
Abstract
The influence of CuO/ZrO2/ZnO (Cu/Zr/Zn) catalyst modification with Mn, Ni, Ga on the physicochemical properties and activity toward hydrogen production in steam reforming of ethanol (SRE) reaction has been evaluated. The increase in hydrogen yield and the lowest selectivity to acetaldehyde were observed upon Cu/Zr/Zn modification with Mn and Ga. The physicochemical characterisation of spent catalysts revealed changes in catalysts phase compositions and reducibility. In the case of Cu/Zr/Zn, Cu/Zr/Zn/Ni and Cu/Zr/Zn/Ga catalysts, the CuO phase was reduced to metallic phase and Cu2O. Therefore, these spent catalysts exhibited lower reduction degree (Rd) in comparison with fresh catalysts. On the other hand, the addition of Mn preserved the copper on + 2 oxidation state during SRE reaction as indicated by XRD and XPS. The µRaman experiments showed that carbon deposit is formed only on the surface of Cu/Zr/Zn/Ni catalyst, which is the reason for the vast deactivation and the lower total activity of this catalyst in SRE. This was also supported by XPS which additionally showed interaction of carbon containing by-products with the surface active sites. In the case of other synthesised catalysts, no carbon formation was stated.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Ethanol is regarded as an attractive potential source of hydrogen generation. The major advantage of ethanol is the fact that it can be produced from biomass fermentation, which is a well-established, renewable and sustainable process. For this purpose, three main types of biomass can be used: starch-containing raw materials [1], sugar-containing raw materials [2], and lignocellulosic biomass (i.e.: straw, wood residues, agricultural waste) [3]. Since starch and sugar containing feedstocks are crucial for the food industry, lignocellulosic biomass conversion to ethanol is an alternative, promising route. Additionally, ethanol can be easily handled, stored and transported. It is less toxic than methanol, which is also used for steam reforming.
Hydrogen, which is considered to be environmentally friendly future energy carrier, can be produced from ethanol via three main reactions: steam reforming (1), partial oxidation (2) and oxidative steam reforming (3). Among mentioned methods, steam reforming is the most efficient in terms of hydrogen yield [4].
When complete steam reforming of ethanol (SRE) reaction is performed, 6 mol of hydrogen are produced from 1 mol of ethanol. This is the highest hydrogen yield possible to reach when compared with steam reforming of other fuels [5].
Unlike methanol, ethanol contains C–C bond which must be activated in order to obtain complete ethanol conversion and high selectivity to hydrogen and CO2 in SRE. This requires appropriate catalysts capable not only to cleavage C–C bond but also to oxidase selectively each of carbon atoms to CO2, limiting the formation of various carbon-containing side products such as acetaldehyde, ethylene, acetone, acetic acid, methane, CO, crotonaldehyde, coke. The occurrence of side reactions, which significantly reduces the theoretical concentration of hydrogen in the outlet stream, depends on the reaction temperature, pressure and steam to ethanol ratio (S/E) [6]. The wide spectrum of possible chemical reaction pathways taking place during SRE includes: ethanol dehydration, ethanol dehydrogenation, cracking, water gas-shift reaction, acetone formation, methanation, and reactions leading to coke formation i.e.: Boudouard reaction, methane decomposition, ethylene transformation [7, 8].
The final selectivity to each particular carbon-containing product is also strongly related to the physicochemical properties of the catalyst. It is known that the ratio of acidic to basic sites at the catalyst surface is a key parameter to determine the selectivity to ethylene and acetaldehyde. This is due to the ability of acidic centres to facilitate the ethanol dehydration reaction whereas basic centres promote dehydrogenation of ethanol. The ethylene, which is formed over acidic catalysts, is considered to be a coke precursor. The formation of carbon deposit due to ethylene polymerization is one of the reasons for catalyst deactivation in SRE [9].
The supported nobles metals (Rh [10, 11], Ru [12], Ir [13], Pd [14], Pt [15]) over various oxides such as ZrO2, Al2O3, MgO, CeO2 have been stated to show high activity toward ethanol conversion. Basagiannis et al. [16] found that catalytic activity and hydrogen selectivity change in the following order: Pt > Pd > Rh > Ru, with Pt and Pd showing significantly better catalytic performance. The authors highlight the fact that both the dispersed metallic phase and γ-Al2O3 support are involved in SRE reaction. The interaction of ethanol with acidic γ-Al2O3 support results in ethanol dehydration while the presence of Pt promotes the reaction of ethanol decomposition and dehydrogenation, especially at low temperature.
On the other hand, there are literature reports [17] saying that Rh supported catalysts with low metal loading exhibit the highest ethanol conversion and hydrogen selectivity in comparison with Ru, Pt, Pd based catalysts. This is also supported by Auprêtre et al. [18] who showed that Ru/γ-Al2O3 catalyst reaches 100% ethanol conversion and hydrogen selectivity equal to 73.5% at 600 °C. The high activity of Rh based catalyst in SRE is due to its high ability to C–C bond cleavage [19]. Breen et al. [20] ordered the importance of alumina-supported metals according to their catalytic activity as follows: Rh > Pd > Ni = Pt. The authors confirmed that alumina-supported catalysts facilitate dehydration of ethanol to ethylene. Moreover, it was found that the order of activity of metals at higher temperature changes when ceria/zirconia supports are used and it can be presented in the following manner: Pt ≥ Rh > Pd.
Nevertheless, the high cost of noble based catalysts seriously limits their application as catalysts for SRE [21]. Therefore, the less expensive, alternative catalysts for SRE are studied. Among them, non-noble metals seem to be the right choice. Different catalytic systems based on these metals (Co, Ni, Cu), have turned out to be active for SRE reaction [22,23,24,25]. Copper-based catalysts are mainly used in methanol steam reforming due to their high selectivity and activity [26, 27] but there are also studies on the application of copper containing catalysts in SRE. Galetti et al. [28] investigated the SRE reaction over quaternary mixed oxide CuCoZnAl catalyst. The performed studies showed very high activity of CuCoZnAl, reaching 100% ethanol conversion and selectivity to hydrogen equal to 87% at 600 °C. At this temperature the major detected products were CO2 and CO. Unfortunately, the formation of two types of carbon structures was confirmed in the spent catalyst by Raman spectroscopy. Mariño et al. [29] carried out SRE reaction at 300 °C over Cu/Ni/K/γ-Al2O3 and suggested that Cu [30] was the active component in the SRE. This is in contradiction to the findings of Ni et al. [31] and Vizcaíno et al. [32] who showed the ability of Ni to rapture C–C bond. CuO/ZnO/Al2O3 catalysts were tested in SRE by Cavallaro et al. [33] showing good activity above 350 °C. However, the formation of acetaldehyde and acetic acid was stated when SRE was performed at a low temperature (< 325 °C). There are also reports saying that the addition of copper to Co–Mg–Al hydrotalcite support improves hydrogen productivity and ethanol conversion [34]. ZnO has been tested as catalysts for SRE by Llorca et al. [35], showing complete conversion of ethanol and reaching a high H2 production amount at 450 °C. Depending on gas hourly space velocity (GHSV) value, the hydrogen selectivity was ranging from 60.4 to 73.4%. Additionally, ZnO exhibited the lowest selectivity to ethylene in comparison with other tested oxides i.e.: Al2O3, V2O5, Sm2O3, CeO2, La2O3, MgO. All these oxides were next used as supports for cobalt catalysts [36]. Among tested catalysts, Co/ZnO yielded the highest value of hydrogen, reached total ethanol conversion, and deactivation was not observed when SRE was performed at 450 °C. Unfortunately, high selectivity to acetone was stated due to aldol condensation of acetaldehyde. Nevertheless, by applying short contact times and a high ratio of Ar to EtOH + H2O it was possible to limit significantly the acetone production and favour the reforming reaction. Moreover, ZnO can promote hydrogen production reactions during SRE since water has a high sticking coefficient on its surface [37, 38]. The addition of this oxide to ZnxZryOz catalysts was found by Sun et al. [39] to limit the ethanol dehydration and promote the ethanol dehydrogenation reaction.
The aim of the studies was to investigate the influence of Cu/Zr/Zn catalyst modification with Mn, Ni, Ga on its physicochemical properties and activity in the low temperature steam reforming of ethanol (SRE). The SRE reaction was performed at 350 °C to favour the water gas shift reaction and limit the formation of CO. The water/ethanol (S/E) ratio was equal to 10. The higher than stoichiometric S/E ratio during SRE was selected to simulate the bioethanol which contains S/E ≈ 13 [40]. The spent catalysts were investigated with respect to their changes in morphology, textural properties and chemical composition. In order to determine these aspects, a broad spectrum of techniques was implemented such as X-ray diffraction analysis, CO2 temperature-programmed desorption, N2O dissociative chemisorption, µRaman spectroscopy, N2 physisorption, temperature-programmed reduction with hydrogen and X-ray photoelectron spectroscopy.
2 Experimental
The CuO/ZrO2/ZnO/X (where X = NiO, MnO, Ga2O3) catalysts were synthesized via co-precipitation method at pH 7, using Na2CO3 as precipitating agent. The CuO/ZrO2 weight ratio in all synthesised catalysts was fixed to 2.3. The concentration of dopants was 5 wt%. Cations in the form of nitrates and Na2CO3 were simultaneously added dropwise into the beaker containing 100 mL of deionized water at 60 °C. The mixture was vigorously stirred during the precipitation. Next, precipitates were washed by five time centrifugation at 4200 rpm, dried at 100 °C and calcined at 550 °C for 3 h. The synthesised catalysts are donated as Cu/Zr/Zn, Cu/Zr/Zn/Ni, Cu/Zr/Zn/Mn, Cu/Zr/Zn/Ga, respectively for CuO/ZrO2/ZnO, CuO/ZrO2/ZnO/NiO, CuO/ZrO2/ZnO/MnO, CuO/ZrO2/ZnO/Ga2O3.
X-ray diffraction patterns (XRD) were recorded on Bruker AXS D505 with CuKα radiation. The analysis was performed between 0.6°–90° 2Θ range with an accumulation time 5 s. Mean crystallite sizes were evaluated according to Sherrer’s equation. The main reflection of the XRD pattern, for each of the individual phases, was used for calculation.
CO2 temperature-programmed desorption (CO2-TPD) measurements were carried out in quartz fixed-bed flow reactor connected online to the mass spectrometer (QMG 220 PRISMA PLUS). Signals: m/z = 2 (H2), m/z = 44 (CO2), m/z = 40 (Ar) were monitored during TPD. Prior to TPD run, sample (50 mg) was reduced in 5%H2/Ar flow (30 mL/min) at 250 °C for 2.5 h. Next, reactor was cooled down to room temperature (RT) and pulses (250 µL) of 5%CO2/Ar were introduced until saturation. Then, the sample was flushed with He flow (40 mL/min) for 0.5 h until obtaining stable CO2 line. TPD was done from RT to 500 °C with ∆T = 10 °C /min under He flow (40 mL/min).
N2O dissociative chemisorption was performed using the same set-up line as for CO2-TPD. Sample (100 mg) was reduced in the same manner as for CO2-TPD. The pulses (100 µL) of N2O were introduced at 90 °C under He flow (30 mL/min) until reoxidation was completed. Signals: m/z = 44 (N2O) and m/z = 28 (N2, N2O) were monitored. The metallic copper surface area (SCu) was calculated assuming atomic copper surface density of 1.47 × 1019 Cu atoms/m2, corresponding to 6.1 × 10–6 O2 moles/m2 for the stoichiometry Cu/O = 2 [41]. The Cu metal dispersion (DCu) was measured as the percentage ratio of surface Cu moles based on N2O chemisorption to the total moles of Cu present in the catalysts. Additionally, Cu° particle size (PSCu) was determined using the following equation:
where ρ is the density of Cu metal (8.92 g/cm3) and wt.% is Cu loading. N2O chemisorption experiments were repeated a few times for each sample, resulting in a relative standard deviation (RSD) below 1%.
The µRaman analyses for spent catalysts were carried out with a Renishaw inVia dispersive spectrometer equipped with a CCD detector and combined with a Leica DMLM confocal microscope. The laser line of 514 nm was used as the excitation source. The spectra were recorded with a resolution of 2 cm−1 at ambient conditions and the scattered light was collected with a 50 × Olympus objective. The six scans were collected to assure a good signal-to-noise ratio.
Temperature-programmed reduction with hydrogen (H2-TPR) was carried out on Chembet-3000 (Quantochrome). The hydrogen consumption was monitored with TCD detector. For a typical H2-TPR experiment, a sample (25 mg) was placed in quartz U-shape tube reactor and conditioned at 100 °C under He flow (30 mL/min) for 1.5 h. Next, the sample was cooled down to RT under He flow and H2-TPR experiment was performed in 5%H2/Ar (30 mL/min) in temperature range RT – 850 °C (∆T = 10 °C /min). For quantitative H2-TPR studies, four point calibration curve was prepared using appropriate amounts of CuO standard (Aldrich) mixed with Al2O3 in % wt. ratio = 1:2, 1:4, 1:16, 1:12 (mechanical mixture). The calibration has covered the hydrogen consumption range corresponding to TPR analysis. The H2-TPR and CO2-TPD profiles have been deconvoluted into Gaussian peaks. Deconvolution was performed in Origin 2018 Pro software.
The BET surface area was measured with nitrogen adsorption at − 196 °C on Quantachrome Autosorb-1. Prior to measurements, samples were degassed for 18 h at 150 °C to remove adsorbed water and other surface impurities. The pores size distribution (PSD) profiles were obtained by Barrett-Joyner-Halenda (BJH) method from the desorption branch.
The X-ray photoelectron spectra (XPS) were recorded using hemispherical analyser EA 15 (PREVAC) equipped with dual anode X-ray source RS 40B1 (PREVAC). The measurements were performed using Mg Kα (1254 eV) radiation and analyser pass energy of 100 eV. The spectra were recorded in normal emission geometry with an energy resolution of 0.9 eV. The powder samples were pressed into indium foil and mounted at the dedicated holder, pumped out to high vacuum then transferred into UHV chamber. The spectra were analysed with the use of CasaXPS 2.3.15 software. The electron binding energy (BE) scale was calibrated for the Fermi edge at 0.0 eV. The Shirley type spectrum background was used. The highly resolved spectra were deconvoluted with Voigt function (Gauss to Lorentz profile ratio of 70:30). The spectra were compared in relation to the background level.
Steam reforming of ethanol (SRE) was performed in the fixed-bed flow reactor connected online to gas chromatograph. H2, CO, CO2 were separated with DB-PS624 capillary column connected to TCD detector while C2H5OH, CH4, C2H4, CH3CHO, CH3COCH3, CH3COOH were separated with Porapak-T packed column connected to FID detector. Argon was used as a carrier for GC analysis. For SRE, catalyst (150 mg mixed with quartz to 0.7 cm3) was loaded into the reactor and reduced in the stream of 5%H2/Ar (50 mL/min) at 250 °C for 1.5 h. Next, in order to remove hydrogen from the lines, the catalyst was flushed with Ar (50 mL/min) and the temperature was increased to 350 °C. Then, the steam of water/ethanol mixture (S/E = 10, molar ratio) was introduced in the Ar stream (50 mL/min) via Bronkhorst® Vapour Delivery System (CEM). The temperature of CEM was 120 °C. All lines from CEM outlet to GC inlet were electrically heated and kept at a temperature of 120 °C in order to prevent water and ethanol from condensation. Ethanol conversion (XEtOH) and selectivity (Si) for carbon-containing products were calculated according to carbon balance where: [EtOH]in is a number of ethanol moles in inlet stream, [EtOH]out is a number of ethanol moles in outlet stream, [Ci] is a number of moles of carbon-containing product i, and ni is a number of carbon moles in carbon-containing product i.
Hydrogen yield was calculated as follows:
where [H2]out is number of hydrogen moles being produced during SRE.
3 Results and Discussion
The XRD analysis was performed for both fresh (Fig. 1) and spent catalysts (Fig. 2). In the case of fresh catalysts, CuO is the main crystalline phase (2θ = 35.5° and 38.9°). The modification with all dopants leads to a decrease in CuO crystallite sizes in comparison with Cu/Zr/Zn catalyst (Table 1). The smallest CuO crystallite sizes (12.2 nm) are observed for Cu/Zr/Zn/Ni catalysts whereas doping with Mn and Ga leads to CuO crystallite sizes equal to 16.7 nm and 15.1 nm, respectively. The XRD peak of low intensity (2θ = 30.5°) assigned to tetragonal ZrO2 phase (t-ZrO2) is present in diffractograms for Cu/Zr/Zn/Ni and Cu/Zr/Zn/Ga. The calculated values of t-ZrO2 crystallite sizes based on this XRD signal are 2.0 nm and 2.1 nm for Cu/Zr/Zn/Ni and Cu/Zr/Zn/Ga, respectively. On the other hand, there is no crystallization of t-ZrO2 in the case of Cu/Zr/Zn and Cu/Zr/Zn/Mn catalysts. Taking into account the low intensity of XRD peaks of ZrO2 phase or their absence, one can say that ZrO2 is present mainly in the amorphous phase in the synthesised catalysts. This is quite surprising since all catalysts were calcined at 550 °C, at which a-ZrO2 should no longer be present [42]. The explanation for this is that in the obtained catalysts a-ZrO2 phase is thermally stabilized, which hinders its transformation into t-ZrO2. A similar effect was observed for Cu/Zr catalysts [43]. The XRD peaks related to MnO, Ga2O3, and NiO are not observed due to their small crystallite sizes (< 2 nm), making them undetectable by means of XRD and/or due to the amorphous state of dopants. Additionally, only in the case of Cu/Zr/Zn catalyst, the XRD signals corresponding to ZnO phase were found, which are partially overlapping with the reflexes assigned to CuO phase.

Diffraction patterns of fresh catalysts

Diffraction patterns of spent catalysts

H2-TPR profiles for fresh and spent catalysts (blue line)
The significant changes in phase composition are observed for spent Cu/Zr/Zn, Cu/Zr/Zn/Ni and Cu/Zr/Zn/Ga catalysts. Firstly, the intensities of reflexes assigned to t-ZrO2 are increased, meaning the transformation of amorphous ZrO2 into t-ZrO2 occurred during SRE reaction. For Cu/Zr/Zn/Ni and Cu/Zr/Zn/Ga catalysts, the crystallite sizes of t-ZrO2 reache 4.0 nm and 3.0 nm, respectively. Secondly, the diffraction peaks of CuO phase disappear and the metallic copper phase (Cu°) is present (2θ = 43.4° and 50.4°). The reduction, which occurs during SRE reaction, leads to Cu° crystallite sizes of higher value than CuO crystallite sizes in fresh catalysts (Table 1). Additionally, the crystallization process of the new Cu° phase strongly depends on additives. The introduction of Ni and Ga in Cu/Zr/Zn catalyst results in lower Cu° crystallite sizes, which are 17.6 nm and 22.7 nm, respectively.
On the other hand, the XRD analysis of spent Cu/Zr/Zn/Mn catalyst revealed only minor changes in phase composition. In both, fresh and spent Cu/Zr/Zn/Mn catalyst, CuO phase is present but the CuO crystallite sizes in spent catalyst are smaller (9.5 nm) in comparison with fresh catalyst (16.7 nm). The presence of CuO phase in spent Cu/Zr/Zn/Mn catalyst suggests that addition of Mn preserves the crystalline CuO phase and hinders the process of its reduction during SRE reaction. This phenomena was also supported by TPR and XPS analyses of spent Cu/Zr/Zn/Mn, which are described further in the manuscript. Similarly to remaining spent catalysts, in the case of Cu/Zr/Zn/Mn the crystallization of a-ZrO2 into t-ZrO2 proceeds.
TPR profiles of synthesized catalysts and spent catalysts (blue line) are depicted in Fig. 3. The reduction degree (Rd) was calculated based on reduction stoichiometry (Cu/H2) and hydrogen consumption in TPR experiments for all catalysts (Table 2). According to scientific literature, the ZnO and ZrO2 do not undergo reduction in the experimental region [44, 45]. In the case of Cu/Zr/Zn/Ni catalyst, the broad peak of very low intensity is visible with maximum at 415 °C (Fig. S1, supplementary information). This signal is ranging from 300 to 515 °C. In order to identify this signal the reduction of pure NiO standard was carried out. The position of this signal matches the position of NiO reduction peak, which can be seen in the Fig S1

Raman spectra of spent catalysts
The remains visible peaks of high intensity in TPR profile have been solely assigned to the reduction of different CuO species to metallic copper. The temperature corresponding to maximum reduction rate (Tmax) is decreased for Cu/Zr/Zn/Ni (Tmax = 235 °C) and Cu/Zr/Zn/Mn (Tmax = 266 °C) in comparison with unmodified Cu/Zr/Zn catalyst (Tmax = 277 °C). In this case, the observed decrease in the Tmax is related with smaller particle sizes of CuO in the Cu/Zr/Zn/Ni and Cu/Zr/Zn/Mn as indicated by XRD and by higher metallic copper dispersion as measured by N2O dissociative chemisorption (Table 2). Not only does the addition of Mn and Ni result in lowering the positon of TPR signal but also leads to increase in Rd value, reaching 99% for Cu/Zr/Zr/Mn. All above mentioned observations show that catalysts doping with Mn and Ni facilities the reduction of CuO. On the other hand, Cu/Zr/Zn/Ga catalyst is reduced at higher temperature (Tmax = 292 °C) and has Rd value equal to 84% which is the lowest among synthesised catalysts. This suggests the existence of strong interaction between Ga2O3 and CuO, which has influence on the amount of reducible CuO species. The decrease in the amount of reducible CuO, as a result of its interaction with other metal oxides was also stated by Guo et al. [46] and Wan et. Al [47].
For all catalysts, very broad reduction signal is visible in the range of 171–320 °C which can be deconvoluted in two (Cu/Zr/Zn/Mn), or three (Cu/Zr/Zn, Cu/Zr/Zn/Ni, Cu/Zr/Zn/Ga) Gaussian peaks. These peaks can be related with two or three different CuO species present in the catalysts. The low-temperature signals can be assigned to well dispersed copper oxide species, whereas the high-temperature signals are related with reduction of bulk-like CuO species of higher particle sizes [48].
The TPR profiles of spent Cu/Zr/Zn, Cu/Zr/Zn/Ni and Cu/Zr/Zn/Ga catalysts show signals of significant lower intensities. The calculated Rd values of these catalysts are 17%, 25%, and 21% for Cu/Zr/Zn, Cu/Zr/Zn/Ni and Cu/Zr/Zn/Ga, respectively. This means that the concentration of CuO phase is greatly reduced. This is in good agreement with XRD results which shows that metallic copper is a dominant phase in the spent Cu/Zr/Zn, Cu/Zr/Zn/Ni Cu/Zr/Zn/Ga catalysts. On the contrary, only a slight decrease in Rd value for spent Cu/Zr/Zn/Mn catalyst is observed in comparison with fresh catalysts. In this case, the concentration of CuO in both fresh and spent catalyst is similar and no metallic copper phase was found in spent Cu/Zr/Zn/Ga catalysts according to XRD analysis. The visible TPR signals for spent catalyst suggest that CuO phase is still present in the spent catalyst in the case of Cu/Zr/Zn, Cu/Zr/Zn/Ni and Cu/Zr/Zn/Ga, which is in contradiction to XRD diffractograms of these catalysts. In order to confirm the presence of the amorphous CuO phase in spent catalysts, the Raman spectra were recorded (Fig. 4). Raman spectroscopy is a well-known technique to provide information about the surface of the sample and it can be also applied for carbon deposit analysis. According to relevant literature, only three modes are Raman active for CuO phase i.e.: Ag (274 cm−1), B1g (328 cm−1), B2g (627 cm−1) [49]. All three bands are visible in the Raman spectra obtained for spent catalysts. The broad shape of the recorded CuO Raman signals and their slight shift toward the low wavenumber region can result from the strong interaction between Cu and ZrO2. On the other hand, the Raman spectra for spent Cu/Zr/Zn, Cu/Zr/Zn/Ni and Cu/Zr/Zn/Ga catalysts do not show signals related with Cu2O phase which presence was confirmed by XRD analysis (Fig. 2) According to scientific literature, Cu2O phase gives rise to five Raman peaks at 201, 300, 406, 489 and 638 cm−1 [49]. In this case, the transformation of Cu2O to CuO can undergo during exposure to a laser beam while collecting Raman spectra. This phenomenon was stated by Tran et al. [50]. It was shown that illumination of CuO nanostructure sample with the high density laser beam triggers the oxidation Cu2O to CuO. This manifests in the disappearance of Cu2O Raman peaks while A1 signal assigned to CuO becomes broader and is shifted toward low wavenumber. Additionally, the broad CuO signals in Raman spectra are overlapping with Raman bands for t-ZrO2, which are reported to appear at 148, 472, 608 and 640 cm−1 [51]. Therefore, the confirmation of t-ZrO2 phase presence in spent catalysts was not possible with Raman spectroscopy. The visible signal above 1000 cm−1 can be assigned to ZnO [52], which is in accordance with XRD results of spent catalysts, whereas peak at 530 cm−1 is related with NiO [53, 54].

N2 adsorption–desorption isotherms: fresh catalysts (black line) and spent catalysts (blue line)
The N2 adsorption–desorption isotherms, BJH pore size distribution (PSD) profiles of fresh (blue line) and spent catalysts (black line) are presented respectively in Figs. 5 and 6 whereas textural properties are summarized in Table 3. The typical type V isotherms with H3-type loop according to IUPAC classification [55] are observed for all catalysts. The visible hysteresis loop above p/p° = 0.7 results from capillary condensation of nitrogen in the mesoporous. The modification with Ni, Ga and Mn does not change the position of the hysteresis loops in comparison with Cu/Zr/Zn catalyst. On the other hand, the shape of the hysteresis loops becomes wider and steeper upon Cu/Zr/Zn catalyst doping. These changes in hysteresis loops result from the increase in total pore volume (Table 3). Based on PSD profiles, one can say that the increase in the total pore volume for the modified catalysts is mainly due to mesoporous generation within the range from 9 to 30 nm. The addition of all dopants leads to the increase in BET surface area in comparisons with unmodified Cu/Zr/Zn catalyst (Table 3). Surprisingly, there is a noticeable increase in BET surface area for spent catalysts. This is due to the appearance of microporous as confirmed by t-plot method. Moreover, PSD is broader and peak maxima in the PSD profiles of spent catalysts are higher which results in an increase in total pore volume.

Pore size distribution: fresh catalysts (black line) and spent catalysts (blue line)

CO2-TPD profiles for fresh catalyst: signal—black line, cumulative curve—red line, deconvoluted peaks used for quantification—green line, temperature—dashed line
The N2O dissociative chemisorption results are presented in Table 2. In the case of all modified catalysts, the increase in metallic surface copper area (SCu) and in metallic copper dispersion (DCu) are observed in comparison with Cu/Zr/Zn catalyst. Among doped catalysts, the strongest effect of surface copper stabilization occurs for Cu/Zr/Zn/Ni catalyst, which manifests in the highest value of SCu (17.8 m2/gcat.) and DCu (5.9%). Additionally, the SCu/BET ratio = 0.28 is the highest for this catalyst. This shows that 28% of the catalyst BET surface is occupied by copper. For other catalysts this ratio is lower, which means that less copper is available on the surface, whereas more copper is in the bulk. According to N2O dissociative chemisorption results, it can be stated that reduction of all catalysts leads to average Cu° particle sizes (PSCu) of higher value than CuO crystallites sizes in fresh catalysts, which is in line with XRD results of spent catalysts. Nevertheless, the differences in PSCu and Cu° crystallites sizes are visible. The explanation for these discrepancies is that Cu° crystallites sizes evaluated according to Sherrer equation refer only to one XRD peak, i.e., one crystal direction defined by (hkl). On the other hand, the N2O dissociative chemisorption technique gives information on average metallic sites, which are located solely in the surface region of the catalyst.
The surface basicity of the catalysts was determined by means of CO2-TPD. The obtained CO2-TPD profiles are shown in Fig. 7 whereas the quantitative data are depicted in Table 4. Desorption profiles were deconvoluted into Gaussian peaks in order to get a better insight into the distributions of basic sites. Based on the literature, CO2 can desorb from weak (50–150 °C), from medium (150–240 °C), and from strong (> 240 °C) basic sites during TPD [56]. The observed in TPD profiles, deconvoluted peak maxima were classified according to the temperature ranges mentioned above. It is well known that weak basic sites can be attributed to the surface –OH groups, medium basic sites to oxygen bonded to metal (Mex–O2−, x = 2 + , 3 + , 4 +) whereas strong basic sites are related with isolated O2− anions [57].
In the case of all synthesised catalysts, the weak and medium basic sites are present. The CO2 desorption is observed in the temperature range RT – 234, when the position of deconvoluted peak maxima (green lines) is considered. The addition of promoters to Cu/Zr/Zn catalyst leads to the increase in total basicity in the following order: Cu/Zr/Zn < Cu/Zr/Zn/Mn < Cu/Zr/Zn/Ga < Cu/Zr/Zn/Ni. For catalyst modified with Ni, the total concentration of basic sites reaches 165.9 µmol/gcat.. Additionally, the changes in the distribution of weak and medium basic sites are also visible. For Cu/Zr/Zn catalyst, the weak basic sites dominate (73.1 µmol/gcat.) whereas for modified catalysts the contribution of medium basic sites in total basicity is significantly higher, resulting in a higher concentration of total basicity as mentioned before.
In all recorded CO2-TPD profiles, the increase in CO2-TPD signal is observed above 275 °C. This is related to the desorption of additional CO2 which originates from the atmosphere. The contribution of this extra CO2 was marked with blue lines and was confirmed in blank TPD (Fig. S2). For these experiments, catalysts were only reduced and later heated up in the same manner as during CO2-TPD, meaning no CO2 was introduced. For all catalysts, signals below 275 °C were not present. Only above this temperature, the increase in the CO2 signals was visible. Moreover, the ratio of desorbed CO2 (Totdes.) to adsorbed CO2 (Totads.), based on the deconvoluted maxima (green lines) up to 234 °C, is close to one for all CO2-TPD experiments (Table 4). This additionally proves that CO2 desorbing above this temperature can be solely assigned to extra atmospheric CO2. Similar results were reported by Smyrnioti et al. [58] who investigated CO2 adsorption on a commercial CuO/ZnO/Al2O3 catalyst with TPD technique. During thermal treatment of reduced sample, the desorption of CO2 was also stated above 275 °C. The authors concluded that this extra amount of CO2 must be taken into consideration during CO2-TPD experiment.
In order to identify the chemical state of the catalysts surface, the XPS analysis was performed. The Cu 2p spectra of fresh and spent catalysts are depicted in Fig. S3a and Fig. S3b, respectively. In the case of fresh catalysts, two maxima of Cu 2p2/3 and Cu 2p1/2 doublet components are visible with BEs value at ca. 932 eV and 952 eV, respectively. The presence of shake-up satellites and BE values indicate that all fresh samples contain CuO species, which is in line with XRD analysis. The deconvolution of Cu 2p core excitation was performed into three components (A, B, C) in order to determine the contribution of different copper species. Peaks A were assigned to Cu+ bonded to oxygen in defected oxides. Peaks B were ascribed to either Cu+ in Cu2O or Cu2+ in CuO. Peaks C were related to Cu2+ in hydroxide or salt type surrounding [59, 60].
On the other hand, based on the shape of deconvoluted Cu 2p peaks (Fig. S3a), it cannot be distinguished what kind of copper species dominate on the surfaces of the fresh catalysts. Therefore, an analysis of Cu L3M45M45 Auger spectra (Fig. S4a) must be performed to reveal the real electronic state of copper. Additionally, the calculated modified Auger parameter (α′) (Table 5) indicates that the surface of fresh catalysts contains CuO as the main copper component, which is supported by XRD. Moreover, in the case of Cu/Zr/Zn catalyst, hydroxylation of the copper species is the most pronounced among all fresh catalysts.
In the case of spent Cu/Zr/Zn, Cu/Zr/Zn/Ni and Cu/Zr/Zn/Ga catalysts, the shake-up satellites peaks are no longer visible. This suggests that the surface of catalysts is reduced after SRE, which is also supported by α’ value (Table 5, Fig. S3b and Fig. S4b). This value indicates that the surface is enriched in Cu2O phase as supported by XRD. However, the XPS spectra revealed the presence of Cu2+ species at the surface layer of these catalysts, mainly in the form of salts (components C and D in Fig. S3b). The observed partial reduction of cooper species in Cu/Zr/Zn, Cu/Zr/Zn/Ni and Cu/Zr/Zn/Ga catalysts correlates with TPR experiments (Rd value). Contrary to the mentioned spent catalysts, copper in the form of CuO is preserved at the surface of Cu/Zr/Zn/Mn. This is also supported by XRD and TPR analyses.
Among all studied samples, Cu/Zr/Zn/Ni catalyst shows the highest ability for adsorption of SRE by-products, which is indicated by higher carbon concentration at the surface (Table 6). This results in the appearance of additional spectra components (Fig. S3b–S11) at higher binding energies (BE), which are observed in C 1s, O 1s, Zr 3d, Zn 2p Cu 2p and Ni 2p core excitations.
In the case of Zr 3d core excitation analysis for fresh catalysts (Fig. S7a), two main components (A, B) in the deconvoluted spectrum are observed. They are assigned to Zr3+ in defected oxide lattice (A) and in oxide lattice (B), respectively [59, 60]. After SRE, C components at higher BE appear indicating the interaction of Zr3+ ions with SRE by-products. Additionally, performing the SRE reaction over Mn and Ga doped catalysts results in the appearance of the X component in C 1 s spectrum (Fig. S5b), ascribed to metal–carbon bonds. For spent Ni modified Cu/Zr/Zn catalyst, large C component is present in the Zr 3d spectrum (Fig. 7b), which indicates pronounced adsorption of organic by-products of SRE reaction at Zr active sites.
In the case of Zn 2p spectrum (Fig. S8b), SRE reaction leads to hydroxylation of Zn containing active sites. This appears as some increase of the C component assigned to hydroxyl groups bonded to Zn2+ ions in the oxide lattice. Additionally, spent Cu/Zr/Zn/Ni catalyst exhibits D component, which can be related to the interaction of organic by-products with Zn containing active sites (Fig. S8b).
The analysis of Ni 2p spectra revealed changes in Ni2+ electronic state caused by SRE. The reaction leads to much more electronegative (electrophilic) properties of Ni surrounding as indicated by D component appearing for spent catalyst (Fig. S9). The spectrum for spent catalyst does not contain A component assigned to NiO compound that suggests possible chemical adsorption of by-products at NiO sites.
In the case of catalysts modified with Mn, the Mn 2p spectra for fresh and spent catalyst do not show significant changes. Two components assigned to Mn2+ and Mn4+ are observed (Fig. S10). This indicates that Mn2+ is mainly present in the form of salts and SRE reaction slightly oxidizes Mn species by increasing Mn4+ surface concentration from ca. 49 to 55%. The surface state of Ga containing catalyst does not undergo significant changes during SRE as it is observed in core excitations for all analysed elements. (Fig. S3–S8 and S11). It was found that gallium in form of oxide exists at the surface of both fresh and spent catalysts.
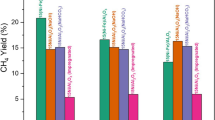
The catalytic results are shown in Table 7. The steady state conditions were reached after time on stream (TOS) = 2 h. Then, the ethanol conversion (XEtOH), hydrogen yield (\({\text{Y}}_{{{\text{H}}_{2} }}\)) and selectivity to particular carbon containing products were measured. The presented values are average from five consecutive online GC analyses. The following carbon containing products were found in the outlet stream during steam reforming of ethanol: CO, CO2, CH4, C2H4 (ethylene), CH3CHO (acetaldehyde), CH3COCH3 (acetone), CH3COOH (acetic acid).
The lowest yield of hydrogen and ethanol conversion were recorded for Cu/Zr/Zn/Ni. Although this catalyst exhibited the initial ethanol conversion of 100%, the rapid deactivation was observed leading to a decrease in ethanol conversion to 67% after TOS = 2 h and the lowest selectivity to CO2 (\({\text{S}}_{{{\text{CO}}_{2} }}\) = 7%) and hydrogen yield (\({\text{Y}}_{{{\text{H}}_{2} }}\) = 34%) among tested catalysts. The Raman spectra (Fig. 4 and S12) of spent Cu/Zr/Zn/Ni catalyst revealed the peak at 1590 cm−1 (G-band), which is related to the typically ordered graphite. On the other hand D-band (ca. 1330 cm−1), assigned to the carbon of disordered structure, is not observed [61]. Based on that, it can be concluded that ordered carbon deposit, formed on the Cu/Zr/Zn/Ni catalyst surface, is responsible for rapid catalyst deactivation. The formation of carbon deposit is also supported by a lower carbon balance (95%), which was calculated based on GC results. The accumulation of by-products at Cu/Zr/Zn/Ni surface, as confirmed by XPS experiments, also contributes to the deactivation process. On the other hand, in the case of other spent catalysts, neither appearance of D-band nor G-band was visible in Raman spectra in the range of 1300–1600 cm−1, suggesting that carbon deposit had not been formed. According to literature, the formation of carbon deposit can be related to Boudouard reaction or dissociation of hydrocarbon molecules [6].
In the product distribution of Cu/Zr/Zn/Ni catalyst, acetaldehyde dominates (\({\text{S}}_{{{\text{CH}}_{3} {\text{CHO}}}}\) = 60%). This means that hydrogen is produced mainly due to ethanol dehydrogenation reaction (4). The presence of CH4 and CO in the outlet stream suggests that a fraction of acetaldehyde, which is formed during SRE, is also decomposed (5). Moreover, ethanol decomposition (6) can undergo too.
It is known that basic sites of higher strength facilitate the ethanol dehydrogenation to acetaldehyde [62]. The measurements of CO2-TPD showed that Cu/Zr/Zn/Ni catalyst possesses the higher concentration of basic sites (165.9 \(\mu {\text{mol}}_{{{\text{CO}}_{2} }} /{\text{g}}_{{{\text{cat}}.}}\)), among which medium basic sites account for 72% of total basicity. Therefore, the selectivity to acetaldehyde is the highest in comparison with other synthesised catalysts. The reaction of acetaldehyde condensation (7) is hindered in comparison with the remaining catalysts since the selectivity to acetone is the lowest and equal to 12%. Similarly, the reaction of ethanol reforming to acetic acid (8) is limited (\({\text{S}}_{{{\text{CH}}_{3} {\text{COOH}}}}\) = 3%).
The addition of Ga and Mn changes significantly the product distribution of SRE reaction. Firstly, due to the lower concentration of medium basic sites in comparison with Cu/Zr/Zn/Ni, the selectivity to acetaldehyde is lower and equal to 6% and 12%, respectively for Cu/Zr/Zn/Mn and Cu/Zr/Zn/Ga. Secondly, the higher rate of acetone formation leads to the increase in the selectivity to acetone for both mentioned catalysts. During acetone synthesis (7), via acetaldehyde condensation, hydrogen is generated. Therefore, one can say that this reaction is the main source of hydrogen when SRE is performed over Cu/Zr/Zn catalyst doped with Mn (\({\text{Y}}_{{{\text{H}}_{2} }}\) = 66%) or Ga (\({\text{Y}}_{{{\text{H}}_{2} }}\) = 67%). Moreover, the lack of CO and CH4 in the products suggests that side reactions like acetaldehyde decomposition (5) and ethanol decomposition (6) do not proceed when Cu/Zr/Zn/Ga and Cu/Zr/Zn/Mn are used for SRE. On the other hand, the improvement in selectivity to CO2 upon Mn and Ga addition is visible, which reaches 21% and 18%, respectively for Cu/Zr/Zn/Mn and Cu/Zr/Zn/Ga. This indicates that CO, which is formed in acetone synthesis (7), is further oxidised to CO2 due to a water gas shift reaction (9). For both catalysts, ethanol conversion is 100% at 350 °C. In the case of unmodified Cu/Zr/Zn catalyst, the acetone (\({\text{S}}_{{{\text{CH}}_{3} {\text{COCH}}_{3} }}\) = 44%) and acetaldehyde (\({\text{S}}_{{{\text{CH}}_{3} {\text{CHO}}}}\) = 34%) dominate in the products but the hydrogen yield is lower (\({\text{Y}}_{{{\text{H}}_{2} }}\) = 58%) in comparison with catalysts doped with Mn and Ga.
Additionally, in the case of all catalysts, the presence of CO2 in products during SRE reaction performed at 350 °C, can also results from the steam reforming of acetaldehyde (10) and/or from the steam reforming of acetone (11) as stated by Dolgikh et al. [63], who investigated the SRE reaction over iron based catalysts in the temperature range from 300 to 500 °C.
4 Conclusions
The modification of Cu/Zr/Zn catalysts with Ga, Mn and Ni leads to an increase in BET surface area and copper dispersion. In the case of fresh catalysts, CuO is the dominant phase whereas ZrO2 is present as an amorphous phase or as t-ZrO2 of very low crystallites sizes. The addition of promoters results in a decrease in CuO crystallite sizes when compared with Cu/Zr/Zn catalyst.
The highest yield to hydrogen is observed for Cu/Zr catalyst modified with Ga (\({\text{Y}}_{{{\text{H}}_{2} }}\) = 66%) and Mn (\({\text{Y}}_{{{\text{H}}_{2} }}\) = 67%). In both cases, the primary source of hydrogen is acetone formation reaction which undergoes during SRE. Moreover, the highest selectivity to CO2 for Cu/Zr/Zn/Ga and Cu/Zr/Zn/Mn is due to the water gas shift reaction. On the other hand, a vast deactivation of Cu/Zr/Zn/Ni catalyst is observed which leads to a decrease in ethanol conversion (XEtOH = 67%) and hydrogen yield (\({\text{Y}}_{{{\text{H}}_{2} }}\) = 34%) in comparison with Cu/Zr/Zn catalyst. This is caused by the formation of carbon deposit and adsorption of organic by-products at the surface. In the case of this catalyst, hydrogen is produced mainly upon ethanol dehydrogenation to acetaldehyde, which results from the highest surface basicity.
The partial reduction of CuO undergoes during SRE reaction for Cu/Zr, Cu/Zr/Zn/Ni and Cu/Zr/Zn/Ga. This is the reason for significantly lower hydrogen consumption during H2-TPR. Moreover, the transformation of amorphous ZrO2 into tetragonal phase and increase in t-ZrO2 crystallites sizes are observed in spent catalysts. For catalyst modified with Mn, copper is stabilised on + 2 oxidation state in spent catalysts. Therefore no changes in phase composition and H2-TPR profile are observed.
References
Bušić A, Marđetko N, Kundas S, Morzak G, Belskaya H, Šantek MI, Komes D, Novak S, Šantek B (2018) Food Technol Biotechnol 56:289
Mussatto SI, Dragone G, Guimarães PMR, Silva JPA, Carneiro LM, Roberto IC, Vicente A, Domingues L, Teixeria JA (2010) Biotechnol Adv 28:817
Zhao L, Zhang X, Xu J, Ou X, Chang S, Wu M (2015) Energies 8:4096
Akdim O, Cai W, Fierro V, Provendier H, Veen A, Shen W, Mirodatos C (2008) Top Catal 51:22
Vargas P, Campos CH, Navarro RM, Fierro JLG, Reyes P (2015) J Mol Catal A 407:169
Mattos LV, Jacobs G, Davis BH, Noronha FB (2012) Chem Rev 112:4094
Sohn H, Ozkan US (2009) Energy Fuels 30:5309
Valant AL, Can F, Bion N, Duprez D, Epron F (2010) Int J Hydrogen Energy 35:5015
Erdőhelyi A, Raskó J, Kecskés T, Toth M, Dömök M, Baan K (2006) Catal Today 116:367
Roh HS, Wang Y, King DL, Platon A, Chin YH (2006) Catal Lett 108:15
Birot A, Epron F, Descorme C, Duprez D (2008) Appl Catal B 79:17
Koh ACW, Leong WK, Chen L, Ang TP, Lin J, Johnson BFG, Khimyak T (2008) Catal Commun 9:170
Wang F, Cai WT, Provendier H, Schuurman Y, Descorme C, Mirodatos C, Shen W (2012) Appl Catal B 125:546
Goula M, Kontou S, Tsiakaras P (2004) Appl Catal B 49:135
Ciambelli P, Palma V, Ruggiero A (2010) Appl Catal B 96:18
Basagiannis CA, Panagiotopoulou P, Verykios XE (2008) Top Catal 51:2
Liguras DK, Kondarides DI, Verykios XE (2003) Appl Catal B 43:345
Auprêtre F, Descorme C, Duprez D (2002) Catal Commun 3:263
Sheng PY, Yee A, Bowmaker GA, Idriss H (2002) J Catal 208:393
Breen JP, Burch R, Coleman HM (2002) Appl Catal B 39:65
Song H, Ozkan US (2010) J Phys Chem A 114:3796
Frusteri F, Freni S, Spadaro L, Chiodo V, Bonura G, Donato S, Cavallaro S (2004) Catal Commun 5:611
Li S, Li M, Zhang C, Wang S, Ma X, Gong J (2012) Int J Hydrogen Energy 37:2940
Han SJ, Song JH, Bang Y, Yoo J, Park S, Kang KH, Song IK (2016) Int J Hydrogen Energy 41:2554
Garbarino G, Riani P, Lucchini MA, Canepa F, Kawale S, Busca G (2013) Int J Hydrogen Energy 38:82
Lorenzut B, Montini T, De Rogatisa L, Canton P, Benedetti A, Fornasiero P (2011) Appl Catal B 101:397
Navarro RM, Peña MA, Fierro JLG (2007) Chem Rev 107:3952
Galetti AE, Gomez MF, Arrua LA, Marchi AJ, Abello MC (2008) Catal Commun 9:1201
Mariño F, Boveri M, Baronetti G, Laborde M (2001) Int J Hydrogen Energy 26:665
Mariño F, Baronetti G, Jobbagy M, Laborde M (2003) Appl Catal A 238:41
Ni M, Leung DYC, Leung MKH (2007) Int J Hydrogen Energy 32:3238
Vizcaíno AJ, Carrero A, Calles JA (2007) Int J Hydrogen Energy 32:1450
Cavallaro S, Freni S (1996) Int J Hydrogen Energy 21:465
Homsi D, Rached JA, Aouad S, Gennequin C, Dahdah E, Estephane J, Tidahy HL, Aboukais A, Aad EA (2017) Environ Sci Pollut Res 24:9907
Llorca J, Ramírez de la Piscina P, Sales J, Homs N (2001) Chem Commun 7:641
Llorca J, Homs N, Sales J, Ramírez de la Piscina P (2002) J Catal 209:306
Kunat M, Girol SG, Burghaus U, Woll C (2003) J Phys Chem B 107:14350
Sun J, Wang Y (2014) ACS Catal 4:1078
Sun JM, Zhu KK, Gao F, Wang CM, Liu J, Peden CHF, Wang Y (2011) J Am Chem Soc 133:11096
Arslan A, Gunduz S, Dogu T (2014) Int J Hydrogen Energy 39:18264
Skrzypek J, Słoczyński J, Ledakowicz S (1994) Methanol synthesis. Polish Scientific Publishers PWN, Warsaw
Wang LC, Liu Q, Chen M, Liu YM, Cao Y, He HY, Fan KN (2007) J Phys Chem C 111:16549
Tada S, Kayamori S, Honma T, Kamei H, Nariyuki A, Kon K, Toyao T, Shimizu K, Satokowa S (2018) ACS Catal 8:7809
Cabrera IM, Granados ML, Fierro JLG (2002) J Catal 210:273
Ribeiro NFP, Souza MMVM, Schmal M (2008) J Power Sources 179:329
Guo X, Mao D, Lu G, Wang S, Wu G (2011) Catal Commun 12:1095
Wan H, Wang Z, Zhu J, Li X, Liu B, Gao F, Dong L, Chen Y (2008) Appl Catal B 79:254
Guo X, Mao D, Lu G, Wang S, Wu G (2010) J Catal 271:178
Chen LC, Chen CC, Liang KC, Chang SH, Tseng ZL, Yeh SC, Chen CT, Wu WT, Wu CG (2016) Nanoscale Res Lett 11:402
Tran TH, Nguyen VT (2016) Mater Sci Semicond Process 46:6
Yamamoto T, Tanaka T, Takenaka S, Yoshida S, Onari T, Takahashi Y, Kosaka T, Hasegawa S, Kudo M (1999) J Phys Chem B 103:2385
Juma AO, Arbab EAA, Muiva M, Lepodise LM, Mola GT (2017) J Alloy Compd 723:866
Ambursa MM, Sudarsanam P, Voon LH, Hamid SBA, Bhargava SK (2017) Fuel Process Technol 162:87
Ambursa MM, Ali TH, Voon LH, Sudarsanam P, Bhargava SK, Hamid SBA (2016) Fuel 180:767
Thommes M, Kaneko K, Neimark AV, Olivier JP, Reinoso FR, Rouquerol J, Sing KSW (2015) Pure Appl Chem 87:1051
Zhong C, Guo X, Mao D, Wang S, Wu G, Lu G (2015) RSC Adv 5:52958
Smoláková L, Frolich K, Troppová I, Kutálek P, Kroft E, Čapek L (2017) J Therm Anal Calorim 127:1921
Smyrnioti M, Tampaxis C, Steriotis T, Ioannidesa T (2020) Catal Today 357:495
NIST X-ray Photoelectron Spectroscopy Database, Version 4.1 (National Institute of Standards and Technology, Gaithersburg, 2012); http://srdata.nist.gov/xps/
Moulder J, Stickle WF, Sobol PE, Bomben K (1992) Handbook of X-ray photoelectron spectroscopy. Perkin-Elmer Corporation, Minnesota
Galetti AE, Gómez M, Arrúa LA, Abello MC (2008) Appl Catal A Gen 348:94
Song H, Zhang L, Ozkan US (2012) Top Catal 55:1324
Dolgikh LY, Staraya LO, Stolyarchuk IL, Pyatnitsky YI (2020) Theoret Exp Chem 56:192
Acknowledgements
We acknowledge the financial support from National Science Centre, Poland (SONATA Grant: 2016/23/D/ST4/02492, 2017-2020).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Śliwa, M., Socha, R.P. Modification of CuO–ZrO2–ZnO Mixed Oxide Catalyst with Mn, Ga, Ni: Impact on Physicochemical Properties and Hydrogen Production via Low Temperature Steam Reforming of Ethanol. Catal Lett 152, 3747–3760 (2022). https://doi.org/10.1007/s10562-022-03947-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-022-03947-y