
Abstract
The relevance of a selection of organic impurities for the initiation of the MTO process was quantified in a kinetic model comprising 107 elementary steps with ab initio computed reaction barriers (MP2:DFT). This model includes a representative part of the autocatalytic olefin cycle as well as a direct initiation mechanism starting from methanol through CO-mediated direct C–C bond formation. We find that the effect of different impurities on the olefin evolution varies with the type of impurity and their partial pressures. The reactivity of the considered impurities for initiating the olefin cycle increases in the order formaldehyde < di-methoxy methane < CO < methyl acetate < ethanol < ethene < propene. In our kinetic model, already extremely low quantities of impurities such as ethanol lead to faster initiation than through direct C–C bond formation which only matters in complete absence of impurities.
Graphic Abstract

Similar content being viewed by others
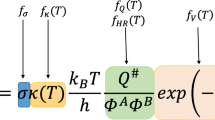


Avoid common mistakes on your manuscript.
1 Introduction
In a sustainable methanol economy the methanol-to-olefins (MTO) process enables the production of hydrocarbons through the formation of carbon-carbon bonds from potentially renewable feed stocks [1]. The industrially applied process is operated with porous acidic zeolite catalysts at temperatures between 350 to \(400\,^\circ {\text{C}}\) [2]. Essentially, the acidic sites of the catalyst transform methanol (MeOH) and its condensation product dimethyl ether (DME) into a variety of olefins. The product distribution is governed both by the intrinsic catalyst properties such as acid strength, acid site density and catalyst topology (framework and shape confinement) as well as by the overall process conditions [3,4,5,6,7].
Advances in the understanding of the mechanistic details of the process lead to the establishment of the hydrocarbon pool (HCP) concept [8,9,10,11,12]. Extensive experimental and theoretical studies have described a dual-cycle concept comprising the olefin and the aromatic cycle. In the former, olefins are formed by repeated alkene methylation [13,14,15] and cracking [2, 7], whereas the latter comprises the methylation of aromatics producing olefins via either the side chain or pairing mechanism.
Density functional theory (DFT) calculations are increasingly employed to gain insight into the reactivity of zeolites [16,17,18,19,20,21], for methanol dehydration [22,23,24,25] as well as subsequent steps in the MTO process involving the reaction of olefins and aromatics [26,27,28,29,30,31,32,33,34]. Kinetic models were developed to study initiation, step-wise methylation via surface methoxy species (SMS), concerted methylation, cracking and deactivation through coking revealing many features of the process [35,36,37,38,39]. Perhaps the most vividly debated issue relates to the early stages of the MTO process and the initial formation of the hydrocarbon pool (i.e. the formation of the first carbon-carbon bond in absence of any olefins) [5, 7, 12, 16, 40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66]. One of the major challenges for the experimental investigation of the initiation mechanism lies in the avoidance of organic impurities that could be present in the reaction feed as well as during the preparation of the zeolite, as they have been proposed to be responsible for initializing the HCP [5, 58, 67].
Theoretical studies have been employed to calculate reaction free energy barriers of possible initiation and C–C coupling steps to shed light on plausible reaction mechanisms [43,44,45,46,47,48,49,50,51,52,53,54, 68,69,70]. Using H-SSZ-13 as the catalyst we recently proposed a viable mechanism of the initiation reaction based on DFT calculations that have been corrected using second order Møller Plesset perturbation theory (MP2) [71] thus comprising highly accurate reaction barriers [72]. This is crucial since non-hybrid DFT calculations, particularly those using PBE-D3 [73, 74], give rise to significantly underestimated activation barriers, effectively making reaction mechanisms appear more favorable than they actually are [68, 72]. The key steps that we have identified for the initiation reaction starting from MeOH or DME using DFT:MP2 [68] are (1) the oxidation of MeOH to formaldehyde (FA) and subsequent oxidation to carbon monoxide (CO) and (2) the methylation of CO to methyl acetate (MA) forming the first C–C bond (see Fig. 1) [68].

Within this reaction sequence, the highest free energy barrier at a reaction temperature of 673 K has been identified for the oxidation of FA to CO ranging from 250 to 275 kJ mol−1, depending on the zeolite employed. [69] In previous studies, the CO methylation pathway has been identified as the most favorable initiation pathway in absence of olefins [51, 68]. Therefore, we only consider this pathway here and do not include other mechanisms such as the carbene, oxonium ylide or methane-formaldehyde mechanisms.
As noted above, higher-level methods are required to obtain accurate activation barriers. Furthermore, the interpretation of activation barriers is particularly complicated in the context of the initiation due to the autocatalytic nature of the olefin cycle. Therefore, we have previously investigated the effect of the above described reaction energetics for H-SSZ-13 on the mechanism of the initiation reaction using a multi-scale modeling approach employing a kinetic batch reactor model comprising both the initiation as well as a substantial part of the autocatalytic olefin cycle [70]. The simulated kinetics showed that the proposed pathway (Fig. 1) is indeed viable and the most likely mechanism. The simulations also showed, however, that the initiation mechanism is only relevant in the very early stages with no to little olefins present, as the autocatalytic cycle becomes the dominant source of olefin formation already at very small pressures of olefins.
In this work we quantify the effect of a variety of impurities in the reaction feed as well as typical experimental co-feeds on the olefin formation lightoff and mechanism by employing the existing kinetic batch reactor model [70] using ab initio rate constants for the initiation [68] and olefin cycle [71]. In addition, we extend our reaction scheme by including the impurity ethanol (EtOH) and we hence compute and consider free energy barriers for the dehydration of EtOH to ethene at the same level of theory (MP2:DFT).
2 Results and Discussion
Methanol (MeOH) synthesized via Cu/Zn/Al\(_2\)O\(_3\) catalysts from syngas (CO, CO\(_2\), and H\(_2\)O) contains trace amounts of DME, methane, EtOH, ethane and propane [75]. Commercially available MeOH [76] contains \(\le 0.05\%\) EtOH, \(\le 0.05\%\) water, \(\le 0.001\%\) acetone, \(\le 0.001\%\) acetaldehyde and \(\le 0.001\%\) formaldehyde. Impurities that are commonly considered to play an important role for the formation of the C–C bond during the initiation phase or within the autocatalytic olefin cycle and that we consider in the present study are: propene (C\(_3\)H\(_6\)), ethene (C\(_2\)H\(_4\)), ethanol (EtOH), methyl acetate (MA), carbon monoxide (CO), di-methoxy methane (DMM) and formaldehyde (FA). In total, our kinetic batch reactor model comprises rate constants of 107 (ab initio) computed elementary reaction steps for the CO-formation, the CO-mediated C–C coupling and the olefin cycle as shown in Fig. 1. We consider both step-wise methylation via surface methoxy species (SMS) as well as concerted methylation via MeOH or DME up to 2-nonene. The free energy barriers of the most important reaction steps that involve the considered impurities are listed in Table 1. A complete list of all reaction barriers is provided in Table S1 of the supporting information (SI). The computational procedure to obtain these barriers at the MP2:DFT level of theory is described in the Methods section (Sect. 4).
All kinetic batch simulations were performed starting from a mixture of MeOH, DME and H\(_2\)O that was equilibrated from 1 bar of pure DME at 673.15 K. Adsorption processes and SMS formation were included in the equilibration, see Fig. S2 in the SI. Consistently with previous studies [70] an active site concentration of 17.9 mol m\(^{-3}\) was chosen. For an in depth discussion of the batch reactor model used we refer to the supporting information of our previous publication [70]. We stress here that our focus lies solely on the olefin cycle of the HCP without considering aromatics formation and the aromatic cycle, as we expect the olefin cycle to be the most relevant post-initiation mechanism during and right after the initiation phase.
The outcome of our kinetic simulations is shown in Fig. 2. Here, the changes of the partial pressures are given as a function of time. We simulated a feed without impurities (Fig. 2a) as well as with 2 mbar of FA (Fig. 2b), ethanol (Fig. 2c) and propene (Fig. 2d).

Results of the kinetic batch reactor model: Partial pressure evolution of an equilibrated DME feed with a no impurities, b 2 mbar of formaldehyde, c 2 mbar of ethanol and d 2 mbar of propene. The black line shows the total partial pressure of the produced olefins. Two lightoff criteria for the olefin production are highlighted: the time after which the produced olefins surpass 1 mbar and their total partial pressure after 3 s
Our kinetics show the general trends observed experimentally. Olefin production has an initiation phase after which production lights off, e.g. at about 3 s in the case where there are no impurities in the feed gas. We also observe that DME is consumed faster than MeOH, which is due to the fact that the methylation of olefins is faster with DME compared to MeOH [13, 38]. Methylation with DME produces MeOH and we hence observe an increase in MeOH pressure shortly after lightoff, which is also observed experimentally [2]. We simulated the effect of three different impurities in Fig. 2b–d. As can be seen, while FA has little effect on the lightoff behavior, the presence of both EtOH and propene greatly reduces the time before olefin formation occurs. EtOH is dehydrated to ethene through which the olefin cycle is started directly. The same analysis with 2 mbar of ethene impurity yields a light off time of 0.43 s. Therefore, the difference in lightoff time between EtOH and propene (0.89 s) is composed of the times needed to convert sufficient amounts of EtOH to ethene (0.58 s) and for subsequent methylation (0.31 s).
We note that the definition of the olefin lightoff criteria is somewhat arbitrary. Here we quantify the effect of impurities on the olefin lightoff using two measures: the time \(t_{\text{lightoff}}\) after which the total pressure of the produced olefins surpasses 1 mbar (Eq. (1)) and the olefin production after 3 s (Eq. (2)).
The relation between the amount of impurity and its effect on the olefin production is shown in Fig. 3. Figure 3a shows that all considered impurities shorten the initiation period \(t_{\text{lightoff}}\) to some extent, but vary largely in the observed effect. Interestingly, over a wide pressure range we observe a linear dependence of the lightoff time on the logarithm of the initial partial pressure of the impurity. The dependence of our second lightoff measure on the initial partial pressure of the impurity is depicted in Fig. 3b. The dashed horizontal line in Fig. 3a and b at 3.04 s, i.e. \(6.32 \times 10^{-4}\) bar represents the limit for a pure feed without impurities where all initial olefin contributions arise from the initiation reaction. The dotted lines crossing the dashed line show the simulation with excluded initiation mechanism that entirely depends on autocatalysis through impurities. Both lightoff measures allow for a ranking of impurity effectiveness. The onset of the olefin formation benefits from these impurities in descending order: C\(_3\)H\(_6\) > C\(_2\)H\(_4\) > EtOH > MA > CO > DMM > FA. This ranking is plausible for several reasons. Propene and ethene are olefins and thus part of the HCP which means autocatalysis is active from the beginning. In general, cracking barriers decrease for higher alkenes. Propene is one methylation step closer to these higher alkenes than ethene and thus more effective. Dehydration of EtOH leads to ethene thereby circumventing the need for initial carbon-carbon bond formation. The ranking of the other impurities can be explained in terms of how advanced they are in the initiation mechanism. In particular, MA and CO are one oxidation step ahead of DMM and FA.

Simulated effect of impurities on olefin formation with (solid lines) and without (dotted lines) consideration of the initiation mechanism using a kinetic batch reactor model employing H-SSZ-13 as a function of the corresponding impurities. The dashed line marks the limit for a pure reactant mixture with the initiation mechanism included. a The lightoff time is defined as the time when the produced olefins surpass a pressure of 1 mbar. b The lightoff pressure is defined as the olefin pressure produced after 3 s of simulation time
We now discuss the cause of the linear decrease of \(t_{\text{lightoff}}\) with increasing logarithm of \(p_{\text{impurity}}\) and provide an explanation with two model reactions for initiation and autocatalysis of first (Eq. 3) and second (Eq. 4) order, respectively.
Here, A represents a C\(_1\) species (MeOH, DME or SMS) and B represents an autocatalytically active species from the HCP which also includes the impurity. Assuming that the rate of autocatalysis (Eq. 4) is significantly faster than that of initiation (Eq. 3) the latter can be neglected and the integrated rate expression of formal kinetics for a second-order autocatalytic reaction [77] leads to:
Assuming \(p_0^\text{A} \gg p_0^\text{B}\) and \(p_0^\text{A} \approx p^\text{A}\) in Eq. 5, the second order autocatalytic reaction (Eq. 4) is of pseudo first order:
Recalling the definition of \(t_\text{lightoff}\) being the time when \(p^\text{B}(t_\text{lightoff}) = p_0^\text{B} + 1\,\text{mbar}\) and assuming \(p_0^\text{B} \ll 1\,\text{mbar}\) leads to:
and assuming the impurity to be equivalent to the autocatalytically active species (\(p_0^\text{B} = p_0^\text{impurity}\)) one finds \(t_\text{lightoff} \propto -\log \left( p_0^\text{impurity}/\text{mbar} \right) \). Under these assumptions, we can divide the dependence on the impurity partial pressure into three regimes: (i) the regime, where the initiation reaction (3) dominates and the solid curves in Fig. 3 approach the dashed line representing the limit lightoff for a pure mixture without impurities, (ii) the regime where autocatalysis dominates and the assumptions of pseudo first order hold and (iii) the regime where autocatalysis dominates but the linear relationship is lost because the assumptions do not hold anymore at higher amounts of HCP species.
The contribution of initial C–C formation to olefin formation is investigated in Fig. 4. The relative contribution of the initiation to the olefin production becomes negligible at extremely low concentrations of ethene. Even with ethene impurities as low as \(2\times 10^{-18}\) bar the olefin formation is dominated by autocatalysis from the beginning. This can be understood when comparing the calculated reaction free energy barriers of the olefin methylation with that of the highest step of the oxidation of methanol to CO. Step-wise olefin methylation has free energy barriers on the order of \(130\,\text{kJ mol}^{-1}\) to \(170\, \text{kJ mol}^{-1}\) (see Table S1). In stark contrast the highest free energy barrier for the formation of CO from MeOH is 251 kJ mol\(^{-1}\) (see reaction no. 15 in Table 1). These differences in the order of 100 kJ mol\(^{-1}\) translate to differences in the rate constants of about seven to eight orders of magnitude at 673.15 K. In addition, olefin methylation is autocatalytic in itself, increasing the overall rate with which olefins are formed.

The relative contribution of initiation and autocatalysis to olefin production is shown as a function of reaction time for varying levels of ethene (upper panels) and CO (lower panels) impurities
Figure 4 also illustrates that, although the effect of CO on olefin production is clearly large (see Fig. 3), this is not due to the direct formation of olefins from CO, but due to the faster initiation of the autocatalytic olefin cycle. It may appear puzzling at first that the time after which the rates for olefin production from initiation and autocatalysis cross over hardly changes in Fig. 4, when the CO pressure is increased from \(2 \times 10^{-10}\) over \(2 \times 10^{-5}\) to 0.2 bar. However, the absolute value of the rates at this crossover does increase dramatically, leading to the reduced lightoff times as shown in Fig. 3.
3 Summary and Conclusions
We have analyzed the effect of various impurities on the initiation of the MTO process based on batch reactor kinetics using ab initio calculated rate constants. Initiation of the autocatalytic olefin cycle through these impurities competes with initiation through direct C–C bond formation starting from methanol. Direct C–C bond formation proceeds through sequential oxidation of the carbon in MeOH (oxidation state −II), over formaldehyde (oxidation state 0) to carbon monoxide (oxidation state +II). Carbon monoxide can then be methylated to form the first C–C bond and subsequently olefins.
The effect of impurities can be directly related to their role in either initiation or olefin cycle. The olefins propene and ethene have clearly the largest effect, since they can directly initiate the olefin cycle. Methyl acetate (MA) and CO are somewhat less active since a few more reaction steps are still required to convert them into olefins. The effect of ethanol, which enters the HCP via dehydration to ethene, is comparable to that of MA. Formaldehyde and DMM are least active, which is due the fact that an oxidation step is required to form CO.
We conclude the more active impurities such as ethanol or MA suffice to be present in sub ppb-levels to allow the initiation of the olefin cycle, without the need for a direct C–C bond forming initiation mechanism.
4 Methods
The rate constants were calculated with harmonic transition state theory according to:
with \(\varDelta {G^\ddagger }\) being extracted from previously computed data [68, 71] which used a hierarchical cluster approach [78, 79] employing periodic density functional theory (DFT) calculations using the dispersion-corrected PBE-D3 [73, 74] density functional and the VASP code with the standard PAWs and an energy cutoff of 400 eV [80,81,82]. Convergence criteria of \(1 \times 10^{-8}\) eV and \(0.01\,\text{e V}\,{\AA }^{-1}\) were applied to SCF cycles and geometry optimization. The Brillouin zone was sampled at the \(\varGamma \)-point only [83] using Gaussian smearing with a width of 0.1 eV. Transition states were optimized using automated relaxed potential surface scans [84] and confirmed to contain one imaginary frequency whose transition vector leads to the correct minima of the reaction. The partial Hessian matrix was computed using a central finite difference scheme including only the adsorbate, the acid site and its adjacent Al- and Si-atom. Energy corrections have been applied with highly accurate non-periodic MP2/def2-TZVPP calculations [85,86,87,88] on T46 cluster models employing Turbomole [89]. Entropic contributions to the free energy barriers have been calculated using the harmonic approximation and a temperature of 673.15 K and a reference pressure of 1 bar where vibrational frequencies of the adsorbate below 10 cm\(^{-1}\) were raised to this value because they can lead to inaccurate entropies otherwise [38]. Additional rate constants for the EtOH dehydration to ethene were computed in this work using the exact same procedure on the same H-SSZ-13 zeolite unit cell.
While the energy contribution to the free energy is quite accurate as it is computed with MP2 calculations on cluster models, entropic contributions are of limited accuracy as they are derived from the harmonic approximation based on DFT calculations. The harmonic approximation is particularly problematic for loosely bound adsorbates in the zeolite [17, 18], whereas molecules in the gas phase are expected to be described accurately. We therefore expect that activation energies and adsorption energies may in some cases be overestimated, when referenced to the gas phase. Another approximation that we employ is to neglect diffusion limitations, which should generally slow down reactions, particular those involving relatively large molecules [90]. An additional limitation of our study is that the reaction network is incomplete, although it contains more than 100 elementary reactions. For example, aromatic molecules are not present. Although aromatic molecules cannot be formed before the first C–C bond during initiation, it cannot be excluded that other relevant reaction pathways are missing. Lastly, we focus on Brønsted acid sites, as they are present in the pores of the zeolite. However, other active site motifs such as surface Brønsted and Lewis acid sites [91,92,93,94], as well as extra-framework aluminum sites [50] may also play an important role.
The kinetic model comprises step-wise and direct methylation with DME and MeOH as well as cracking up to 2-nonene (C\(_9\)). Olefin isomerization barriers that only shift the double bond (for example 1-butene to 2-butene) are expected to be small compared to methylation and cracking barriers [71] and are therefore treated implicitly. The kinetic simulations of the batch reactor were carried out based on the mean field approximation using simple Euler integration with a maximum time step smaller than \(5 \times 10^{-8}\) s as implemented in the in-house code used in previous work [70]. Since we only consider the initial part of the MTO reaction mechanism we neglect diffusion limitations in the reactor simulations, thus assuming perfect mixing. Further details on the kinetic model can be found in the SI of our previous publication [70].
References
Olah GA (2013) Angew Chem Int Ed 52:104
Olsbye U, Svelle S, Bjørgen M, Beato P, Janssens TVW, Joensen F, Bordiga S, Lillerud KP (2012) Angew Chem Int Ed 51:5810
Teketel S, Olsbye U, Lillerud KP, Beato P, Svelle S (2010) Microporous Mesoporous Mater 136:33
Haw JF, Song W, Marcus DM, Nicholas JB (2003) Acc Chem Res 36:317
Olsbye U, Svelle S, Lillerud KP, Wei ZH, Chen YY, Li JF, Wang JG, Fan WB (2015) Chem Soc Rev 44:7155
Müller S, Liu Y, Kirchberger FM, Tonigold M, Sanchez-Sanchez M, Lercher JA (2016) J Am Chem Soc 138:15994
Yarulina I, Chowdhury AD, Meirer F, Weckhuysen BM, Gascon J (2018) Nat Catal 1:398
Dahl IM, Kolboe S (1993) Catal Lett 20:329
Dahl I, Kolboe S (1994) J Catal 149:458
Dahl IM, Kolboe S (1996) J Catal 161:304
Wragg DS, O’Brien MG, Bleken FL, Di Michiel M, Olsbye U, Fjellvåg H (2012) Angew Chem Int Ed 51:7956
Wang W, Jiang Y, Hunger M (2006) Catal Today 113:102
Svelle S, Kolboe S, Swang O, Olsbye U (2005) J Phys Chem B 109:12874
Bleken F, Svelle S, Lillerud KP, Olsbye U, Arstad B, Swang O (2010) J Phys Chem A 114:7391
Redekop EA, Lazzarini A, Bordiga S, Olsbye U (2020) J Catal 385:300
Hemelsoet K, Van der Mynsbrugge J, De Wispelaere K, Waroquier M, Van Speybroeck V (2013) ChemPhysChem 14:1526
Van Speybroeck V, De Wispelaere K, Van der Mynsbrugge J, Vandichel M, Hemelsoet K, Waroquier M (2014) Chem Soc Rev 43:7326
Van Speybroeck V, Hemelsoet K, Joos L, Waroquier M, Bell RG, Catlow CRA (2015) Chem Soc Rev 44:7044
Liu C, Tranca I, van Santen RA, Hensen EJM, Pidko EA (2017) J Phys Chem C 121:23520
Li G, Pidko EA (2019) ChemCatChem 11:134
Chizallet C (2020) ACS Catal 10:5579
Ghorbanpour A, Rimer JD, Grabow LC (2014) Catal Commun 52:98
Ghorbanpour A, Rimer JD, Grabow LC (2016) ACS Catal 6:2287
Arvidsson AA, Plessow PN, Studt F, Hellman A (2020) J Phys Chem C 124:14658
Hoffman AJ, Bates JS, Di Iorio JR, Nystrom SV, Nimlos CT, Gounder R, Hibbitts D (2020) Angew Chem 132:18845
De Wispelaere K, Hemelsoet K, Waroquier M, Van Speybroeck V (2013) J Catal 305:76
Moors SLC, De Wispelaere K, Van der Mynsbrugge J, Waroquier M, Van Speybroeck V (2013) ACS Catal 3:2556
Wang CM, Wang YD, Xie ZK (2013) J Catal 301:8
Wang CM, Wang YD, Xie ZK (2014) Catal Sci Technol 4:2631
Wang CM, Wang YD, Du YJ, Yang G, Xie ZK (2015) Catal Sci Technol 5:4354
DeLuca M, Kravchenko P, Hoffman A, Hibbitts D (2019) ACS Catal 9:6444
Rey J, Bignaud C, Raybaud P, Bučko T, Chizallet C (2020) Angew Chem 132:19100
Fečík M, Plessow PN, Studt F (2020) ACS Catal 10:8916
DeLuca M, Janes C, Hibbitts D (2020) ACS Catal 10:4593
Janssens TV, Svelle S, Olsbye U (2013) J Catal 308:122
Kumar P, Thybaut J, Teketel S, Svelle S, Beato P, Olsbye U, Marin G (2013) Catal Today 215:224
Kumar P, Thybaut JW, Svelle S, Olsbye U, Marin GB (2013) Ind Eng Chem Res 52:1491
Brogaard RY, Henry R, Schuurman Y, Medford AJ, Moses PG, Beato P, Svelle S, Nørskov JK, Olsbye U (2014) J Catal 314:159
Standl S, Kirchberger FM, Kühlewind T, Tonigold M, Sanchez-Sanchez M, Lercher JA, Hinrichsen O (2020) Chem Eng J 402:126023
Hutchings GJ, Gottschalk F, Hunter R (1987) Ind Eng Chem Res 26:635
Hutchings GJ, Gottschalk F, Hall MVM, Hunter R (1987) Faraday transactions 1: physical chemistry in condensed phases. J Chem Soc 83:571
Salehirad F, Anderson MW (1996) J Catal 164:301
Blaszkowski SR, van Santen RA (1996) J Am Chem Soc 118:5152
Sinclair PE, Catlow CRA (1997) J Phys Chem B 101:295
Blaszkowski SR, van Santen RA (1997) J Am Chem Soc 119:5020
Tajima N, Tsuneda T, Toyama F, Hirao K (1998) J Am Chem Soc 120:8222
Lesthaeghe D, Van Speybroeck V, Marin GB, Waroquier M (2006) Angew Chem Int Ed 45:1714
Lesthaeghe D, Van Speybroeck V, Marin G, Waroquier M (2006) Chem Phys Lett 417:309
Li J, Wei Z, Chen Y, Jing B, He Y, Dong M, Jiao H, Li X, Qin Z, Wang J, Fan W (2014) J Catal 317:277
Comas-Vives A, Valla M, Copèret C, Sautet P (2015) ACS Central Sci 1:313
Liu Y, Müller S, Berger D, Jelic J, Reuter K, Tonigold M, Sanchez-Sanchez M, Lercher JA (2016) Angew Chem Int Ed 55:5723
Wei Z, Chen YY, Li J, Guo W, Wang S, Dong M, Qin Z, Wang J, Jiao H, Fan W (2016) J Phys Chem C 120:6075
Liu Y, Müller S, Berger D, Jelic J, Reuter K, Tonigold M, Sanchez-Sanchez M, Lercher JA (2017) Angew Chem 129:7448
Rasmussen DB, Christensen JM, Temel B, Studt F, Moses PG, Rossmeisl J, Riisager A, Jensen AD (2017) Catal Sci Technol 7:1141
Dewaele O, Geers VL, Froment GF, Marin GB (1999) Chem Eng Sci 54:4385
Stöcker M (1999) Microporous Mesoporous Mater 29:3
Wang W, Buchholz A, Seiler M, Hunger M (2003) J Am Chem Soc 125:15260
Jiang Y, Wang W, Reddymarthala V, Huang J, Sulikowski B, Hunger M (2006) J Catal 238:21
Wang W, Hunger M (2008) Acc Chem Res 41:895
Chen X, Neidig ML, Tuinstra R, Malek A (2010) J Phys Chem Lett 1:3012
Yamazaki H, Shima H, Imai H, Yokoi T, Tatsumi T, Kondo JN (2011) Angew Chem Int Ed 50:1853
Hwang A, Prieto-Centurion D, Bhan A (2016) J Catal 337:52
Wei Z, Chen YY, Li J, Wang P, Jing B, He Y, Dong M, Jiao H, Qin Z, Wang J, Fan W (2016) Catal Sci Technol 6:5526
Chowdhury AD, Houben K, Whiting GT, Mokhtar M, Asiri AM, Al-Thabaiti SA, Basahel SN, Baldus M, Weckhuysen BM (2016) Angew Chem Int Ed 55:15840
Wu X, Xu S, Zhang W, Huang J, Li J, Yu B, Wei Y, Liu Z (2017) Angew Chem Int Ed 56:9039
Chowdhury AD, Paioni AL, Houben K, Whiting GT, Baldus M, Weckhuysen BM (2018) Angew Chem Int Ed 57:8095
Song W, Marcus DM, Fu H, Ehresmann JO, Haw JF (2002) J Am Chem Soc 124:3844
Plessow PN, Studt F (2017) ACS Catal 7:7987
Plessow PN, Studt F (2018) Catal Lett 148:1246
Plessow PN, Smith A, Tischer S, Studt F (2019) J Am Chem Soc 141:5908
Plessow PN, Studt F (2018) Catal Sci Technol 8:4420
Goncalves TJ, Plessow PN, Studt F (2019) ChemCatChem 11:4368
Perdew JP, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:3865
Grimme S, Antony J, Ehrlich S, Krieg H (2010) J Chem Phys 132:154104
van den Berg R, Prieto G, Korpershoek G, van der Wal LI, van Bunningen AJ, Lægsgaard-Jørgensen S, de Jongh PE, de Jong KP (2016) Nat Commun 7:13057
ACS-grade MeOH by Merck KGaA (2020). https://www.merckmillipore.com
Moore JW, Pearson RG, Frost AA (1981) Kinetics and mechanism, 3rd edn. Wiley, New York
Svelle S, Tuma C, Rozanska X, Kerber T, Sauer J (2009) J Am Chem Soc 131:816
Hansen N, Kerber T, Sauer J, Bell AT, Keil FJ (2010) J Am Chem Soc 132:11525
Blöchl PE (1994) Phys Rev B 50:17953
Kresse G, Furthmüller J (1996) Phys Rev B 54:11169
Kresse G, Joubert D (1999) Phys Rev B 59:1758
Monkhorst HJ, Pack JD (1976) Phys Rev B 13:5188
Plessow PN (2018) J Chem Theory Comput 14:981
Schäfer A, Horn H, Ahlrichs R (1992) J Chem Phys 97:2571
Schäfer A, Huber C, Ahlrichs R (1994) J Chem Phys 100:5829
Weigend F, Ahlrichs R (2005) Phys Chem Chem Phys 7:3297
Weigend F (2006) Phys Chem Chem Phys 8:1057
TURBOMOLE V.7.1 2016, a development of Karlsruhe Institute of Technology, Karlsruhe, 1989-2019, TURBOMOLE GmbH, since 2007.http://www.turbomole.com (Turbomole GmbH, 2016)
Smith AT, Plessow PN, Studt F (2021) Chem Phys 541:111033
Chizallet C, Raybaud P (2010) ChemPhysChem 11:105
Leydier F, Chizallet C, Chaumonnot A, Digne M, Soyer E, Quoineaud AA, Costa D, Raybaud P (2011) J Catal 284:215
Schallmoser S, Ikuno T, Wagenhofer M, Kolvenbach R, Haller G, Sanchez-Sanchez M, Lercher J (2014) J Catal 316:93
Chu Y, Yi X, Li C, Sun X, Zheng A (2018) Chem Sci 9:6470
Acknowledgements
The authors gratefully acknowledge support by the GRK 2450, by the state of Baden-Württemberg through bwHPC (bwUniCluster and JUSTUS, RV bw17D01) and by the Helmholtz Association.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Amsler, J., Plessow, P.N. & Studt, F. Effect of Impurities on the Initiation of the Methanol-to-Olefins Process: Kinetic Modeling Based on Ab Initio Rate Constants. Catal Lett 151, 2595–2602 (2021). https://doi.org/10.1007/s10562-020-03492-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-020-03492-6