when taking these approaches.
Diseases benefitting from in vivo gene editing
As HDR products always have the correct sequence restored, this can theoretically be used to restore any genetic disorder. Indeed, most therapeutic gene editing research thus far relies on HDR to correct diseased mutations (Cox et al. 2015; Wang et al. 2017; Cornu et al. 2017; Nishitani et al. 2019; Yin et al. 2014a). For example, HDR is particularly suitable to correct in-frame nonsense mutations, or multiple clustered mutations simultaneously. However, the relative inefficiency of HDR limits its applications to diseases where low-efficiency editing can still significantly improve gene function and disease pathology. For example, a small percentage of gene correction to exhibit functional restoration is beneficial to diseases, such as cystic fibrosis (Schwank et al. 2013; Hodges and Conlon 2019), Duchenne muscular dystrophy (Duan 2015; Long et al. 2014, 2016; Bogdanovich et al. 2002), Huntington’s disease (Kolli et al. 2017; Carroll et al. 2011; Kordasiewicz Holly et al. 2012), retinal dysfunction (Min et al. 2005; Narfström et al. 2003), severe combined immunodeficiency (Gaspar et al. 2004), and tyrosinemia (Yin et al. 2014a, 2016).
The NHEJ mechanism could provide an efficient therapeutic option for single-gene autosomal dominant disorders. Such disorders are caused by mutations of a sole gene on one of the autosomal (i.e. non-sex) chromosomes. Huntington’s disease (Glorioso et al. 2015) and epidermolysis bullosa simplex (Lewin et al. 2005) are two such examples. Because patients of these diseases retain one copy of the wild-type gene, knockout of the mutated version via NHEJ will infer the recessive wild-type copy to regain normal protein function. This general concept of silencing the single disease-causing allele has been proven to be effective in a number of knockdown studies by antisense oligonucleotides (Seyhan 2011). Although the use of Cas9 to silence a gene has not been fully explored (Kolli et al. 2017; Christie et al. 2017; Bakondi et al. 2016), it is theoretically possible to use Cas9-mediated knockout to replace antisense oligonucleotides for gene silencing.
Alternatively, DNA repair by the NHEJ approach can be used to restore a gene silenced by certain indels. One-nucleotide insertion is the most prevalent outcome (> 20%) of NHEJ (Cradick et al. 2013; Sürün et al. 2018; Chen et al. 2018a), so this repair mechanism can restore the open reading frame of genes containing specific indels (i.e., …, − 4, − 1, 2, 5, …). Indeed, this concept has been proven in cellular models of X-linked chronic granulomatosis disease, where the production of full-length cytochrome b-245 heavy chain increased by 25% as the result of non-templated NHEJ repair (Sürün et al. 2018).
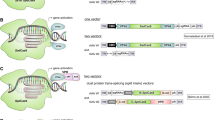
NHEJ can also be used to produce a functional protein by exon skipping, and this approach has been explored as a new gene therapy in animal models (Long et al. 2016; Chen et al. 2018b; Amoasii et al. 2018; Xu et al. 2016; Aartsma-Rus et al. 2017; Turczynski et al. 2016; Touznik et al. 2014). Many mammalian genes contain several exons that together code for the amino acid sequence in the final protein product (Fig. 4a). If a disease-causing mutation or frame shift occurs in a region not critical for protein function, exon skipping can be used to produce a shorter, but functional, protein. This can be achieved by using two gRNA molecules to direct Cas9 to cut each end of the mutant exon, followed by non-templated NHEJ to connect the adjacent DNA, resulting in removal of the mutant exon (Fig. 4b). Alternatively, exon skipping can be achieved by disrupting the intron-exon junction (Fig. 4c). Both strategies have been successfully demonstrated in mouse models of Duchenne muscular dystrophy (Long et al. 2016; Chen et al. 2018b; Amoasii et al. 2018; Xu et al. 2016). Furthermore, exon skipping approaches have already been investigated in other genetic diseases, including muscular dystrophy (Aartsma-Rus et al. 2017), dystrophic epidermolysis bullosa (Turczynski et al. 2016), and neuromuscular diseases (Touznik et al. 2014). Therefore, it is foreseeable that development of Cas9-mediated exon skipping will be of clinical interest.

Exon skipping by NHEJ to restore an open reading frame. a Protein production from a normal or disease state DNA. b Exon skipping by cutting out the mutant exon. c Exon skipping by disrupting the intron-exon boundary
Gene editing can involve exploitation of either of the endogenous cellular repair pathways, and both have been studied in vivo for genetic mutation correction (Table 1). A key advantage of HDR is that the entire section of DNA can be replaced with a corrected sequence, restoring the gene to its wild-type sequence. This contrasts with NHEJ which produces indels that may restore the open reading frame, induce exon skipping, or lead to gene knockout. While both may result in resolution of disease phenotype, only HDR can result in a DNA sequence indistinguishable from the wild-type sequence. For both repair mechanisms, prediction tools are available to analyze any target gene. For HDR, the most promising gRNA sequences for high-efficiency Cas9 editing can be generated (O’Brien et al. 2018). The nature of indels resulting from NHEJ can be predicted using tools which analyze the most likely outcome for a particular nucleotide sequence (Chen et al. 2018a). These tools can be used with target gene sequences to select suitable splice sites that will increase the probability of a preferred indel, and therefore the desired outcome. However, as repair via NHEJ is more efficient than that of HDR (Wyman and Kanaar 2006; Pawelczak et al. 2018), it would be beneficial to employ NHEJ whenever possible.
Delivering Cas9 and gRNA for in vivo gene editing
The practicality of in vitro gene editing using Cas9 is relatively well established (Wang et al. 2017). For gene therapy, both ex vivo and in vivo methods can be used to deliver technologies to cells. Ex vivo delivery involves removing cells or tissues from patients for editing, then engrafting the edited cells back into the patient. However, this review limits the discussion to only in vivo editing. While the potential applications of its in vivo use are apparent, delivery of Cas9 and gRNA into the target cells of the patient is the major obstacle (Wang et al. 2017; Yin et al. 2017; Mout et al. 2017). The required components can be delivered together or separately as either DNA, RNA, or ribonucleoprotein through viral or non-viral approaches. Ideally, components required for gene editing are only delivered to necessary cells in patients, although many current approaches lack the required target specificity. Here, we discuss the features of nucleic acid delivery by either viral or non-viral vectors as well as ribonucleoprotein delivery.
Viral vectors are designed to deliver a payload to the target cells by utilizing the viral infection pathway, while most of the non-essential viral genome is removed from the vector. Integration-deficient lentiviruses, adenoviruses, and adeno-associated viruses are the most popular viral vectors for gene therapy due to their non-integrating nature, eliminating the risks of mutagenesis and tumorigenicity associated with gene insertion (Yin et al. 2017; Lukashev and Zamyatnin 2016; Lundstrom 2018). Among them, adeno-associated viruses are particularly attractive for their low immunogenicity and broad ability to target specific tissues, including liver, brain, skeletal, kidney, retina, lung, and vascular tissue (Mingozzi and High 2011). In comparison, adenoviral vectors suffer from high immunogenicity, while lentiviral vectors normally lack tissue specificity (Yin et al. 2017; Escors and Breckpot 2010). However, the large size of the SpCas9 gene (4.3 kilobases) poses a challenge to pack into a single adeno-associated virus vector along with the required gRNA (Wu et al. 2010; Senis et al. 2014). One option is to use two vectors, one encoding SpCas9 and the other encoding the gRNA. The two vectors will be delivered simultaneously, an approach requiring efficient co-transduction of the target cells (Long et al. 2016; Amoasii et al. 2018; Swiech et al. 2015). Alternatively, the smaller Staphylococcus aureus Cas9 (3.2 kilobases) can be used, allowing a single vector to encode both the SaCas9 and gRNA (Ran et al. 2015). However, viral systems can induce long-term transgene expression in humans with a single injection; thus, the potential induction of immunogenicity against Cas9 protein and increase in off-target editing due to sustained endonuclease expression need to be taken into consideration (Yin et al. 2017; Fu et al. 2013).
Non-viral delivery methods, such as hydrodynamic injection and electroporation, have the potential to transfer large genetic payloads with the advantage of a transient expression pattern (Yin et al. 2014b). Hydrodynamic injection is the rapid delivery of a large volume of DNA-containing solution via intravenous injection (Liu et al. 1999). This has been used to deliver components required for Cas9-mediated gene editing in mouse (Yin et al. 2014a; Zhen et al. 2015) and rat models (Bakondi et al. 2016). However, as a large injection volume (about 10% of the animal’s body weight) is required, hydrodynamic injection is unlikely to be suitable for human applications. Alternatively, electroporation, the stimulation of cells via electrical pulse, can also facilitate cellular uptake of foreign components specific to particular tissue, and its use has also been demonstrated in animal models (Bakondi et al. 2016; Xu et al. 2016). However, due to the large amount of cell death induced in the treatment area by this method, electroporation has yet to be employed in human clinical trials. In addition, neither hydrodynamic injection nor electroporation shows cell specificity.
Ribonucleoprotein, composed of Cas9 and gRNA, can be directly delivered into cells for genetic modification. In comparison to the delivery of DNA or RNA to generate ribonucleoprotein in vivo, delivery of ribonucleoprotein is appealing as the molecules are immediately active, resulting in rapid editing (Kim et al. 2014). This method is also recognized for its limited half-life, which reduces potential off-target editing (Liang et al. 2015). A major challenge of ribonucleoprotein delivery is packaging Cas9 protein with gRNA. Cationic lipids such as RNAiMAX (Zuris et al. 2015) have enabled gene editing of up to 20% in mouse models and have entered clinical testing for other gene therapies (Fitzgerald et al. 2014; Coelho et al. 2013). Gold nanoparticles have also been used to deliver the ribonucleoprotein in mouse models (Lee et al. 2017a). The nanoparticles were complexed with donor DNA, Cas9 RNP, and PAsp(DET). PAsp(DET) is a polymer that induces both endocytosis and later endosomal disruption to release CRISPR components into the cytosol. Although target-specific delivery by these means are yet limited to local injections, recent development of receptor-mediated ribonucleoprotein delivery has shown cell specificity in vitro (Rouet et al. 2018), indicating the possibility of targeted ribonucleoprotein delivery.
Many factors need to be considered when selecting the delivery vector. An ideal delivery method should have high specificity to the diseased cells and tissues, be non-immunogenic and non-toxic to the host, and enable transient Cas9 activity to minimize potential off-target editing. Although none of the currently available delivery methods fulfill all these criteria (Table 2), it is possible to use a switchable Cas9 variant to control tissue specificity and the duration of Cas9 activity.
Switchable Cas9
Cas9 variants that can be regulated by an external stimulus are of great interest to therapeutic development, as they allow an extra level of spatial and temporal control over gene editing (Gangopadhyay et al. 2019; Nihongaki et al. 2018; Richter et al. 2017; Zhou and Deiters 2016). Improved spatial resolution limits activity to specific cells and tissues, if this is not already conferred by the delivery vector, whereas control over temporal resolution can minimize off-target editing by confining the duration of active Cas9 in cells (Hu et al. 2018; Yin et al. 2014a). To date, different switchable Cas9 variants have been developed (Gangopadhyay et al. 2019; Nihongaki et al. 2018; Richter et al. 2017; Zhou and Deiters 2016), and their activity can be controlled by temperature (Richter et al. 2016; Jiang et al. 2019), light (Nihongaki et al. 2015; Hemphill et al. 2015; Richter et al. 2016; Jain et al. 2016; Zhou et al. 2018), or small molecules (Suzuki et al. 2018; Zetsche et al. 2015; Davis et al. 2015; Oakes et al. 2016; Liu et al. 2016; Nguyen et al. 2016; Tang et al. 2017; Senturk et al. 2017; Rose et al. 2017). These variants work well in vitro; however, their applicability for controlling in vivo gene editing is likely to greatly depend on the nature of the stimuli.
Temperature-sensitive Cas9 variants (Richter et al. 2016; Jiang et al. 2019) are unlikely to be suitable for controlling gene editing inside a human body. The body temperature of humans is normally maintained at 37 °C with minimal fluctuation. It will therefore be challenging to maintain a target tissue at an alternative temperature for an extensive period for gene editing to take place. This holds even for a Cas9 variant which is active at 29 °C instead of 37 °C (Richter et al. 2016). Conversely, light is a unique stimulus that offers superior spatial control to subcellular levels. However, the major drawback of using light to modulate Cas9 function is the limited tissue penetrability (Ash et al. 2017), making this approach unfeasible when targeting internal organs. To date, light-responsive Cas9 variants can be controlled by either 365 nm (Hemphill et al. 2015; Jain et al. 2016), 470 nm (Nihongaki et al. 2015; Richter et al. 2016), or 500 nm (Zhou et al. 2018) wavelength light, which can penetrate tissues at about 700, 1600, or 2500 μm depth respectively (Ash et al. 2017). Therefore, their uses may be limited to skin diseases, such as epidermolysis bullosa simplex (Lewin et al. 2005), as the depth of the epidermis is within 130 μm (Sandby-Moller et al. 2003).
Small molecules arguably hold the greatest potential for controlled in vivo activation of Cas9. High temporal control is achieved by the time of administration and dosage of the small molecule. Although high spatial resolution with this approach can only be achieved by local administration of the modulator molecules (Han et al. 2017), small molecules can theoretically reach any tissues within a human body, unlike regulation by temperature or light. Ideally, a small-molecule Cas9 modulator should have no effects on other proteins or biomolecules, preventing disturbance of other cellular processes. Unfortunately, most switchable Cas9 variants developed to date are responsive to drug molecules, such as antibiotic rapamycin (Zetsche et al. 2015), estrogen receptor modulator 4-hydroxytamoxifen (Davis et al. 2015; Oakes et al. 2016; Liu et al. 2016; Nguyen et al. 2016), Bcl-XL inhibitor A-385358 (Rose et al. 2017), and respiratory drug theophylline (Tang et al. 2017). Nevertheless, there are two approaches to control Cas9 activity by non-drug molecules, Shield-1 (Senturk et al. 2017), and Lys(Boc) (Suzuki et al. 2018).
Shield-1, a ligand that binds to and stabilizes an FKBP12-derived destabilizing protein domain, is highly cell permeable, and has no in vivo toxicity (Banaszynski et al. 2008). By fusing the destabilizing domain to SpCas9, the resultant destabilized Cas9 protein variant was not detectable in the absence of Shield-1 (Fig. 5a). Upon addition of Shield-1 into the culture media, Cas9 protein was detected within 2 h, whereas subsequent removal of Shield-1 from the culture media led to depletion of Cas9 protein within 12 h (Senturk et al. 2017).

Regulation of SpCas9 by non-drug molecules. a Stability of the fusion protein containing SpCas9 and a FKBP12-derived destabilizing domain can be regulated by Shield-1 so that in the absence of Shield-1, all fusion proteins are rapidly degraded. b Genetic code expansion for site-specific non-canonical amino acid incorporation is used to control the production of full-length, functional SpCas9
Lys(Boc) is an economic, non-canonical amino acid that does not show any observable toxicity to cell lines and embryos (Suzuki et al. 2018). It can be site-specifically incorporated into a protein of interest in mammalian cells using genetic code expansion. In mammalian cells, proteins composed of 20 canonical amino acids are produced by ribosome, which employs aminoacyl-tRNAs to decode the information on mRNA to generate the corresponding protein. To expand the genetic code, an orthogonal aminoacyl-tRNA synthetase/tRNA pair is introduced into the cell. The orthogonal synthetase specifically acylates the orthogonal tRNA with a designated non-canonical amino acid, such as Lys(Boc), to generate the required aminoacylated tRNA (Nödling et al. 2019). Here, orthogonality means that the orthogonal synthetase does not use any of 20 canonical amino acids nor any of the endogenous tRNA as substrate, and the non-canonical amino acid is not a substrate of any of the endogenous synthetases. The orthogonal tRNA recognizes a blank codon on the mRNA to direct incorporation of the non-canonical amino acid into the target protein. Amber stop codon (UAG) is usually used as the blank codon due to its rarity among the three stop codons in most organisms. Pyrrolysyl-tRNA synthetase/tRNA pair from the archaea Methanosarcina species is an orthogonal pair in mammalian cells and can direct Lys(Boc) incorporation in response to an amber codon (Nödling et al. 2019).
An SpCas9 gene harboring a centrally located amber codon alongside a pyrrolysyl-tRNA synthetase/tRNA pair has been successfully used to control gene editing in mouse embryos (Suzuki et al. 2018). In the absence of Lys(Boc), truncated and non-functional SpCas9 is obtained (Fig. 5b), whereas supplementation of the non-canonical amino acid led to production of full-length and functional SpCas9 protein. This approach enables heritable Cas9-mediated mammalian genome editing that is acutely controlled by the economic and readily available lysine derivative (Suzuki et al. 2018). However, amber suppression may interfere with translation of endogenous genes ending with the amber stop codon. In addition, the system requires multiple components to be delivered to the cell and, as previously discussed, vectors capable of delivering a large cargo are limited.
Currently, duration of Cas9 activity in vivo depends on the delivery methods as described in Table 2. Switchable Cas9 variants can offer a solution to long-term transgene expression and subsequent off-target editing associated with viral delivery vectors (Yin et al. 2017; Fu et al. 2013). The great temporal control of switchable Cas9 variants enables accurate regulation of genetic modification, circumventing the concerns over extended activity timeframes associated with delivery of Cas9 DNA. Cas9 variants regulated by Shield-1 (Senturk et al. 2017) and Lys(Boc) (Suzuki et al. 2018) can be switched on and off easily by the presence or absence of the required small molecule, enabling delicate regulation of Cas9 activity and greater tissue specificity through local administration of the molecule. Despite the advantages offered by switchable Cas9 variants, their uses in disease animal models and therapeutic applications are still very limited. Considerable further development of these switchable variants is therefore needed before they can be applied to the clinical setting.
Conclusions
Progress in gene editing techniques have improved rapidly in recent years and benefited by the discovery of CRISPR/Cas9. The versatile and highly specific gene editing achieved by Cas9 is so far the most promising approach for correction of genetic diseases. The potential for many genetic diseases to be resolved in a permanent and heritable fashion by this technology is clear and may be achievable in the near future especially for diseases that could benefit from repair by the NHEJ mechanism. However, significant improvement in HDR efficiency is required before the power of this repair mechanism can be fully exploited for the therapeutic applications. Regardless of the repair mechanism, a major hindrance of in vivo gene editing thus far has been the lack of a suitable delivery vector with cell specificity, while providing transient Cas9 delivery and low immunogenicity.
Fortunately, switchable Cas9 variants offer solutions to some of the obstacles. Specifically, the great temporal control of switchable Cas9 variants enables rapid and accurate regulation of genetic modification, circumventing the extended activity timeframe associated with some means of Cas9 delivery, whereas the spatial control of switchable Cas9 variants could provide target specificity if not conferred by delivery vectors. However, detailed in vivo investigations of switchable Cas9 variants are required to translate their use into clinical applications. Unfortunately, protein scientists working on Cas9 engineering often lack in vivo expertise. Thus, it will be necessary that scientists working on tool and therapeutic development closely collaborate, so the true potential of Cas9-mediated gene therapy can be transformed into clinical settings.
References
Aartsma-Rus A, Straub V, Hemmings R, Haas M, Schlosser-Weber G, Stoyanova-Beninska V, et al. Development of exon skipping therapies for Duchenne muscular dystrophy: a critical review and a perspective on the outstanding issues. Nucleic Acid Ther. 2017;27(5):251–9.
Amoasii L, Hildyard JCW, Li H, Sanchez-Ortiz E, Mireault A, Caballero D, et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362(6410):86–90.
Anguela XM, High KA. Entering the modern era of gene therapy. Annu Rev Med. 2019;70:273–88.
Ash C, Dubec M, Donne K, Bashford T. Effect of wavelength and beam width on penetration in light-tissue interaction using computational methods. Lasers Med Sci. 2017;32(8):1909–18.
Bakondi B, Lv W, Lu B, Jones MK, Tsai Y, Kim KJ, et al. In vivo CRISPR/Cas9 gene editing corrects retinal dystrophy in the S334ter-3 rat model of autosomal dominant retinitis Pigmentosa. Mol Ther. 2016;24(3):556–63.
Banaszynski LA, Sellmyer MA, Contag CH, Wandless TJ, Thorne SH. Chemical control of protein stability and function in living mice. Nat Med. 2008;14(10):1123–7.
Bogdanovich S, Krag TOB, Barton ER, Morris LD, Whittemore L-A, Ahima RS, et al. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420(6914):418–21.
Carroll JB, Warby SC, Southwell AL, Doty CN, Greenlee S, Skotte N, et al. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene/allele-specific silencing of mutant huntingtin. Mol Ther. 2011;19(12):2178–85.
Chen W, McKenna A, Schreiber J, Yin Y, Agarwal V, Noble WS, et al. Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. bioRxiv. 2018a;481069.
Chen D, Tang J-X, Li B, Hou L, Wang X, Kang L. CRISPR/Cas9-mediated genome editing induces exon skipping by complete or stochastic altering splicing in the migratory locust. BMC Biotechnol. 2018b;18(1):60.
Christie KA, Courtney DG, DeDionisio LA, Shern CC, De Majumdar S, Mairs LC, et al. Towards personalised allele-specific CRISPR gene editing to treat autosomal dominant disorders. Sci Rep. 2017;7.
Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819–29.
Cornu TI, Mussolino C, Cathomen T. Refining strategies to translate genome editing to the clinic. Nat Med. 2017;23(4):415–23.
Courtney DG, Moore JE, Atkinson SD, Maurizi E, Allen EHA, Pedrioli DML, et al. CRISPR/Cas9 DNA cleavage at SNP-derived PAM enables both in vitro and in vivo KRT12 mutation-specific targeting. Gene Ther. 2015;23:108.
Cox DBT, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nat Med. 2015;21(2):121–31.
Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/Cas9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013;41(20):9584–92.
Dangi AK, Sinha R, Dwivedi S, Gupta SK, Shukla P. Cell line techniques and gene editing tools for antibody production: a review. Front Pharmacol. 2018;9:630.
Davis KM, Pattanayak V, Thompson DB, Zuris JA, Liu DR. Small molecule-triggered Cas9 protein with improved genome-editing specificity. Nat Chem Biol. 2015;11(5):316–8.
Duan D. Duchenne muscular dystrophy gene therapy in the canine model. Hum Gene Ther Clin Dev. 2015;26(1):57–69.
Escors D, Breckpot K. Lentiviral vectors in gene therapy: their current status and future potential. Arch Immunol Ther Exp. 2010;58(2):107–19.
Fitzgerald K, Frank-Kamenetsky M, Shulga-Morskaya S, Liebow A, Bettencourt BR, Sutherland JE, et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet. 2014;383(9911):60–8.
Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31(9):822–6.
Gangopadhyay SA, Cox KJ, Manna D, Lim D, Maji B, Zhou QX, et al. Precision control of CRISPR-Cas9 using small molecules and light. Biochemistry. 2019;58(4):234–44.
Gaspar HB, Parsley KL, Howe S, King D, Gilmour KC, Sinclair J, et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet. 2004;364(9452):2181–7.
Glorioso JC, Cohen JB, Carlisle DL, Munoz-Sanjuan I, Friedlander RM. Moving toward a gene therapy for Huntington’s disease. Gene Ther. 2015;22(12):931–3.
Guan S, Rosenecker J. Nanotechnologies in delivery of mRNA therapeutics using nonviral vector-based delivery systems. Gene Ther. 2017;24(3):133–43.
Gupta SK, Shukla P. Gene editing for cell engineering: trends and applications. Crit Rev Biotechnol. 2017;37(5):672–84.
Han S, Yang A, Lee S, Lee HW, Park CB, Park HS. Expanding the genetic code of Mus musculus. Nat Commun. 2017;8:14568.
Hemphill J, Borchardt EK, Brown K, Asokan A, Deiters A. Optical control of CRISPR/Cas9 gene editing. J Am Chem Soc. 2015;137(17):5642–5.
Hodges CA, Conlon RA. Delivering on the promise of gene editing for cystic fibrosis. Genes Dis. 2019;6(2):97–108.
Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556:57–63.
Jain PK, Ramanan V, Schepers AG, Dalvie NS, Panda A, Fleming HE, et al. Development of light-activated CRISPR using guide RNAs with Photocleavable protectors. Angew Chem Int Ed. 2016;55(40):12440–4.
Jiang F, Doudna JA. CRISPR–Cas9 structures and mechanisms. Annu Rev Biophys. 2017;46(1):505–29.
Jiang FG, Liu JJ, Osuna BA, Xu M, Berry JD, Rauch BJ, et al. Temperature-responsive competitive inhibition of CRISPR-Cas9. Mol Cell. 2019;73(3):601–10.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–21.
Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014;15(5):321–34.
Kim S, Kim D, Cho SW, Kim J, Kim J-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24(6):1012–9.
Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529(7587):490–5.
Kolli N, Lu M, Maiti P, Rossignol J, Dunbar GL. CRISPR-Cas9 Mediated gene-silencing of the mutant huntingtin gene in an in vitro model of Huntington’s disease. Int J Mol Sci. 2017;18(4).
Kordasiewicz Holly B, Stanek Lisa M, Wancewicz Edward V, Mazur C, McAlonis Melissa M, Pytel Kimberly A, et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of Huntingtin synthesis. Neuron. 2012;74(6):1031–44.
Lee K, Conboy M, Park HM, Jiang F, Kim HJ, Dewitt MA, et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat Biomed Eng. 2017a;1:889–901.
Lee CS, Bishop ES, Zhang R, Yu X, Farina EM, Yan S, et al. Adenovirus-mediated gene delivery: potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes Dis. 2017b;4(2):43–63.
Lewin AS, Glazer PM, Milstone LM. Gene therapy for autosomal dominant disorders of keratin. J Investig Dermatol Symp Proc. 2005;10(1):47–61.
Liang X, Potter J, Kumar S, Zou Y, Quintanilla R, Sridharan M, et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol. 2015;208:44–53.
Liu F, Song YK, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258–66.
Liu KI, Ramli MNB, Woo CWA, Wang YM, Zhao TY, Zhang XJ, et al. A chemical-inducible CRISPR-Cas9 system for rapid control of genome editing. Nat Chem Biol. 2016;12(11):980–7.
Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science. 2014;345(6201):1184–8.
Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351(6271):400–3.
Lukashev AN, Zamyatnin AA Jr. Viral vectors for gene therapy: current state and clinical perspectives. Biochemistry. 2016;81(7):700–8.
Lundstrom K. Viral vectors in gene therapy. Diseases. 2018;6(2):42.
Mention K, Santos L, Harrison PT. Gene and base editing as a therapeutic option for cystic FibrosisLearning from other diseases. Genes. 2019;10(5):387.
Milone MC, O’Doherty U. Clinical use of lentiviral vectors. Leukemia. 2018;32(7):1529–41.
Min SH, Molday LL, Seeliger MW, Dinculescu A, Timmers AM, Janssen A, et al. Prolonged recovery of retinal structure/function after gene therapy in an Rs1h-deficient mouse model of X-linked juvenile Retinoschisis. Mol Ther. 2005;12(4):644–51.
Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet. 2011;12(5):341–55.
Mout R, Ray M, Lee YW, Scaletti F, Rotello VM. In vivo delivery of CRISPR/Cas9 for therapeutic gene editing: progress and challenges. Bioconjug Chem. 2017;28(4):880–4.
Naldini L. Gene therapy returns to centre stage. Nature. 2015;526(7573):351–60.
Narfström K, Katz ML, Bragadottir R, Seeliger M, Boulanger A, Redmond TM, et al. Functional and structural recovery of the retina after gene therapy in the RPE65 null mutation dog. Invest Ophthalmol Vis Sci. 2003;44(4):1663–72.
Nguyen DP, Miyaoka Y, Gilbert LA, Mayerl SJ, Lee BH, Weissman JS, et al. Ligand-binding domains of nuclear receptors facilitate tight control of split CRISPR activity. Nat Commun. 2016;7:12009.
Nihongaki Y, Kawano F, Nakajima T, Sato M. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat Biotechnol. 2015;33(7):755–60.
Nihongaki Y, Otabe T, Sato M. Emerging approaches for spatiotemporal control of targeted genome with inducible CRISPR-Cas9. Anal Chem. 2018;90(1):429–39.
Nishitani N, Ohmura Y, Nagayasu K, Shibui N, Kaneko S, Ohashi A, et al. CRISPR/Cas9-mediated in vivo gene editing reveals that neuronal 5-HT1A receptors in the dorsal raphe nucleus contribute to body temperature regulation in mice. Brain Res. 2019;1719:243–52.
Nödling AR, Spear LA, Williams TL, Luk LYP, Tsai Y-H. Using genetically incorporated unnatural amino acids to control protein functions in mammalian cells. Essays Biochem. 2019;63(2):237-66.
O’Brien AR, Wilson LOW, Burgio G, Bauer DC. Unlocking HDR-mediated nucleotide editing by identifying high-efficiency target sites using machine learning. bioRxiv. 2018;464610.
Oakes BL, Nadler DC, Flamholz A, Fellmann C, Staahl BT, Doudna JA, et al. Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch. Nat Biotechnol. 2016;34(6):646–51.
Pankowicz FP, Barzi M, Legras X, Hubert L, Mi T, Tomolonis JA, et al. Reprogramming metabolic pathways in vivo with CRISPR/Cas9 genome editing to treat hereditary tyrosinaemia. Nat Commun. 2016;7:12642.
Paulk NK, Loza LM, Finegold MJ, Grompe M. AAV-mediated gene targeting is significantly enhanced by transient inhibition of nonhomologous end joining or the proteasome in vivo. Hum Gene Ther. 2012;23(6):658–65.
Pawelczak KS, Gavande NS, VanderVere-Carozza PS, Turchi JJ. Modulating DNA repair pathways to improve precision genome engineering. ACS Chem Biol. 2018;13(2):389–96.
Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520(7546):186–91.
Richter F, Fonfara I, Bouazza B, Schumacher CH, Bratovic M, Charpentier E, et al. Engineering of temperature- and light-switchable Cas9 variants. Nucleic Acids Res. 2016;44(20):10003–14.
Richter F, Fonfara I, Gelfert R, Nack J, Charpentier E, Moglich A. Switchable Cas9. Curr Opin Biotechnol. 2017;48:119–26.
Robert F, Barbeau M, Ethier S, Dostie J, Pelletier J. Pharmacological inhibition of DNA-PK stimulates Cas9-mediated genome editing. Genome Med. 2015;7:93.
Rose JC, Stephany JJ, Valente WJ, Trevillian BM, Dang HV, Bielas JH, et al. Rapidly inducible Cas9 and DSB-ddPCR to probe editing kinetics. Nat Methods. 2017;14(9):891–6.
Rouet R, Thuma BA, Roy MD, Lintner NG, Rubitski DM, Finley JE, et al. Receptor-mediated delivery of CRISPR-Cas9 endonuclease for cell-type-specific gene editing. J Am Chem Soc. 2018;140(21):6596–603.
Sandby-Moller J, Poulsen T, Wulf HC. Epidermal thickness at different body sites: relationship to age, gender, pigmentation, blood content, skin type and smoking habits. Acta Derm Venereol. 2003;83(6):410–3.
Schwank G, Koo B-K, Sasselli V, Dekkers Johanna F, Heo I, Demircan T, et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13(6):653–8.
Senis E, Fatouros C, Grosse S, Wiedtke E, Niopek D, Mueller AK, et al. CRISPR/Cas9-mediated genome engineering: an adeno-associated viral (AAV) vector toolbox. Biotechnol J. 2014;9(11):1402–12.
Senturk S, Shirole NH, Nowak DG, Corbo V, Pal D, Vaughan A, et al. Rapid and tunable method to temporally control gene editing based on conditional Cas9 stabilization. Nat Commun. 2017;8:14370.
Seyhan AA. RNAi: a potential new class of therapeutic for human genetic disease. Hum Genet. 2011;130(5):583–605.
Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351(6268):84–8.
Sürün D, Schwäble J, Tomasovic A, Ehling R, Stein S, Kurrle N, et al. High efficiency gene correction in hematopoietic cells by donor-template-free CRISPR/Cas9 genome editing. Mol Ther Nucleic Acids. 2018;10:1–8.
Suzuki T, Asami M, Patel SG, Luk LYP, Tsai Y-H, Perry ACF. Switchable genome editing via genetic code expansion. Sci Rep. 2018;8(1):10051.
Swiech L, Heidenreich M, Banerjee A, Habib N, Li YQ, Trombetta J, et al. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol. 2015;33(1):102–U286.
Tang WX, Hu JH, Liu DR. Aptazyme-embedded guide RNAs enable ligand-responsive genome editing and transcriptional activation. Nat Commun. 2017;8:15939.
Touznik A, Lee JJ, Yokota T. New developments in exon skipping and splice modulation therapies for neuromuscular diseases. Expert Opin Biol Ther. 2014;14(6):809–19.
Turczynski S, Titeux M, Tonasso L, Decha A, Ishida-Yamamoto A, Hovnanian A. Targeted exon skipping restores type VII collagen expression and anchoring fibril formation in an in vivo RDEB model. J Investig Dermatol. 2016;136(12):2387–95.
Wang HX, Li M, Lee CM, Chakraborty S, Kim HW, Bao G, et al. CRISPR/Cas9-based genome editing for disease modeling and therapy: challenges and opportunities for nonviral delivery. Chem Rev. 2017;117(15):9874–906.
Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol Ther. 2010;18(1):80–6.
Wyman C, Kanaar R. DNA double-strand break repair: all’s well that ends well. Annu Rev Genet. 2006;40:363–83.
Xu L, Park KH, Zhao LX, Xu J, El Refaey M, Gao YD, et al. CRISPR-mediated genome editing restores dystrophin expression and function in mdx mice. Mol Ther. 2016;24(3):564–9.
Yang Y, Wang L, Bell P, McMenamin D, He Z, White J, et al. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat Biotechnol. 2016;34:334–8.
Yang S, Chang R, Yang H, Zhao T, Hong Y, Kong HE, et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington's disease. J Clin Invest. 2017;127(7):2719–24.
Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol. 2014a;32(6):551–3.
Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014b;15(8):541–55.
Yin H, Song C-Q, Dorkin JR, Zhu LJ, Li Y, Wu Q, et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol. 2016;34:328–33.
Yin H, Kauffman KJ, Anderson DG. Delivery technologies for genome editing. Nat Rev Drug Discov. 2017;16(6):387–99.
Zetsche B, Volz SE, Zhang F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat Biotechnol. 2015;33(2):139–42.
Zhen S, Hua L, Liu YH, Gao LC, Fu J, Wan DY, et al. Harnessing the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated Cas9 system to disrupt the hepatitis B virus. Gene Ther. 2015;22(5):404–12.
Zhou WY, Deiters A. Conditional control of CRISPR/Cas9 function. Angew Chem Int Ed. 2016;55(18):5394–9.
Zhou XX, Zou X, Chung HK, Gao Y, Liu Y, Qi LS, et al. A single-chain photoswitchable CRISPR-Cas9 architecture for light-inducible gene editing and transcription. ACS Chem Biol. 2018;13(2):443–8.
Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol. 2015;33(1):73–80.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mills, E.M., Barlow, V.L., Luk, L.Y.P. et al. Applying switchable Cas9 variants to in vivo gene editing for therapeutic applications. Cell Biol Toxicol 36, 17–29 (2020). https://doi.org/10.1007/s10565-019-09488-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10565-019-09488-2