
Abstract
Monodispersible Co3O4 nanoparticles were prepared via a facile solvothermal route using polyvinylpyrrolidone (PVP) as capping agent and the porous silica shell was then coated by means of the Stöber process to fabricate Co3O4@porous-SiO2 (Co3O4@p-SiO2) nanocomposites. The particle size of Co3O4 and porous silica shell thickness of Co3O4@p-SiO2 nanocomposites could be easily controlled through changing the amount of PVP and tetraethoxysilane, respectively. The high resolution transmission electron microscopy results, together with the X-ray diffraction results, indicated that monodispersible Co3O4 nanoparticles were successfully prepared and uniformly encapsulated by porous silica. During the growth of silica shell, the PVP was trapped and dispersed in the silica shell. As a result, the porous silica shell was obtained after burning off the PVP and a positive correlation existed between the Brunauer–Emmett–Teller surface area of the porous silica shell and quantity of PVP in the original Co3O4 nanoparticles. Compared with the Co/SiO2 reference catalyst, CO conversion of the Co@p-SiO2 model catalyst was more stable and higher in a period of 240 h, and hydrocarbon selectivity towards C5–C18 fraction was also higher than that of the Co/SiO2 catalyst. The results of analysis for the Co@p-SiO2 catalyst showed that core@shell structure could maintain high dispersion of Co particles so as to provide higher number of Co active sites, and enhance selectivity towards C5–C18 fraction due to confined structure of porous channel in the silica shell.
Graphical Abstract
Schematic diagram of the procedure for synthesizing Co3O4@p-SiO2 nanocomposites.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Fischer–Tropsch (F–T) synthesis has attracted much research attention nowadays because of the severe oil crisis and the tight fuel specifications [1, 2]. One of the goals of modern F–T synthesis research is to design a catalyst capable of highest selectivity to the desired product without sacrificing activity and lifetime [3]. Cobalt F–T synthesis catalysts are preferred due to their low activity for the water–gas shift reaction, slow deactivation rates and highly selective to linear paraffin than iron catalysts [4]. Since cobalt is ~1,000 times more expensive than iron, therefore optimal design of a cobalt catalyst is essential for its utilization. Decreasing the Co particle size to increase the exposed surface areas per unit mass of the Co [5] and hindering Co particles from agglomerating during reaction would be reasonable pathway to achieve this goal. Although up to now, many methods were designed to prepare cobalt-based catalysts with different Co particle size, and morphologies as well. It had been difficult to translate these advances into successful application in F–T synthesis as to be the drawback of easily agglomeration and sintering of the Co particles under harsh catalytic reaction conditions. In order to prevent Co particles from agglomerating and decrease the deactivation rates of the cobalt catalysts, a novel cobalt catalyst with core@shell structure was preferable to be adopted, where a porous shell played dual roles as: permitting syngas molecules to pass through the channel freely and preventing Co cores from coming into direct contact each other and thus hindering Co cores from agglomerating. This confined structure enabled cobalt catalyst showing special catalytic performances and product distribution in F–T synthesis. For example, Tsubaki co workers [6–9] had prepared core@shell cobalt catalysts by coating H-ZSM-5 zeolite membranes on the Co/SiO2 catalysts, which showed an excellent selectivity to the C5–C11 isoparaffins (gasoline distillates) benefiting from the confined reaction conditions constructed by H-ZSM-5 membranes. Moreover, the article of Trépanier et al. [10] has also showed that carbon nanotubes, when used as a cobalt catalyst support, allowed a better Co dispersion and minimized the Co metal phase interaction with the support. The Co particles located inside the tubes were more active than the ones on the outer surface of the CNT owing to confined effect. Thus, how to synthesize cobalt catalysts with confined structure via a facile method became a big challenge.
The present work developed a procedure that permitted the preparation of the Co3O4@porous-SiO2 (Co3O4@p-SiO2) nanocomposites with different particle sizes of Co3O4 core and thickness of porous silica shell via solvothermal route using polyvinylpyrrolidone (PVP) as capping agent. Compared with the conventional Co/SiO2 catalyst prepared via precipitation method, the Co@p-SiO2 catalyst, together with high dispersion of Co particles due to confined structure, could exhibit higher and more stable CO conversion and higher selectivity towards C5–C18 fraction in F–T synthesis.
2 Experimental Section
2.1 Nanocomposites Preparation
2.1.1 Preparation of the Co3O4@p-SiO2 Nanocomposites
The synthesis procedure for the Co3O4@p-SiO2 nanocomposites was illustrated in Scheme 1. In a typical synthesis, 7.09 g Co(NO3)2·6H2O and 3.54 g PVP were dissolved in 800 mL ethanol solution under magnetic stirring at room temperature. The concentrations of the solution were calculated in order to obtain 20 % cobalt loadings in the final catalysts. After the solution turned clear, it was transferred into a Teflon-lined stainless autoclave with the total volume of 1,000 mL, which afterwards was loaded in an oven at 453 K for 4 h and then naturally cooled to room temperature. The suspension was again transferred to the beaker. 2,076 mL ethanol was added into the suspension. After that, 1,656 mL distilled water and 144 mL 25 % ammonia were subsequently added into the suspension. After magnetic stirring for 2 h, 20 mL tetraethoxysilane (TEOS) was added dropwise into the suspension during 12 h. Magnetic stirring was continued for 48 h. The solid products were collected by filtration, washed thoroughly with distilled water and ethanol, and dried in an oven at 333 K. The template was removed by calcination at 773 K for 6 h in flowing air. The prepared nanocomposites were denoted as Co3O4@p-SiO2, which were selected as model catalysts for the subsequent F–T synthesis. Meanwhile, by controlling Co/PVP mass ratio, Co3O4@p-SiO2 nanocomposites with different core sizes and thickness of silica shell were also prepared using same method.

Schematic diagram of the procedure for synthesizing Co3O4@p-SiO2 nanocomposites
2.1.2 Preparation of the Co3O4/SiO2 Nanocomposites
The Co3O4/SiO2 nanocomposites (cobalt content 20 %) were prepared as follows: Firstly, 60 mL distilled water and 60 mL NH3·H2O were dissolved in 648 mL ethanol solution in sequence with magnetic stirring at room temperature. After that, 31.8 mL TEOS was added dropwise into above solution. Magnetic stirring was continued for 2 h. The spherical SiO2 support were collected by filtration, washed thoroughly with distilled water and ethanol, and dried in an oven at 333 K. Secondly, the Co3O4 nanoparticles were precipitated on the as-made SiO2 support by adding a highly concentrated NH3·H2O solution to a dilute cobalt nitrate solution, After aging for 2 h, the precipitate was separated from the suspension by filtration, washed thoroughly with distilled water and ethanol. Finally, the products were dried at 333 K and calcined at 773 K for 6 h. The as-made nanocomposites were denoted as Co3O4/SiO2, which were used as reference catalysts.
2.2 Nanocomposites Characterization
High resolution transmission electron microscopy (HRTEM) images were obtained on a Philips CM200FEG operating at 200 kV. The powder X-ray diffraction (XRD) patterns were recorded on a Bruker diffractometer using a CuKα radiation. The texture properties of nanocomposites were determined by N2 adsorption/desorption isotherms at 77 K using a Micromeritics Tristar 3000 sorptometer. The pore size distribution and pore volume were determined by the BJH method. X-ray photoelectron spectroscopy spectra (XPS) were performed on a PerkinElmer PHI-5300 spectrometer. Hydrogen temperature programmed reduction (H2-TPR) was carried out in a quartz microreactor heated by an electrical furnace. The reactor was loaded with 25 mg of calcined sample and heated at a rate of 10 K/min to 1,233 K with a gas consisting of 5 % H2 in N2. The gas flow rate was 60 mL/min. The mount of H2 consumed by the sample was measured by analysis of the effluent gas with a thermal conductivity detector (TCD).
2.3 Catalysts Test
F–T synthesis was carried out at 2 Mpa, 1,000 h−1 (the gas flow rate: 33.3 mL/min) and a H2/CO ratio of 2 in a stainless-steel fixed-bed reactor (i.d. = 10 mm) with a catalyst loading of 2 mL. The catalyst was reduced in a flow of hydrogen at 673 K for 10 h and then naturally cooled to room temperature before switching to syngas. Data were taken at steady state after 24 h on-stream. Wax was collected with a hot trap and the liquid products were collected in a cold trap. The gas effluents were analyzed on-line by using Carbosieve-packed column with TCD. The gas hydrocarbons were analyzed on-line using Porapack-Q column with FID. Oil and wax were analyzed offline in OV-101 capillary columns. 5 % N2 was added to syngas as an internal standard. The carbon balance and mass balance were 100 ± 5 %.
3 Results and Discussion
3.1 Synthesis and Characterization of Co3O4@p-SiO2 Nanocomposites
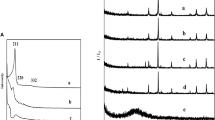
PVP is an amphiphilic polymer and usually employed as stabilizer for the preparation of various metal nanoparticles [11–15]. As reported previously, the Co3O4 nanoparticles with different particle size could be prepared via solvothermal route using PVP as stabilizer [16]. The PVP was chosen as stabilizer for the following reasons. The hydroxyl groups located on the surface of prepared Co3O4 nanoparticles, the hydrogen bond could form between PVP and Co3O4 nanoparticles [18]. By adsorbing PVP onto the surface of Co3O4 nanoparticles, this PVP layer could help to stabilize Co3O4 nanoparticles and avoid aggregation. It has been known that the coating of the host materials by PVP was very effective for the subsequent silica shell formation [19]. Figure 1a showed the XRD pattern of the Co3O4 nanoparticles prepared via solvothermal methods. It was found that all the diffraction peaks matched Co3O4 well with standard JCPDS data (42–1,467). After coating with porous silica shell, the Co3O4@p-SiO2 nanocomposites, as shown in Fig. 1b, exhibited similar diffraction peaks to that of the Co3O4 nanoparticles, suggesting that the Co3O4 phase was retained. However, it was found that the diffraction peaks of Co3O4@p-SiO2 nanocomposites had relatively low intensity, because of the shielding effect of the porous silica shell to the detecting X-rays [17]. Moreover, the Co3O4/SiO2 reference nanocomposites, as shown in Fig. 1c, also exhibited similar diffraction peaks to that of the Co3O4 nanoparticles, indicating that cobalt phase was also present in the form of Co3O4 spinel.

XRD patterns of a Co3O4 nanoparticles; b Co3O4@p-SiO2 nanocomposites; c Co3O4/SiO2 nanocomposites
Seen from Fig. 2a–c, it could be clearly observed that the PVP-stabilized Co3O4 nanoparticles were monodispersible and uniform. By controlling the amount of PVP, the Co3O4 nanoparticles with different size could be prepared. Moreover, these PVP-stabilized Co3O4 nanoparticles favored uniform growth of silica shell owing to interaction between PVP and Si–OH groups. To fabricate Co3O4@p-SiO2 nanocomposites, the stöber process would be applied to coat the silica onto the PVP-stabilized Co3O4 nanoparticles, which acted as seeds or nuclei for silica shell formation. Furthermore, in order to ensure uniform coating and to avoid formation of individual silica particles, the reaction conditions had to be properly controlled. For example, a lower TEOS concentration was indispensable of preventing aggregation of the particles during the growth of the initial silica shell [20]. Furthermore, by varying the amount of PVP and TEOS, Co3O4@p-SiO2 nanocomposites with different particle size of Co3O4 and silica shell thickness could also be prepared. Seen from Fig. 2d–i, monodispersible and uniform silica coating of Co3O4@p-SiO2 nanocomposites could be clearly observed in HRTEM photographs. It was remarkable that nearly perfect core@shell morphology was obtained, with rather uniform size distributions. Such results demonstrated the high efficiency of this procedure, despite its simplicity, as compared to other silica-coating techniques previously reported [21–25].

HRTEM photographs of a–c different particle sizes of Co3O4 nanoparticles [uniform 7.09 Co(NO3)2·6H2O was provided under different PVP mass from d (PVP = 14.17 g), e (PVP = 7.09 g) to f (PVP = 3.54 g)]; d–f Co3O4@p-SiO2 nanocomposites with different particle sizes of Co3O4 nanoparticles [uniform 20 mL TEOS was provided under different PVP mass from d (PVP = 14.17 g), e (PVP = 7.09 g) to f (PVP = 1.77 g)]; g–i Co3O4@p-SiO2 nanocomposites with different thicknesses of porous silica shell [uniform 3.54 g PVP was provided under different TEOS from d (TEOS = 20 mL), e (TEOS = 40 mL) to f (TEOS = 60 mL)]
Since the major contribution of surface area and porous structures results from PVP, it is therefore necessary to correlate the quantity of PVP in the reacting system with final specific surface area of Co3O4@p-SiO2 nanocomposites. Figure 3 showed the Brunauer–Emmett–Teller (BET) surface area of Co3O4@p-SiO2 nanocomposites after calcinations at various PVP/Co mass ratios. It could be clearly observed that the BET surface area of the Co3O4@p-SiO2 nanocomposites increased with increasing PVP/Co mass ratio from 0.25 to 2.0. This result indicated that a positive correlation existed between the BET surface area of the porous silica shell and quantity of PVP.

The BET surface area of Co3O4@p-SiO2 nanocomposites after calcinations at various PVP/Co mass ratios
In addition, Fig. 4 also showed HRTEM images of the Co3O4@p-SiO2 model catalysts and Co3O4/SiO2 reference catalysts before and after reaction, respectively. Seen from Fig. 4a, the fresh Co3O4@p-SiO2 catalysts showed nearly perfect core@shell morphology, Co3O4 nanoparticles were completely coated by porous silica. After F–T reaction, the HRTEM image of the spent Co@p-SiO2 model catalysts (see Fig. 4b; Table 1) illustrated that the core@shell structure was perfectly preserved due to higher hydrothermal stability of porous silica shell. The coated Co particles did not agglomerate owing to protection of porous silica shell during the reaction. However, the HRTEM image of the fresh Co3O4/SiO2 catalysts prepared via a controlled coprecipitation of cobalt nitrate with ammonia showed the fluffy Co3O4 nanoparticles were synthesized onto outer surface of spherical SiO2 support (see Fig. 4c; Table 1). For the spent Co/SiO2 catalysts, it was clear that serious aggregation of the Co particles were observed on the external surface of spherical SiO2 support (see Fig. 4d), indicating that small Co particles located on outer surface of the nonporous spherical SiO2 support were easier to agglomerate into large Co clusters without protection.

HRTEM photographs of a fresh Co3O4@p-SiO2 nanocomposites, b spent Co@p-SiO2 model catalysts, c fresh Co3O4/SiO2 nanocomposites, d spent Co/SiO2 reference catalysts
In Fig. 5a, it was not found Co2p or Co(LMM) peaks in XPS spectra of Co3O4@p-SiO2 nanocomposites, whereas XPS spectra was sensitive only to the Co ions localized at the surface and subsurface layer of the nanocomposites (depth of analysis ~6 nm) [26]. This result indicated that the Co3O4 nanoparticles did not located on the surface of porous silica shell and were completely coated by porous SiO2. Whereas, in the case of Co3O4/SiO2 nanocomposites, Co2p or Co(LMM) peaks were presented in XPS spectra in Fig. 5b, suggesting that the Co3O4 nanoparticles located on the external surface of nonporous silica spheres.

XPS spectra of the a Co3O4@p-SiO2 nanocomposites and b Co3O4/SiO2 nanocomposites
3.2 Reducibility of the Co3O4@p-SiO2 Nanocomposites
Hydrogen temperature programmed reduction is a very convenient technique for studying the reduction behaviors of catalysts qualitatively. In some case it is also possible to obtain useful information about the degree of interaction of the supported phase with the support from the reduction profiles of supported oxides. Figure 6 showed the H2-TPR profiles for the Co3O4@p-SiO2 and Co3O4/SiO2 nanocomposites. For the Co3O4@p-SiO2 nanocomposites (see Fig. 6a), the first peak was apparent and this was attributed to the reduction of Co3O4 to CoO. However, the first peak was significantly shifted to higher temperature than that of the Co3O4/SiO2 nanocomposites. This result could be attributed to the protective effects of porous silica shell which inhibited H2 to diffuse along the porous channels to access Co3O4 nanoparticles [27, 28]. Moreover, the second peak was also gradually shifted to higher temperatures and this was attributed to the reduction of CoO to metallic Co0. This result indicated that the reduction of CoO to metallic Co0 was difficult owing to H2 diffusion limitation in porous channels. A reduction pattern in the temperature range of about 820–1,015 K was observed for the Co3O4@p-SiO2 nanocomposites. Such a high reduction temperature might be assigned to the reduction of cobalt silicate species formed during the H2-TPR experiment by reaction of CoO with Si–OH groups [26]. However, for the Co3O4/SiO2 nanocomposites (see Fig. 6b), two peaks were apparent and these were attributed to the reduction of Co3O4 to CoO, which then reduced at lower temperature to metallic Co0. Significantly increasing area of the second peak indicated that the CoO particles were much easier to be reduced to active Co species. Thus first and second peaks were shifted to lower temperatures due to the lower interaction between CoO and Si–OH groups and higher contact area with H2. As to the reduction of cobalt silicate species, a small reduction peak in the temperature range of about 595–695 K was also observed for the Co3O4/SiO2 nanocomposites. Thus, the reducibility of the Co3O4/SiO2 nanocomposites was higher than that of the Co3O4@p-SiO2 nanocomposites, as shown in Table 1.

H2-TPR profiles of the Co3O4@p-SiO2 and Co3O4/SiO2 catalysts
3.3 FTS Performances
The performances of the Co@p-SiO2 and Co/SiO2 catalysts for F–T synthesis were evaluated in a fixed-bed reactor. Figure 7 depicted the variations of CO conversion with the time on-stream (TOS) under the same reaction temperature (503 K). CO conversion of the Co@p-SiO2 catalyst was more stable and higher than that of Co/SiO2 catalyst. In the case of the Co@p-SiO2 catalyst, CO conversion decreased slightly from 71.6 to 56.1 % in a period of 240 h, whereas, the Co/SiO2 catalyst showed a quick drop in CO conversion from 61.7 to 30.5 %. It is well known that dispersion and reducibility of the cobalt catalyst play an important role in catalytic activity. Although the Co@p-SiO2 catalyst had the lower reduction degree than the Co/SiO2 catalyst, it maintained a high dispersion of Co particles and number of active sites due to the protection of porous silica shell (see Fig. 4b; Table 1), which inhibited the agglomeration of Co particles during the reaction process. Therefore, more Co active sites were available for the CO hydrogenation. However, Co particles of the Co/SiO2 catalyst agglomerated easily among them during the reaction without the protection of porous silica shell (see Fig. 4d), thus decreasing the number of active sites and catalytic activity. To conclude, it is known that the CO conversion not only related to the reducibility of catalysts, but also to the dispersion of Co active sites. A high and stable CO conversion of the Co@p-SiO2 catalyst could therefore be attributed to a combination of high Co dispersion and appropriate reducibility in comparison to that of the Co/SiO2 catalyst.

Change in CO conversion with TOS for the Co@p-SiO2 and Co/SiO2 catalysts (reaction conditions: H2/CO = 2, GHSV = 1,000 h−1, P = 2 MPa, T = 503 K)
The hydrocarbon selectivity of both Co@p-SiO2 and Co/SiO2 catalysts were summarized in Table 2 under the different reaction temperatures (503 and 513 K). The data showed that selectivity towards both CH4 and C5–C18 fraction of the Co@p-SiO2 catalyst was higher than that of the Co/SiO2 catalysts. Whereas, the Co/SiO2 catalyst was more selective towards C19+ fraction while its selectivity towards both CH4 and C5–C18 fraction were lower. Reuel and Batholomew [29] reported that high CH4 selectivity observed on the Co/SiO2 catalyst with narrow pore was likely to be attributed to the presence of either unreduced cobalt species or the small cobalt particles which produced relatively high CH4 fraction at the expenses of the C19+ fraction than large cobalt particles. In our case, a part of unreduced CoO in the Co@p-SiO2 catalyst, which was discussed in H2-TPR session, resulted in high CH4 selectivity. Moreover, it is known that large Co particles lead to more selective for long-chain hydrocarbons under the same cobalt loadings [30, 31]. Khodakov et al. [27] had shown that large Co particles were more active in F–T synthesis and produced higher selectivity towards long-chain hydrocarbons than small Co particles. In addition, confined effect of the porous channel could inhibit the growth of long-chain hydrocarbons, thus increasing the selectivity towards C5–C18 fraction in the Co@p-SiO2 catalyst. The results of product selectivity analysis for Co@p-SiO2 catalysts showed that high CH4 selectivity and low selectivity towards C19+ fraction of the Co@p-SiO2 catalyst resulted from partly unreduced CoO species and the small Co particles. It was also reasonable to suggest that the confined structure of porous channel in the Co@p-SiO2 catalyst could enhance product selectivity towards C5–C18 fraction. Whereas, high selectivity towards both CH4 and C19+ fraction of the Co/SiO2 catalyst could be ascribed to high reducibility and formation of the large Co clusters (see Fig. 4d), respectively. This result in turn led to decreasing of C5–C18 selectivity.
4 Conclusions
Our work showed that Co3O4 nanoparticles could be synthesized via a facile solvothermal route using PVP as capping agent. Novel core@shell Co3O4@p-SiO2 nanocomposites with different Co3O4 particle sizes and silica shell thickness had been subsequently prepared via the Stöber process. CO conversion of the Co@p-SiO2 model catalyst was more stable and higher than that of the Co/SiO2 reference catalyst due to confined structure of porous silica shell, which could maintain high Co dispersion and number of active sites during harsh reaction condition. Selectivity towards both CH4 and C5–C18 fraction of the Co@p-SiO2 catalyst were higher than that of the Co/SiO2 catalyst due to more unreduced CoO and confined effect of porous channels, respectively. Compared with the Co@p-SiO2 catalyst, the large Co clusters formed during reaction owing to agglomeration among small Co particles was in favor of enhancing selectivity towards C19+ fraction for the Co/SiO2 catalyst.
References
Khodakov AY, Chu W, Fongarland P (2007) Chem Rev 107:1692
Khodakov AY (2009) Catal Today 144:251
Lee B, Koo HM, Park M-J, Lim B, Moon DJ, Yoon KJ, Bae JW (2013) Catal Lett 143:18
Xiong HF, Zhang YH, Liew KY, Li JL (2008) J Mol Catal A 295:68
Wang ZJ, Skiles S, Yang F, Yan Z, Goodman DW (2012) Catal Today 181:75
He JJ, Yoneyama Y, Xu B, Nishiyama N, Tsubaki N (2005) Langmuir 21:1699
Liu ZW, Li X, Asami K, Fujimoto K (2005) Catal Today 104:41
He JJ, Liu ZL, Yoneyama Y, Nishiyama N, Tsubaki N (2006) Chem Eur J 12:8296
Yang GH, He JJ, Zhang Y, Yoneyama Y, Tan YS, Han YZ, Vitidsant T, Tsubaki N (2008) Energy Fuels 22:1463
Trépanier M, Dalai AK, Abatzoglou N (2010) Appl Catal A 374:79
Graf C, Vossen DLJ, Imhof A, Blaaderen AV (2003) Langmuir 19:6693
Graf C, Dembski S, Hofmann A, Rühl E (2006) Langmuir 22:5604
Song H, Rioux RM, Hoefeimeyer JD, Komor R, Niesz K, Grass M, Yang PD, Somorjai GA (2006) J Am Chem Soc 128:3027
Li KT, Hsu MH, Wang I (2008) Catal Commun 9:2257
Lee CL, Wu RB, Syu CM (2009) Electrochem Commun 11:270
Xie RY, Li DB, Hou B, Wang JG, Jia LT, Sun YH (2011) Catal Commun 12:380
Guo XH, Deng YH, Gu D, Che RC, Zhao DY (2009) J Mater Chem 19:6706
Zhang YJ, Lu JJ (2008) Cryst Growth Des 8:2101
Lin KJ, Chen LJ, Prasad MR, Cheng CY (2004) Adv Mater 16:1845
Meng YD, Chen DR, Jiao XL (2006) J Phys Chem B 110:15212
Yang CQ, Wang G, Lu ZY, Sun J, Zhuang JQ, Yang WS (2000) J Mater Chem 15:4252
Salgueirino-Maceira V, Correa-Duarte MA (2006) J Mater Chem 16:3593
Kobayashi Y, Horie M, Konno M, Rodriguez-González B, Liz-Marzán LM (2003) J Phys Chem B 107:7420
Fu WY, Yang HB, Chang LX, Li MH, Bala H, Yu QJ, Zou GT (2005) Colloids Surf A 262:71
Eggeman AS, Petford-Long AK, Dobson PJ, Wiggins J, Bromwich T, Dunin-Borkowski R, Kasama T (2006) J Magn Magn Mater 301:336
Rosynek MP, Polansky CA (1991) Appl Catal 73:97
Khodakov AY, Becharay R, Griboval-Constant A (2003) Appl Catal A 254:273
Bechara R, Balloy D, Vanhove D (2001) Appl Catal A 207:343
Reuel RC, Batholomew CH (1984) J Catal 84:78
Tsubaki N, Sun SL, Fujimoto K (2001) J Catal 199:236
Okabe K, Li XH, Wei MD, Arakawa H (2004) Catal Today 89:431
Acknowledgments
Project supported by the National Natural Science Foundation for Young Scientists of China (Grant No. B010204).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Xie, R., Wang, C., Xia, L. et al. Controlled Preparation of Co3O4@porous-SiO2 Nanocomposites for Fischer–Tropsch Synthesis. Catal Lett 144, 516–523 (2014). https://doi.org/10.1007/s10562-013-1187-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-013-1187-z