
Abstract
Tuning the reaction parameters to maximize products yield is one of the major needs for any process. The goal of this research is to increase the reduction of CO2 with water by examining the operating parameters of a meso-scale continuous-flow type photochemical reactor over hydrothermally synthesized photocatalysts such as Pt/TiO2 and Pt/TiO2/RGO. Effects of catalyst type, weight of catalyst utilized, photochemical reactor temperature, retention time by variating the liquid water flow rate, and cocatalyst loading were investigated to increase the concentration of total organic carbon compounds including HCHO and CH3OH. The effect of titanium dioxide phase ratio (anatase: rutile) presence at the Pt/TiO2/RGO photocatalysts was also studied. The results revealed that the 0.3 wt.% Pt/TiO2/RGO5% photocatalyst which includes a phase ratio of 81:19 for anatase: rutile respectively has the superior photocatalytic activity to other studied photocatalysts. The physciochemical properties of different prepared photocatalytic samples were determined using various characterization techniques. Analyzing the liquid products on gas chromatography, it was found that CH3OH represents the major product whereas HCHO was the minor one. This reactor exhibits a great performance towards CO2 photocatalytic reduction under the optimized conditions.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The global warming effect is the most serious environmental problem confronting humanity. One of the greenhouse gases (GHG) that contributes to climate change is carbon dioxide (CO2). The earth’s temperature increases as a result of infrared light absorption by CO2 and other GHG then re-emitting this radiation to the surface of earth leading to the global warming effect [1, 2].
However, most of the emitted CO2 occurs from the combustion of fossil fuel which facing a depletion risk. Thus, the photoconversion of CO2 which is a mimic of the plant photosynthesis was a reasonable solution to turn CO2 in the existence of water and solar energy into hydrocarbon fuels. This method considers a very promising method not only to eliminate CO2 emissions but also to produce useful hydrocarbons such as formic acid (HCOOH), formaldehyde (HCHO), methanol (CH3OH), methane (CH4) and ethanol (C2H5OH), etc. [2].
The photo-conversion of CO2 has many limitations which control the conversion efficiency and the yield of products. Some of those limitations are related to the semiconductor utilized and others are directly related to the design of the used reactor.
Titanium dioxide (TiO2) is a common semiconductor applied in the CO2 photoreduction due to its corrosion resistance, long-term stability, nontoxicity and low cost [3, 4]. However, it has many disadvantages not only the recombination of electron–hole pairs but also it needs UV irradiation to activate. Thus, many trials had been performed to overcome those limitations by noble metals loading and the usage of carbonaceous nanomaterials such as carbon nanotube, carbon nanowalls, and graphene [5]. Among the carbonaceous nanomaterials, graphene based photocatalysts have found to improve the visible light absorbance which consequently enhances the rate of CO2 reduction [3, 6].
Many previous studies examined the usage of noble metals such as (Au, Rh, Ag, Pt) and their loading amounts on TiO2 photocatalyst. They confirmed that the most effective one among those metals was Pt.
Xie et al. [7] investigated the effect of different noble metals loading on the TiO2 and found that the CH4 formation rate enhanced with the following order Pt > Pd > Au > Rh > Ag. Also, Li et al. [8] studied the effectiveness of noble metals loading on the photoreduction of CO2 and found that efficiency followed the sequence of Pt > Au > Ag.
In addition, many trials were conducted to enhance the yield of products from CO2 photoreduction especially using continuous mode reactors. The use of water vapor with CO2 gas was commonly utilized to form a single phase of reactants therefore increasing of products yield. Sim et al. [9] examined the use of water vapor and the ternary composite of reduced graphene oxide/Pt–TiO2 nanotube arrays in a gas- solid photoreactor to produce CH4 after the photocatalytic reduction of CO2. Moreover, Tan et al. [10] studied the photoreduction of CO2 using various noble metals anchored on reduced graphene oxide/TiO2, however; the most efficient one was Pt which has further been used to form CH4 via a continuous gas flow reactor.
Wang et al. [11] tested the usage of compressed CO2 with water vapor in a continuous flow film reactor then the effluent gas was recorded in relation to the irradiation time duration. The results revealed that the CH4 rates were dropped after 5 h of irradiation, and they attributed that to the decline of adsorption power of the used catalyst.
Kometani et al. [12] examined the photoreduction of CO2 with H2O over Pt loaded TiO2 in a flow type reactor as a function of temperature and pressure up to 400 °C and 30 MPa. They proposed using liquified CO2 to create such homogenous single-phase reactants, and they discovered that increasing the temperature from 300 °C to 400 °C at a constant pressure of 30 MPa enhanced the yields of CO, CH4, HCOOH, and HCHO.
The use of high temperature and pressure to form single-phase reactants is economically unfavorable. Moreover, there are few reports discussed the photoreduction of CO2 in continuous mode reactors especially with suspension photocatalyst.
In a prior work, our group introduced the utilization of an advanced meso-scale continuous-flow photochemical reactor to reduce CO2 with liquid water. [13]. Findings revealed that the use of this reactor resulted in an outstanding performance for CO2 photoreduction where its selectivity was focused toward methanol production and suppressing water reduction into H2. Therefore, this study aims to optimize the operating conditions of the previously mentioned photochemical reactor with a reduced graphene oxide supported on a low platinum content (0.3 wt.%) over the TiO2 semiconductor. Moreover, the effect of photocatalyst type, co-catalyst loading as well as (anatase: rutile) ratio were investigated in this work to improve the concentration of the total organic carbon (TOC) by comparing between the pre-synthesized binary and ternary composites (PTG1 and PTG2).
The performance of CO2 photoreduction has been studied in relation to the following parameters: (1) the catalyst type including the effect of (anatase: rutile) ratio, (2) the weight of catalyst utilized, (3) the photochemical reactor temperature, (4) the liquid water flow rate, and (5) the cocatalyst loading.
2 Experimental
2.1 Materials
TiO2 was bought from two different suppliers one from (ACROS) organics and the other one from Sigma-Aldrich. The two kinds of TiO2 are differ than each other in (anatase: rutile) ratio. In order to distinguish between the two kinds of TiO2, it will be referred to them as a T1 and a T2. (ACROS) organics provided the metal precursor of platinum salt (H2PtCl6.H2O), while Fisher Scientific provided graphite powder and sodium sulphate (Na2SO4). All other chemicals were analytical grade and commercially available, such as iodine (99.99%) and potassium permanganate (KMnO4).
Merck, Germany supplied the nitric acid (HNO3). Sigma-Aldrich provided the hydrochloric acid (HCl), hydrogen peroxide (H2O2), and sulphuric acid (H2SO4), while Fisher Scientific provided the acetone (C3H6O) and ethanol (C2H5OH). All solvents were of the (HPLC) grade.
2.2 Samples preparation
After graphite oxide synthesis by the modified Hummers method, ternary composites of 0.3 wt.% Pt/TiO2/RGO (PTG) with both forms of TiO2 were synthesized in a one-step procedure utilizing the hydrothermal method [14].
Briefly, strong acids such as nitric acid (HNO3) were used to convert graphite powder to graphite oxide, followed by the slow addition of sulphuric acid (H2SO4) in a vessel immersed in an ice bath under stirring and sonication, and then potassium permanganate (KMnO4) was progressively added. To avoid temperature increases during the exothermic reaction, the temperature was kept below 20 °C using an ice bath, and the mixture was then allowed for 24 h with continuous stirring to finalize the oxidation process. Hydrogen peroxide was added to quench the reaction until a yellow–brown cake of graphite oxide (GO) was produced, which was then washed with HCl and distilled water multiple times by decantation.
Exfoliating the GO with distilled water was the first step in creating the ternary composite. Then, under sonication and vigorous stirring, both TiO2 suspended in ethanol solution and metal precursor of platinum salt (H2PtCl6.H2O) were blended with the GO solution. Finally, the mixture was placed in a Teflon-lined autoclave and held at 200 °C for 12 h. The powder was then cleaned and dried in a drying oven at 80 °C for 24 h.
2.3 Characterization of the as-prepared photocatalysts
The properties of the nanocomposites synthesized to be further used as photocatalysts in carbon dioxide reduction were studied by different techniques such as Transmission Electron Microscopy (TEM), the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), Energy-Dispersive X-ray Spectroscopy (EDX), X-ray Diffraction Spectroscopy (XRD), Fourier Transform Infrared Spectroscopy (FTIR), Brunauer–Emmett–Teller (BET), UV–Vis diffuse reflectance spectroscopy (DRS), and photoelectrochemical (PEC) measurements.
The field-emission transmission electron microscope (FE-TEM) (JEOL JEM-2100F) was also used in order to identify the shape, distribution, size, and surface morphology of the photocatalysts.
The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was done on a (JEOL JEM-2100F) TEM/STEM with a spherical aberration corrector.
In order to study the elemental analysis and the weight percentage of the samples, EDX was performed using energy-dispersive X-ray spectroscopy (JEOL JEM-2100F).
The phase and crystalline composition of the synthesized photocatalysts were studied by XRD pattern. Before the analysis, the samples were firstly milled into very fine powder. A 2theta scan ranging from 5° to 80° at a scanning pitch of 0.02° was performed using an X-ray diffractor (XRD-6100 Shimadzu, Japan) coupled with a Cu-Kα radiation source with a wavelength of 1.55 Å.
The functional groups contained in each photocatalyst were determined and specified using Fourier Transform Infrared Spectroscopy (FTIR). The FTIR spectrometer concurrently captures high-spectral-resolution data over a wide spectral range and analyses it, based on the infrared spectrum of absorption or emission of the materials. The spectrometer used was Vertex 70 IR (Bruker Scientific Instruments, Germany).
To investigate the specific surface area and pore size distribution, the Brunauer–Emmett–Teller (BET) method was used. The N2 adsorption/desorption isotherms were investigated for the as-prepared photocatalysts. The photocatalysts’ Brunauer–Emmett–Teller (BET) specific surface areas were measured at 77 K using a Belsorp-mini II (BEL JAPAN) nitrogen adsorption equipment. Prior to surface area analysis, the powders were degassed at 300 °C for 3 h under vacuum.
UV–vis spectrometer (JASCO V-630) with an integrating sphere was used to measure diffuse reflectance spectra. The band gap was calculated using the Kubelka–Munk function and reflectance data.
In the presence of 0.1 M Na2SO4 electrolyte, photoelectrochemical (PEC) measurements were performed using a potentiostat (Gamry instrument, reference 3000) in one compartment cell with a Pyrex window. The platinum rod and saturated Ag/AgCl were employed as counter electrode (C.E) and reference electrode (R.E), respectively, in a three-electrode setup while the working electrode (W.E.) was made from various photocatalysts powder using electrophoretic deposition method (EPD) on the fluorine-doped tin oxide (FTO) glass surface. The light irradiation source was a UV LED lamp from (Suruga Seiki-Co, Japan) with a wavelength of 365 nm and an intensity of up to 50 mW/cm2. After deposition of numerous catalysts on the surface of FTO glass, the weight and coated area of each working electrode were approximately 4 mg and 6 cm2, respectively.
2.4 Photocatalytic reduction of CO2 with H2O
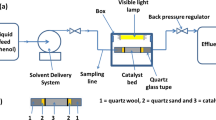
The photoreduction of CO2 with water was conducted in a continuous-flow photochemical reactor system (Vapourtec UV-150) under inner UV light irradiation, as indicated in Fig. 1. The used reactor was constructed from channels with 1.3 mm inner diameter are made of thin-walled Fluorinated Ethylene Propylene (FEP) tubing which allow the penetration of light and its volume is equal to 10 ml. In order to save energy, the light source was a UV (LED) lamp with λ = 365 nm. The used reactor provides many advantages as the uniform inner irradiation, high surface area to volume ratio which improves photon and mass transfer also it could be operated with heating or cooling modes.

The experimental setup scheme
Different types and amounts of catalyst powders were dispersed first in 65 ml deionized water by using ultrasonication. Prior to illumination and to ensure complete saturation, pure CO2 was bubbled at an average flow rate of 35 ml min−1 for 30 min through the dispersed catalyst under a controlled chilled water bath to enhance the solubility of CO2 in the mixture. After the flow was stabilized, the photochemical reactor started on and the pump feeds the suspension into the photochemical reactor with the desired flow rate, the UV LED lamp was turned on to commence the experiment.
Helium (He) was utilized as a carrier gas with a constant flow rate of 20 ml min−1 to separate the produced gases into a gas bag purchased from (GL Sciences Inc., Japan) for the offline analysis. Then, it was analyzed by a GC (Shimadzu 2014, Japan) coupled with ShinCarbon ST micropacked column (Restek, length 2 m × ID 2 mm, U.S.A) and a thermal conductivity detector (TCD). After the analysis of the gas bag, neither CO nor CH4 were detected. Also, the GC did not identify hydrogen gas (H2), indicating that its amount was too little to be identified or that it was probably reacted.
In addition to that the cold trap was used to ensure that all organic vapors produced were condensed in the liquid phase. After filtering and extraction, the liquid products were evaluated qualitatively using GC/flame ionization detector (FID) (Shimadzu 2014 with Rt-Qplot capillary column [length 30 m, ID 0.32 mm]) and GC/mass spectroscopy (MS) (Thermo-Scientific with TG-5MS capillary column [length 30 m, ID 0.25 mm]). Second, the total organic carbon analyzer (Shimadzu, TOC-L series, Japan) was used to quantitatively analyze the obtained liquid samples at the end of each run [15,16,17], and the Nash approach [12, 18, 19] was employed to determine the formaldehyde quantity at an absorbance wavelength of 414 nm. There were no TOC or formaldehyde identified in the blank samples.
3 Results and Discussion
3.1 Photocatalysts Characterization
Figure 2a–d and f–h present the morphology of the synthesized photocatalysts with both TEM and HAADF-STEM images.

a,e,i are TEM, HRTEM, EDX for T1 while typical TEM, HAADF-STEM, EDX characterizations of b,f,j for PT1, c,g,k for PTG1, and d,h,l for PTG2, respectively
It is well noticed that the shape of T1 is typical to the spherical nanoparticles with a particle size varying from 14.04 nm to 26.78 nm as exhibited in Fig. 2a. Figure 2e is the HRTEM image for the T1 nanoparticles which illustrates the lattice fringes with a spacing of 0.35 nm at scale 2 nm which is corresponding to (101) anatase TiO2.
Figure 2b shows the well disperse of Pt NPs on the surface of TiO2 nanoparticles and the particle size of Pt NPs were varying from 4.35 nm to 6.13 nm. In addition, Fig. 2c, d illustrates the uniform and homogenous distribution of both Pt NPs and RGO sheets on the surface of T1 and T2, respectively. The HAADF-STEM images confirm the results obtained by TEM.
Furthermore, to study the elemental analysis and weight percentage of each nanocomposite, an energy-dispersive X-ray spectroscope (JEOL JEM-2100F) was used to perform EDX. The analysis proved the existence of titanium, oxygen, carbon, and platinum in the photocatalysts as demonstrated in Fig. 2i–l. The Ti and O elements were derived from TiO2 nanoparticles while C element was from the presence of RGO sheets and Pt peaks confirmed the existence of Pt element in the nanocomposites. The actual weight percentage of the doped Pt in each nanocomposite is shown in Table 1.
XRD patterns were used to investigate the phase and crystalline composition of all produced nanocomposites, and the findings are exhibited in Fig. 3. For both the tetragonal anatase and tetragonal rutile phases of TiO2, all peaks were compared to the Joint Committee on Powder Diffraction Standards (JCPDS) Card File no. 00-002-0387 and Card File no. 00-001-1292, respectively. The anatase phase of TiO2 diffraction peaks exist at 2θ = 25.18°, 37.72°, 38.42°, 47.95°, 53.90°, 54.26°, 62.60°, 68.73°, 70.14°, 74.99° matches with (101), (103), (004), (200), (105), (211), (204), (116), (220), (215) crystal facets of anatase phase, respectively. Whereas the diffraction peaks of rutile TiO2 appears at 2θ = 27.32°, 36.82°, 38.82°, 41.18°, 55.90° corresponding to (110), (101), (200), (111), (211) crystal facets of rutile phase, respectively. Pt NPs were insignificant to be detected using XRD detection limit so that diffraction peaks of TiO2 did not change after Pt NPs were loaded on its surface.

XRD pattern for a T1, bT2, c 0.3 wt% PT1, d 0.3 wt% PTG1 and e 0.3 wt% PTG2
Scherrer’s equation was used to measure the average crystallite size of samples based on the maximum three peaks of the anatase and rutile phases.
where L represents the crystallite size (nm), K expresses a constant which equals to 0.9 in this case and it depends on the crystallite shape, whereas the X-ray wavelength (nm) is referred to by λ, β means the full width at half maximum (FWHM) and θ demonstrates the Bragg’s angle.
The relative peak intensity of anatase and rutile was used to calculate the phase composition of the samples [20, 21].
where the fraction of anatase phase is referred to by FA whereas IR and IA are the intensity of the rutile (110) and anatase (101) diffraction peaks, respectively. Table 1 summarizes the calculated crystallite size and phase composition for both anatase and rutile phases of all photocatalysts.
It is well obvious from Table 1 that the crystallite size i.e. the degree of crystallinity for anatase phase increased with the addition of Pt NPs and RGO to TiO2 samples. The same result was mentioned by Ola et al. [22] for the noble metal loading. Meanwhile, for 0.3 wt.% PTG2, the degree of crystallinity for rutile phase decreased comparing with T2.
Also, the percentage of the rutile phase in 0.3 wt.% PTG1 composite was slightly increased after the introduction of RGO and Pt NPs compared with T1. The same observation of the anatase transformation to rutile was previously reported during other metals loading and the introduction of RGO on TiO2 surface especially with the temperature elevation such as in this case with the hydrothermal reaction [23,24,25,26]. While in the case of 0.3 wt.% PTG2 composite the (anatase: rutile) ratio was maintained at the same value as T2.
This may be related to the microstructure of TiO2 which has three types such as the individual structure of anatase and rutile phases, heterojunction structure involving rutile clusters overlayered on anatase nanoparticles and the inter-particles structure [23]. It was mentioned that the (anatase/rutile) ratio would decrease during the hydrothermal reaction if they existed individually [23].
To further test the chemical bonds and functional groups of catalysts, FTIR for catalysts were preformed and the results are displayed in Fig. 4.

FTIR for a T1, b 0.3 wt.% PT1, c 0.3 wt.% PTG1, d 0.3 wt.% PTG2, and e GO
It is well seen that there are eight distinguished absorption bands in the FTIR spectrum region from 450 cm−1 to 4000 cm−1. The Ti–O–Ti and Ti–O–C stretching vibrations are laid in the absorption bands below 1000 cm−1. The existence of absorption band at 1075 cm−1 is due to the stretch vibrations for C-O group which attributed to epoxy or alkoxy groups whereas the peak at 1250 cm−1 was assigned to epoxide C–O–C or phenolic C-O–H stretching vibrations. The peaks at 1395 cm−1, 1730 cm−1 and 3400 cm−1 belong to carboxyl group O = C–O, carbonyl C = O and hydroxyl group OH, respectively. Finally, the absorption band at 1630 cm−1, in case of TiO2 and Pt/TIO2 catalysts, it may refer to the associated water adsorbed on the particle surfaces while in case of Pt/TiO2 /RGO, it is well obvious that the intensity of the band increased which may be attributed to C = C stretching vibration which represent the skeletal vibrations of unoxidized graphitic domains located at the same absorption band.
The existence of Ti–O–C bond confirms that through the synthesis of Pt/TiO2/RGO using hydrothermal reduction method, GO with the residual O = C–O groups reacted with the surface hydroxyl groups O–H of TiO2 NPs resulting in forming a chemical bonded TiO2/RGO, similar results are reported elsewhere [4, 27].
The effect of RGO and Pt NPs addition on the composites’ specific surface area, BET surface area and Barrett-Joyner-Halenda (BJH) porosity of T1, T2, 0.3 wt. % PT1, 0.3 wt. % PTG1 and 0.3 wt. % PTG2 had been performed. Results are illustrated as demonstrated in Fig. 5.

N2 adsorption/desorption isotherms at 77 K (a) and pore size distribution curve (b) for all synthesized photocatalysts
Table 2 illustrates both the Brunauer–Emmett–Teller (BET) specific surface area and pore volume for all photocatalysts.
It is well noticed from Table 2 that the specific surface area rises with increasing both of metal loaded nanoparticles and RGO sheets that was in agreement with previous studies [22].
As shown in Fig. 5-a, the initial portion of adsorption isotherms at low relative pressure (P/Po) represents the formation of monolayer adsorption on the photocatalyst surfaces then the increment of adsorption volume with the relative pressure increase exhibits the formation of multilayer adsorption leading to spontaneously condensation of gas into liquid-like state inside the photocatalyst pores which is called capillary condensation phenomenon. Therefore, it is well obvious that all the photocatalysts exhibit IV adsorption–desorption isotherm type representative of mesoporous materials agreeing with IUPAC classification with H1 hysteresis loops except the 0.3 wt.% PTG2 photocatalyst shows a H3 hysteresis loop.
The pore diameter of T1, T2, 0.3 PT1, 0.3 wt.% PTG1 and 0.3 wt.% PTG2 were 46.184 nm, 35.54 nm, 28.369 nm, 26.64 nm and 14.56 nm respectively.
The optical features of photocatalysts were examined using the UV–Vis (diffuse reflectance spectra) as demonstrated in Fig. 6. From (DRS) figure (a), it is clear that adding of noble metal (Pt NPs) slightly improves the visible light absorbance. Furthermore, introducing of RGO sheets further boosts the red shift of the absorption edge. This may be owing to the existence of Ti–O–C bond between TiO2 and RGO [14]. Similar observations were reported in literature [4, 10, 14, 28]. The band gap calculations were performed from the plots of Kubelka–Munk function versus the photon energy as exhibited in Fig. 6b and Table 2. It was noticed that the band gap of T1 is smaller than T2 catalyst. As mentioned in Table 2, the band gaps were 3.00 eV and 3.11 eV for T1 and T2, respectively. Even after the introduction of Pt NPs and RGO sheets, the band gap was 2.69 eV for 0.3 wt.% PTG1 and 2.72 eV for 0.3 wt.% PTG2. As reported in the literature, the band gap of pure anatase is equal (3.23 eV) and for pure rutile is (3.02 eV). It is well noted that (anatase: rutile) ratio influences the position of band gaps of metal-TiO2 samples. In this case, the 0.3 wt.% PTG1 has a ratio of (anatase: rutile) equal to (75:25) and its band gap is 2.69 eV while 0.3 wt.% PTG2 has (81:19) ratio and a band gap of 2.72 eV. The same observation was reported by Dubey et al. [28].

UV–Vis spectra (a) and transformed Kubelka–Munk function versus the photonic energy (b) for all samples
3.2 Photoelectrochemical Measurements (PEC)
The optoelectronic performance of the synthesized photocatalysts was investigated using the photoelectrochemical measurements. The photocurrent-potential response (linear sweep voltammetry, LSV), Transient photocurrent response (chronoamperometry CAM), and (electrochemical impedance spectroscopy EIS) were conducted in a three-electrode cell filled with 0.1 M aqueous Na2SO4 solution after 30 min purging with Ar gas. After deposition of catalysts using the (EPD) technique, the working electrode (W.E) was utilized in the PEC measurements even with no further treatment such as necking or heating.
The LSV was carried out in the presence of chopped illumination within range of (−1.5 V to 1 V) and the results are displayed in Fig. 7. The instantaneous photocurrent response via several light on cycles and the subsequent discharge due to the light off cycles confirms the stability of electrodes and the effective separation of electron–hole pairs of catalysts [14, 29, 30].

Photocurrent-potential response (LSV) for the prepared photocatalysts under numerous on/off cycles
Figure 7 shows that all composites exhibit an anodic photocurrent response which proves the n-type semiconductor characteristics. Also, adding of Pt NPs and RGO sheets increase the photocurrent thus improve the photoactivity.
Meanwhile, it is well clear that the 0.3 wt.% PTG2 composite exhibits an increase in the photocurrent response approximately 2 times higher than 0.3 wt.% PTG1. Moreover, the anodic photocurrent transient profile of 0.3 wt.% PTG2 composite shows a triangular pattern (after turning the light off) that differs from the rectangular pattern of the other composites. The same triangular pattern in the charge–discharge cycles was observed for the ideal capacitors [14] which reveals that the 0.3 wt.% PTG2 composite has a superior activity to other photocatalysts. This could be ascribed to the better reproducibility of electrons and effective separation of the photogenerated charges and holes.
To evaluate the transient photocurrent responses of the photocatalysts, the chronoamperometry measurement was performed at (+ 0.6 vs. Ag/AgCl) for 800 s divided into three sections. The first section included 60 s in dark then the second section involved lighting on for 600 s and finally shutting off the light until the measurement was completed as displayed in Fig. 8.

Chronoamperometry (CAM) of the prepared photocatalysts at + 0.6 vs. Ag/AgCl in 0.1 M Na2SO4 electrolyte under UV LED light irradiation (365 nm)
As shown from Fig. 8, all photocatalysts samples exhibited an efficient response towards the transient photocurrent while the superior photocurrent density was recorded by the ternary composites of PTG1 and PTG2 that confirm results observed in the LSV. Also, similar results were reported for the binary and ternary composites with [29] and [9].This may be assigned to the efficient electron–hole separation due to electrons trapping by Pt NPs and the decrease of the recombination in the ternary nanocomposites.
Moreover, the ternary composite of PTG2 had an efficient photocurrent density comparing to PTG1 which confirms the proficiency of the T2 over T1 photocatalyst. This result could be ascribed to the increase of BET surface area of the T2 over T1.
The (EIS) is a convenient way to examine the electrons transfer and the recombination process at the interface of semiconductor/electrolyte [31]. The values of (EIS) of all catalysts in both the dark and light illumination conditions (λ = 365 nm) are exhibited in Nyquist plots and shown in Fig. 8. The analyses were conducted in 0.1 M Na2SO4 solution at frequencies varying from 100 kHz to 0.01 Hz and at 0.0 V (vs. Ag/AgCl).
As previously explained in literatures [14, 31, 32], the intersection between the first point of the arc which located at the region of high frequency and the real impedance (Zreal) represents the bulk resistance between (W.E) and (R.E) or the equivalent series resistance (ESR). While the intermediate section of frequency where the semicircular arc is located, the intercept with (Zreal) shows the charge transfer resistance (RCT). Thus, a smaller impedance arc radius indicates a smaller (RCT) across the electrode/electrolyte interface [31]. Therefore, a higher electronic conductivity, the maximum photocatalytic efficiency and a minimal electron–hole recombination could be achieved [9, 30, 33].
As well seen from Fig. 8, the 0.3 wt.% PTG2 composite has the superior photocatalytic efficiency to the other catalysts. This illustrates that 0.3 wt.% PTG2 composite has the capability of the fast electrons transfer consequently decreasing the recombination process (Fig. 9).

Nyquist plots of EIS for T1, T2, 0.3 wt.% PT1, 0.3 wt.% PTG1, 0.3 wt.% PTG2 in 0.1 M Na2SO4 solution, in dark (a), and light illumination (λ = 365 nm) (b) at 0.0 V (vs. Ag/AgCl)
3.3 Parameters Affecting the Photoreduction of CO2
The CO2 photoreduction is highly influenced by numerous parameters such as (i) type of photocatalyst used, (ii) weight of photocatalyst utilized, (iii) photochemical reactor temperature, (iv) retention time inside the photochemical reactor, and (v) content of cocatalyst loading. Here in this study, all these factors were examined using the previously mentioned flow type photochemical reactor as in Fig. 1.
3.4 Effect of the Catalyst Type
In order to investigate the effect of catalyst type, first we will examine the effect of the presence of both RGO and Pt as a cocatalyst then we will investigate the effect of (anatase: rutile) ratio. Figure 10 shows the variation in both TOC and formaldehyde concentrations with different catalysts.

Effect of the catalyst type on a TOC concentration b Formaldehyde concentration and formaldehyde% in TOC, (Photocatalyst weight = 30 mg, deionized water volume = 65 ml, liquid pump flow rate = 500 µl/min and reactor temperature = 30 °C)
It is well notice that the introduction of Pt NPs and RGO sheets increases the concentration of TOC as well as formaldehyde which enhances the photoreduction of CO2. As shown with 0.3 wt.% PTG1 composite, the Pt loading as a cocatalyst improves the photoactivity owing to Schottky barrier formation between TiO2 and Pt NPs which acts as electrons trap and hinders the recombination [34] thus increases the photoreduction of CO2. Also, the introduction of RGO sheets contributes to the simple movement of photogenerated electrons from TiO2 to RGO as well as the perfect electronic conductivity of RGO because of the 2D and planar π-conjugation structure [5, 14].
Meanwhile, the percentage of formaldehyde in TOC was still too low, and the maximum percentage was (2.7) of TOC. This confirms the results obtained by GC/MS and GC/ FID which illustrates that methanol was the main product as shown in Fig. 11

Analysis for one of the samples confirms the presence of methanol a The chromatographic peak corresponding to methanol on GC/MS, b MS spectrum on GC/MS, and c The chromatographic peak corresponding to methanol on GC/FID
From Fig. 10, it is well obvious that 0.3 wt.% PTG2 enhances the concentration of both TOC and formaldehyde more than those obtained by 0.3 wt.% PTG1 while the formaldehyde percentage in TOC was still too low which may be attributed to the formation of other organic compounds i.e. methanol. The superior photoactivity of 0.3 wt.% PTG2 photocatalyst was previously noted in the PEC measurements section.
This superior activity of 0.3 wt.% PTG2 might be explained according to the results obtained from the previous sections of XRD and BET surface area which demonstrated that (anatase: rutile) ratio and the surface area were (81: 19), 78.14 (m2/g) while they were (75:25), 53.38 (m2/g) for 0.3 wt.% PTG2 and 0.3 wt.% PTG1, respectively.
It seems that (anatase: rutile) ratio shows an essential role in photocatalytic activity. It was previously reported that rich anatase/rutile content had a superior photoactivity for CO2 photocatalytic reduction [35] which is in agreement with our case.
Also, it was confirmed that the anatase phase is highly activity compared with the rutile phase in many reactions [36]. It was also found that a small amount of rutile added to the anatase phase may hinder the recombination of photogenerated holes and electrons, therefore, enhances the photocatalytic activity [37].
Furthermore, it was reported that the surface area of TiO2 has an effective role in the CO2 photoreduction as the increase of TiO2 BET surface area specially the anatase (101) facet could improve the adsorption capacity of CO2 on TiO2 surface [38, 39].
We could conclude that the rich of (anatase: rutile) ratio and the increase of BET surface area are the major reasons for the enhancement of CO2 reduction in the case of 0.3 wt.% PTG2. This may be attributed to the lower recombination of the photogenerated electrons and holes as well as the high adsorption capability of the surface of anatase phase [35, 38].
3.4.1 Effect of the Weight of Catalyst Utilized
Figure 12 illustrates the effect of catalyst weight on TOC and formaldehyde concentrations.

Effect of the catalyst weight on TOC and formaldehyde concentration, (0.3 wt.% PTG2, deionized water volume = 65 ml, liquid pump flow rate = 500 µl/min and reactor temperature = 30 °C)
As shown from the previous figure, the increase of the catalyst weight increases the TOC and formaldehyde concentrations up to 30 mg then any additional increase of the catalyst weight decreases both TOC and formaldehyde concentrations. This could be explained due to the formation of an adhesive layer from the catalyst on the inner surface of the photochemical reactor channels (ID = 1.3 mm) which limits the penetration of light and consequently the light absorption especially with the use of low liquid flow rate, therefore, decreases the photocatalytic reduction of CO2.
3.4.2 Effect of the Photochemical Reactor Temperature
Figure 13 depicts the effect of reactor temperature on the concentrations of TOC and formaldehyde. As the temperature of the reaction rises by 10 °C, from 30 °C to 40 °C, the TOC concentration remains constant. This result is in consistent with Fox et al. [40] and Kočí et al. [41], who confirmed that the photocatalytic reactions are not affected by the small variations in temperature.

Effect of the reactor temperature and on TOC and formaldehyde concentration, (0.3 wt.% PTG2, photocatalyst weight = 30 mg, deionized water volume = 65 ml and liquid pump flow rate = 500 µl/min
While increasing the temperature to higher values may greatly influence on the photocatalytic reaction of CO2. To explain the present results, it should be mentioned that the elevation of temperature in any gas–liquid–solid system leads to two consequences: on one hand, the rate of the chemical reaction and the kinetic rate constants are usually increased exponentially with temperature. On the other hand, the elevation of temperature determines the equilibrium amount of dissolved CO2 [41]. It is well obvious that the predominating effect at 50 °C is the adverse effect so that the TOC concentration decreased.
Whereas the formaldehyde concentration is generally decreasing with the elevation of temperature which indicates the formation of other organics.
3.4.3 Effect of Liquid Flow Rate
We choose 0.3 wt.% PTG2 as a catalyst, 30 mg for the weight of catalyst and 30 °C for the photochemical reactor temperature now we will examine the effect of retention time by changing the liquid pump flow rate. The retention time inside the flow type photochemical reactor could be calculated by this equation:
Figure 14 shows the effect of liquid flow rate and consequently the retention time in the photochemical reactor on TOC and formaldehyde concentrations.

Effect of liquid flow rates on TOC and formaldehyde concentrations, (0.3 wt.% PTG2, photocatalyst weight = 30 mg, deionized water volume = 65 ml and photochemical reactor temperature = 30 °C)
As noted in Fig. 14, the maximum concentrations for both TOC and formaldehyde were obtained at 500 µl/min (retention time = 20 min.).
As known, increasing the retention time by decreasing the liquid flow rate may be beneficial for the photoreduction of CO2. Meanwhile, in this case lowering the liquid flow rate to 300 µl/min decreases the TOC as well as formaldehyde concentrations. This may be attributed to the insufficient CO2 and water (OH−) supplied to the photochemical reactor so that decreasing TOC and formaldehyde. Moreover, increasing time inside the reactor may cause oxidation for the generated organics lowering the TOC and formaldehyde concentrations as well. The same result was reported by Cheng et al. [42] and Kalamaras et al. [43].
3.4.4 Effect of Cocatalyst Loading
Herein, we will investigate the effect of loading amounts of Pt as a cocatalyst by using T1 as a semiconductor. The operating conditions in this case would be 30 °C for the photochemical reactor temperature, 500 µl/min for the liquid flow rate and 30 mg form each catalyst. Figure 15 shows the effect of Pt loading as a cocatalyst.

Effect of cocatalyst loading on TOC and formaldehyde concentrations, (Photocatalyst weight = 30 mg, deionized water volume = 65 ml and photochemical reactor temperature = 30 °C and liquid pump flow rate = 500 µl/min)
As noted from Fig. 15 the excess of Pt NPs loading on TG1 composite surface negatively affects CO2 photocatalytic reduction. This finding is agreement with the previously reported studies [7, 8, 44]. These results may be due to the increase of the recombination of the photo-excited charges which hinder the reduction of CO2 as the too large amount of Pt NPs may lead to a shorter distance between the photogenerated electrons and holes [7]. It was also reported that a certain amount of noble metal loaded on TiO2 photocatalyst could enhance the photoreduction of CO2. However, the excess of loading covers the TiO2 surface thus inhibit the excitation of electrons therefore declines the photoreduction of CO2 [8].
The optimum amount of Pt loading which enhances the concentration of TOC and formaldehyde in this case was 0.5 wt.% PTG1 as depicted in Fig. 15.
4 Photocatalytic CO2 Reduction Mechanism
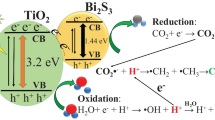
The mechanism of CO2 photoreduction firstly starts with the absorption of CO2 gas in liquid H2O. Secondly, the adsorption of CO2 and H2O molecules takes place on TiO2 surface. As the light strikes on the TiO2 surface with sufficient photonic energy (hν > band gap energy of the semiconductor), electrons jump from the valence band (VB) [O 2p orbital] to the conduction band (CB) [Ti 3d orbital] [5, 10], as displayed schematically in Fig. 16 and illustrated in Eq. (4):

Proposed CO2 photoreduction mechanism over Pt/TiO2 and Pt/TiO2/RGO nanocomposites
Holes in (VB) react with the adsorbed H2O molecules on the surface of TiO2 to form both hydroxyl radicals (\({\text{OH}}^{ \cdot }\)) and hydrogen ions (H+) then further oxidation occurs by \({\text{OH}}^{ \cdot }\) radicals to emit O2 and H+.
As shown in Fig. 16, in case of Pt/TiO2 composite, the presence of Pt NPs on the surface of TiO2 leads to Schottky barrier formation at the interface which aids the separation of photoexcited electrons and improves the trapping of electrons resulted in preventing the recombination of charges. In case of Pt/TiO2/RGO composite, the photogenerated charges from the (CB) of TiO2 could transfer to RGO sheets across the two-dimensional carbon network then trapped by Pt NPs. The RGO sheets offer a large surface area and enhance the conductivity of electrons. Moreover, RGO sheets acted as a support to provide a high Pt NPs dispersion which inhibits the aggregation of Pt NPs thus enhance the CO2 photocatalytic reduction [10].
The possible reactions of the excited electrons with the adsorbed CO2 to produce liquid organic carbons such as formic acid, formaldehyde, and methanol are described by Eqs. (7, 8, 9) [19]:
5 Conclusion
In this study, graphene oxide (GO) was prepared using modified Hummers’ method. A hydrothermal method was employed to synthesize binary (PT) and ternary composites (PTG1 and PTG2). The use of graphene in PTG1 photocatalyst enhanced the TOC by 3.5 and 11.4 times compared with PT1 and T1, respectively using the proposed photochemical reactor in a continuous mode. Furthermore, the effect of (anatase: rutile) ratio was examined by comparing the two kinds of ternary composites. Results revealed that compared to other photocatalysts, the 0.3 wt.% PTG2 photocatalyst, which possessed the (anatase: rutile) ratio of (81:19) and a surface area of 78.14 (m2/g) gave the supreme photocatalytic activity approximately 2 times higher than PTG1, in both the photocurrent generation via PEC measurements and CO2 photoreduction.
Additionally, to achieve higher TOC concentrations, the operating parameters for the advanced meso-scale continuous flow photochemical reactor were investigated. The optimum conditions were found as 500 µl/min for the water flow rate, 30 °C and 40 °C for the reactor temperature, and 30 mg for the weight of catalyst utilized. The maximum amount of Pt loading to enhance CO2 photocatalytic reduction was 0.5 wt.% Pt over TG1.
Finally, we could conclude that the rich of (anatase: rutile) ratio, the increase of BET surface area along with operating the photochemical reactor under the optimum conditions are the key reasons for the enhancement of CO2 reduction and the production of valuable organic compounds.
Data Availability
All data generated or analyzed during this study are included in this published article.
References
Zhuo Xiong CZ, Wang Haibing, Nuoyan Xu, Li Hailong, Fang Baizeng, Zhao Yongchun, Zhang Junying (2015) Photocatalytic reduction of CO2 on Pt2þePt0/TiO2 nanoparticles under UV/Vis light irradiation: a combination of Pt2þ doping and Pt nanoparticles deposition. Int J Hydrogen Energy 40:10049–10062. https://doi.org/10.1016/j.ijhydene.2015.06.075
Wang B et al (2018) Recent progress in the photocatalytic reduction of aqueous carbon dioxide. Catal Today 311(October):23–39. https://doi.org/10.1016/j.cattod.2017.10.006
Li K, An X, Park KH, Khraisheh M, Tang J (2014) A critical review of CO2 photoconversion: catalysts and reactors. Catal Today 224:3–12. https://doi.org/10.1016/j.cattod.2013.12.006
Zhao Y et al (2018) (2017) Graphene-wrapped Pt/TiO2 photocatalysts with enhanced photogenerated charges separation and reactant adsorption for high selective photoreduction of CO2 to CH4. Appl. Catal. B Environ. 226:360–372. https://doi.org/10.1016/j.apcatb.2017.12.071
Dargahi Z, Asgharzadeh H, Maleki-Ghaleh H (2018) Synthesis of Mo-doped TiO2/reduced graphene oxide nanocomposite for photoelectrocatalytic applications. Ceram Int 44(11):13015–13023. https://doi.org/10.1016/j.ceramint.2018.04.120
Ambrožová N, Reli M, Šihor M, Kuśtrowski P, Wu JCS, Kočí K (2018) Copper and platinum doped titania for photocatalytic reduction of carbon dioxide. Appl Surf Sci 430:475–487. https://doi.org/10.1016/j.apsusc.2017.06.307
Xie S, Wang Y, Zhang Q, Deng W, Wang Y (2014) MgO- and Pt-promoted TiO2 as an efficient photocatalyst for the preferential reduction of carbon dioxide in the presence of water. ACS Catal 4(10):3644–3653. https://doi.org/10.1021/cs500648p
Li X, Pan H, Li W, Zhuang Z (2012) Photocatalytic reduction of CO 2 to methane over HNb 3 O 8 nanobelts. Applied Catal A, Gen 413–414(301):103–108. https://doi.org/10.1016/j.apcata.2011.10.044
Sim LC, Leong KH, Saravanan P, Ibrahim S (2015) Rapid thermal reduced graphene oxide/Pt-TiO 2 nanotube arrays for enhanced visible-light-driven photocatalytic reduction of CO 2. Appl Surf Sci 358:122–129. https://doi.org/10.1016/j.apsusc.2015.08.065
Tan LL, Ong WJ, Chai SP, Mohamed AR (2015) Noble metal modified reduced graphene oxide/TiO2 ternary nanostructures for efficient visible-light-driven photoreduction of carbon dioxide into methane. Appl Catal B Environ 166–167:251–259. https://doi.org/10.1016/j.apcatb.2014.11.035
Wang WN et al (2012) Size and structure matter: Enhanced CO 2 photoreduction efficiency by size-resolved ultrafine Pt nanoparticles on TiO 2 single crystals. J Am Chem Soc 134(27):11276–11281. https://doi.org/10.1021/ja304075b
Kometani N, Hirata S, Chikada M (2017) Photocatalytic reduction of CO2 by Pt-loaded TiO2 in the mixture of sub- and supercritical water and CO2. J Supercrit Fluids 120:443–447. https://doi.org/10.1016/j.supflu.2016.05.031
Nabil S, Hammad AS, El-Bery HM, Shalaby EA, El-Shazly AH (2021) The CO2 photoconversion over reduced graphene oxide based on Ag/TiO2 photocatalyst in an advanced meso-scale continuous-flow photochemical reactor. Environ Sci Pollut Res. https://doi.org/10.1007/s11356-021-13090-7
El-Bery HM, Matsushita Y, Abdel-moneim A (2017) Fabrication of efficient TiO 2 -RGO heterojunction composites for hydrogen generation via water-splitting: comparison between RGO, Au and Pt reduction sites. Appl Surf Sci 423:185–196. https://doi.org/10.1016/j.apsusc.2017.06.130
Yang X, Xiao T, Edwards PP (2011) The use of products from CO2 photoreduction for improvement of hydrogen evolution in water splitting. Int J Hydrogen Energy 36(11):6546–6552. https://doi.org/10.1016/j.ijhydene.2011.02.116
Chen W-Y, Shi G, Hailey AK, Tsai EST, Hammer N, Wu Z (2012) Photocatalytic conversion of carbon dioxide to organic compounds using a green photocatalyst: an undergraduate research experiment. Chem Educ 17(4):166–172. https://doi.org/10.1007/s00897122438
Dong WH et al (2017) Coupling of photodegradation of RhB with photoreduction of CO2 over rGO/SrTi0.95Fe0.05O3−Δ catalyst: a strategy for one-pot conversion of organic pollutants to methanol and ethanol. J Catal 349:218–225. https://doi.org/10.1016/j.jcat.2017.02.004
Nash T (1953) The colorimetric estimation of formaldehyde by means of the Hantzsch reaction. Biochem J 55(3):416–421. https://doi.org/10.1042/bj0550416
Hong J, Zhang W, Ren J, Xu R (2013) Photocatalytic reduction of CO2: a brief review on product analysis and systematic methods. Anal Methods 5(5):1086–1097. https://doi.org/10.1039/c2ay26270c
Spurr RA, Myers H (1957) Quantitative analysis of anatase-rutile mixtures with an x-ray diffractometer. Anal Chem 29(5):760–762. https://doi.org/10.1021/ac60125a006
Rajalakshmi K, Jeyalakshmi V, Krishnamurthy KR, Viswanathan B (2012) Photocatalytic reduction of carbon dioxide by water on titania: role of photophysical and structural properties. Indian J. Chem. Sect A Inorg Phys. Theor. Anal. Chem. 51:411–419
Ola O, Maroto-Valer M, Liu D, MacKintosh S, Lee CW, Wu JCS (2012) Performance comparison of CO2 conversion in slurry and monolith photoreactors using Pd and Rh-TiO 2 catalyst under ultraviolet irradiation. Appl Catal B Environ 126:172–179. https://doi.org/10.1016/j.apcatb.2012.07.024
Jiang X et al (2018) Anatase and rutile in evonik aeroxide P25: Heterojunctioned or individual nanoparticles? Catal. Today 300(December):12–17. https://doi.org/10.1016/j.cattod.2017.06.010
Tian B, Li C, Zhang J (2012) One-step preparation, characterization and visible-light photocatalytic activity of Cr-doped TiO 2 with anatase and rutile bicrystalline phases. Chem Eng J 191:402–409. https://doi.org/10.1016/j.cej.2012.03.038
Bouras P, Stathatos E, Lianos P (2007) Pure versus metal-ion-doped nanocrystalline titania for photocatalysis. Appl Catal B Environ 73(1–2):51–59. https://doi.org/10.1016/j.apcatb.2006.06.007
Liu L, Zhang Y, Zhang B, Feng Y (2017) A detailed investigation on the performance of dye-sensitized solar cells based on reduced graphene oxide-doped TiO2 photoanode. J Mater Sci 52(13):8070–8083. https://doi.org/10.1007/s10853-017-1014-9
Vasilaki E, Georgaki I, Vernardou D, Vamvakaki M, Katsarakis N (2015) Ag-loaded TiO 2 /reduced graphene oxide nanocomposites for enhanced visible-light photocatalytic activity. Appl Surf Sci 353:865–872. https://doi.org/10.1016/j.apsusc.2015.07.056
Dubey PK, Tripathi P, Tiwari RS, Sinha ASK, Srivastava ON (2014) Synthesis of reduced graphene oxide-TiO2 nanoparticle composite systems and its application in hydrogen production. Int J Hydrogen Energy 39(29):16282–16292. https://doi.org/10.1016/j.ijhydene.2014.03.104
Melvin AA, Illath K, Das T, Raja T, Bhattacharyya S, Gopinath CS (2015) M-Au/TiO2 (M = Ag, Pd, and Pt) nanophotocatalyst for overall solar water splitting: role of interfaces. Nanoscale 7(32):13477–13488. https://doi.org/10.1039/c5nr03735b
Wang P, Zhan S, Xia Y, Ma S, Zhou Q, Li Y (2017) The fundamental role and mechanism of reduced graphene oxide in rGO/Pt-TiO2 nanocomposite for high-performance photocatalytic water splitting. Appl Catal B Environ 207:335–346. https://doi.org/10.1016/j.apcatb.2017.02.031
Hasan MR, Abd Hamid SB, Basirun WJ, Meriam Suhaimy SH, Che Mat AN (2015) A sol-gel derived, copper-doped, titanium dioxide-reduced graphene oxide nanocomposite electrode for the photoelectrocatalytic reduction of CO2 to methanol and formic acid. RSC Adv. 5(95):77803–77813. https://doi.org/10.1039/c5ra12525a
Mei BA, Munteshari O, Lau J, Dunn B, Pilon L (2018) Physical interpretations of nyquist plots for EDLC electrodes and devices. J Phys Chem C 122(1):194–206. https://doi.org/10.1021/acs.jpcc.7b10582
Tian H, Wan C, Zheng W, Hu X, Qiao L, Wang X (2016) Construction of a ternary hybrid of CdS nanoparticles loaded on mesoporous-TiO2/RGO for the enhancement of photocatalytic activity. RSC Adv 6(88):84722–84729. https://doi.org/10.1039/c6ra16094h
Tasbihi M, Kočí K, Edelmannová M, Troppová I, Reli M, Schomäcker R (2018) Pt/TiO2 photocatalysts deposited on commercial support for photocatalytic reduction of CO2. J Photochem Photobiol A Chem 366:72–80. https://doi.org/10.1016/j.jphotochem.2018.04.012
Liu L, Li Y (2014) Understanding the reaction mechanism of photocatalytic reduction of CO2 with H2O on TiO2-based photocatalysts: a review. Aerosol Air Qual Res 14(2):453–469. https://doi.org/10.4209/aaqr.2013.06.0186
Sclafani A, Herrmann JM (1996) Comparison of the photoelectronic and photocatalytic activities of various anatase and rutile forms of titania in pure liquid organic phases and in aqueous solutions. J Phys Chem 100(32):13655–13661. https://doi.org/10.1021/jp9533584
Li G et al (2008) Synthesizing mixed-phase TiO2 nanocomposites using a hydrothermal method for photo-oxidation and photoreduction applications. J Catal 253(1):105–110. https://doi.org/10.1016/j.jcat.2007.10.014
Chang X, Wang T, Gong J (2016) CO2photo-reduction: insights into CO2activation and reaction on surfaces of photocatalysts. Energy Environ Sci 9(7):2177–2196. https://doi.org/10.1039/c6ee00383d
Shehzad N, Tahir M, Johari K, Murugesan T, Hussain M (2018) A critical review on TiO2 based photocatalytic CO2 reduction system: strategies to improve efficiency. J CO2 Util. 26:98–122. https://doi.org/10.1016/j.jcou.2018.04.026
Fox MA, Dulay MT (1993) Heterogeneous photocatalysis. Chem Rev 93(1):341–357. https://doi.org/10.1016/B978-0-444-53178-0.00004-3
Kočí K, Obalová L, Plachá D, Lacný Z (2008) Effect of temperature, pressure and volume of reacting phase on photocatalytic CO2 reduction on suspended nanocrystalline TiO 2. Collect Czechoslov Chem Commun 73(8–9):1192–1204. https://doi.org/10.1135/cccc20081192
Cheng X et al (2017) An optofluidic planar microreactor for photocatalytic reduction of CO2 in alkaline environment. Energy 120(41):276–282. https://doi.org/10.1016/j.energy.2016.11.081
Kalamaras E, Maroto-valer M, Xuan J, Wang H (2017) A Microfluidic reactor for solar fuel production from photocatalytic CO2 reduction -the 15th international symposium on district heating and cooling photocatalytic CO2 reduction. Energy Procedia 142:501–506. https://doi.org/10.1016/j.egypro.2017.12.078
Zhang QH, Han WD, Hong YJ, Yu JG (2009) Photocatalytic reduction of CO2 with H2O on Pt-loaded TiO2 catalyst. Catal Today 148(3–4):335–340. https://doi.org/10.1016/j.cattod.2009.07.081
Acknowledgements
The author, S. Nabil, gratefully acknowledges the Egypt-Japan University of Science and Technology (E-JUST), especially the TMD (Technology Management Department), for providing the facilities and support to accomplish this work.
Funding
Open access funding provided by The Science, Technology &. The analysis of all samples as well as all experiments were conducted in the laboratories of Egypt-Japan University of Science and Technology (E-JUST).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by [SN]. Monitoring and advice to accomplish this research was done by [Prof. MFE]. [Prof. YM] supplied the UV lamp and its setup to perform the PEC measurements. The supervision, discussion of results and suggestion of the research point were performed by [Prof. EAS], and [Prof. AHE-S]. The first draft of the manuscript was written by [SN] and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical Approval and Consent to Participate
The manuscript does not involve the use of any animal or human data or tissue. “Not applicable”.
Consent for Publication
The manuscript does not contain data from any individual person. “Not applicable”.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nabil, S., Shalaby, E.A., Elkady, M.F. et al. Optimizing the Performance of the Meso-Scale Continuous-Flow Photoreactor for Efficient Photocatalytic CO2 Reduction with Water Over Pt/TiO2/RGO Composites. Catal Lett 152, 3243–3258 (2022). https://doi.org/10.1007/s10562-021-03915-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-021-03915-y