
Abstract
Gold deposition on Hombikat UV100 was found to negatively affect the activity of this Anatase catalyst in selective photo-oxidation of cyclohexane. By ammonia TPD and DRIFT spectroscopy it was determined that the Au deposition procedure leads to a significant decrease in OH-group density (mol m−2 BET) on Hombikat, suggesting that the amount of surface OH-groups, rather than the presence or absence of Au, is determining the catalytic performance. The importance of surface OH-groups was demonstrated by comparing the performance of Hombikat (with and without Au deposition) to surface propoxylated TiO2, synthesized by a sol–gel method from titanium (IV) isopropoxide. The effect of the deposition recipe of noble metals on the surface composition of TiO2 should thus be taken into account in evaluating and explaining photocatalytic performance of TiO2 modified by noble metals (Au), in particular in non-aqueous phase reactions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Liquid phase oxidation of cyclohexane is an important reaction for the conversion of cyclohexane via cyclohexanone in caprolactam, which is the monomer for nylon-6 production. The current commercial process is very energetically intensive, and several studies have focused on the direct use of photon energy to selectively oxidize cyclohexane at room temperature using a photocatalyst [1–11]. Although a high ketone over alcohol selectivity is obtained in photocatalytic cyclohexane oxidation, a low conversion (quantum efficiency) and rapid catalyst deactivation prevent application in practice. In order to overcome these problems several improvements can be made in catalyst, reactor [12], and process design [13]. Of the many options to enhance the efficiency of TiO2 photocatalyst, noble metal deposition (Pt, Pd) has been shown to be effective in many occasions. Various studies show that metal deposition enhances the photocatalytic activity of TiO2 in photo degradation reactions in the aqueous phase [14–16], as well as in gas phase applications [17, 18]. Besides modification by Pt or Pd, modification of Titania by Au has recently been reported to enhance performance in photocatalytic applications involving waste water treatment [19–22]. Most of these studies attribute the enhancement in activity to a decreasing charge carrier recombination velocity [19–21, 23, 24]: a result of localization of electrons on the metal particles.
To the best of our knowledge, the effect of Au on the performance of TiO2 in the selective photocatalytic oxidation of cyclohexane has not been previously investigated. We will demonstrate that the Au deposition procedure causes dramatic changes in TiO2 surface composition (OH-group population), which we evaluate to be dominant in controlling photocatalyst performance in cyclohexane oxidation, rather than the presence or absence of Au.
2 Experimental
2.1 Catalyst Preparation
Hombikat UV100 TiO2 (Sachtleben GmbH), was used as received. Hombikat is 100% anatase as determined by XRD, and has a surface area of 337 m2 g−1 with a primary particle size of 5 nm (determined by Scherrer’s equation), and a mean agglomerate size in cyclohexane after ultrasonification of ±3 μm [4]. Hombikat was used to prepare a Au–TiO2 photo-catalyst by the deposition precipitation method described by Yu et al. [25], aiming at a Au loading of 1 wt%. An excess amount of HAuCl4 was dissolved in 230 mL of deionised water, yielding a concentration of 1.46 × 10−3 M (HAuCl4 3H2O, Merck). The pH of the gold solution was adjusted to 6 with a 0.1 M NaOH aqueous solution. Then 2 g Hombikat was added and the suspension stirred for 1 h, at 80 °C. This was followed by filtration and washing with 500 mL of deionised water. The obtained catalyst was dried at 120 °C, and used without calcination. Throughout the paper Hombikat will be identified by H, and after Au deposition with Au–H. With the objective to test the effect of the preparation conditions on the performance of Hombikat, a reference material (HT HCl) was prepared using HCl in comparable Cl-amounts to the HAuCl4 solution. Again the pH of the solution was adjusted to 6 with a 0.1 M NaOH aqueous solution, followed by Hombikat addition, treatment at 80 °C for 1 h, filtration, washing and drying at 120 °C.
To further evaluate the effect of Au modification and the importance of surface OH-groups in TiO2 catalyzed photo-oxidation of cyclohexane, propoxylated TiO2 was synthesized by a sol–gel method. In the preparation procedure, 20 mL of Titanium (IV) isopropoxide was placed in a beaker and 160 mL of propan-2-ol added. The solution was placed in an ice bath and vigorously stirred. De-ionized water (pre-adjusted to pH 3 using dilute nitric acid) was added to the solution dropwise until complete precipitation occurred. The gel formed was treated in a water bath at 353 K for 2 h under reflux, allowed to cool, filtered, and dried at 393 K overnight. The material was then ground, screened to less than 107 μm diameter and calcined in air at 673 K for 4 h applying a heating rate of 2K min−1 [26]. A solution of 4 mL HAuCl4 (Au concentration of 50 g/L) in 130 mL of high purity water was heated to 343 K and its pH adjusted to 7.5 using a 2 M Na2CO3 solution. 10 g of support was then added to the solution and aged for 60 min with continuous stirring at 400 rpm. During the ageing period the temperature of the slurry was maintained at 343 K and the pH at 7.5, using 2 M Na2CO3 or 0.5 M HNO3. At the end of the ageing time, the slurry was filtered and washed five times; each by re-slurrying in 335 mL of high purity water for 5 min followed by filtration. The resulting material was dried at 393 K overnight, ground and screened to less than 107 μm diameter. These two catalysts are referred to as SG (the bare propoxylated TiO2), and AuSG after Au deposition. Please note that the Au deposition pH used for the Au–SG and Au–H catalysts was in the pH range of 6–8, where Au morphology is expected to be similar [27].
2.2 Catalyst Characterization
Powder X-Ray diffraction (XRD) analysis was performed on a Phillips PW 1840 diffractometer equipped with a graphite monochromator using Cu–Ka radiation (λ = 0.1541 nm). The Scherrer equation was used to calculate the crystal particle size of the samples.
XRF analyses were performed with a Philips PW2400 X-ray fluorescence spectrometer (XRF). The data evaluation was performed with the program Uniquant 5.49.
UV/Vis spectra were recorded on a Perkin–Elmer Lambda 900 spectrometer equipped with an integrating sphere (Labsphere) on porous films of the catalysts, deposited by solvent (water) evaporation under vacuum, onto a 1 mm thick quartz plate.
Temperature-programmed desorption of ammonia (NH3–TPD) was carried out on a Micromeritics TPR/TPD 2900 apparatus equipped with a thermal conductivity detector (TCD). Approximately 25 mg of each sample was pretreated at 398K and rapidly cooled to 373K followed by loading with ammonia applying a flow of 30 mL min−1 for about 1 h. A He flow of 30 mL min−1 was applied to remove weakly adsorbed NH3. A linear temperature program was started (373–873 K at 10K min−1), and the desorbed amount of ammonia was analyzed by the TCD. The TPD spectra were used to analyze the amount of acidic hydroxyl groups present in the catalyst samples.
Nitrogen adsorption and desorption isotherms were recorded on a QuantaChrome Autosorb-6B at 77 K. Samples were previously evacuated at 623 K for 16 h (at a ramp rate of 10K min−1). The BJH model was used to calculate the inter-particle pore size distribution from the adsorption branch, and the BET method was used to calculate the surface area (S BET).
Transmission electron microscopy (TEM) was performed using a Philips CM30UT electron microscope with a FEG (field emission gun) as the source of electrons operated at 300 kV. Samples were mounted on a Quantifoil microgrid carbon polymer supported on a copper grid.
IR absorption spectra of the solid samples were recorded using a Bruker IFS66 spectrometer with a DTGS detector and equipped with a Spectratech Diffuse Reflectance Accessory, including a high temperature cell. Water was removed from the catalyst surface to facilitate the characterization of the OH-group composition by recording the spectra at 120 °C after equilibration for 1 h in He flow (20 mL min−1). All spectra were obtained by collecting 128 scans with 4 cm−1 resolution, against a KBr background (also measured at 120 °C in flowing He).
2.3 Photo-activity Measurements
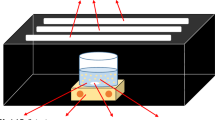
To evaluate catalyst performance in the selective oxidation of cyclohexane, reactions were carried out in a Top Illumination Reactor [4]. The catalysts were dried for 2 h at 120 °C to remove adsorbed water and impurities. In a typical experiment 100 mL of cyclohexane containing 1 g L−1 of catalyst was used (slurry system). The suspension was illuminated from the top of the reactor through a Pyrex window that cuts off the highly energetic UV radiation [4]. A high pressure mercury lamp of 50 W was used (HBO50W from ZEISS). The light intensity of the lamp used in the wavelength absorption range of TiO2 (275–388 nm) is 5.5 W m−2 entering the suspension. Air, dried over Molsieve 3 A (Acros Organics) and pre-saturated with cyclohexane, was bubbled through the TiO2 suspension at a rate of 30 mL min−1. During the reaction, liquid was withdrawn and analyzed by GC. Organic compounds were quantatively analyzed twice using a gas chromatograph with a flame ionization detector (Chrompack, CPwax52CB). Hexadecane was used as an internal standard.
3 Results
3.1 Catalyst Characterization
Figure 1 shows the NH3–TPD profiles obtained for each TiO2 sample. The profile of the Hombikat catalyst is in good agreement with the profile obtained in a previous study in our laboratory [4]. By integration of the TPD curves over the whole temperature range (375–775K), the total amount of surface OH-groups was determined as indicated in Table 1. The Table also contains the corresponding pH values used in the last step of the preparation procedure, and the surface area as determined by the N2-BET method. Since Hombikat (H) is a commercial catalyst, the pH of preparation is not known. The Au–H sample contains only half the amount of hydroxyl groups as compared to commercial Hombikat, while the surface area did not change significantly. Figure 1 shows that the decrease in amount of OH-groups was due to a decrease of both strong and weak acidic sites, suggesting that Au deposition leads to a non-selective loss of surface OH-groups. For the SG catalyst, a high surface area was measured, combined with a relatively small amount of surface OH-groups. The reason for this observation will be discussed later by comparing the DRIFT spectra of the various samples.

NH3 –TPD profiles for the four catalysts studied with corresponding illustration of the ammonia coordination: weakly adsorbed ammonia at low temperatures, and strongly adsorbed at high temperatures
Regarding the Au–SG catalyst, the N2-physisorption results indicate a surface area similar to Au–H, with a higher amount of surface OH-groups being present as deduced from NH3–TPD profiles. In the case of the TiO2 prepared by the sol–gel method, Au deposition apparently leads to an increase of the amount of surface OH-groups, as shown in Table 1. Summarizing, using the preparation variables applied here, a diverse set of TiO2 catalysts was obtained, containing a wide range of OH-group densities (mmol g−1) and surface areas.
To further analyze the surface composition of the catalysts, we show in Fig. 2a the DRIFT spectra of SG TiO2 before and after Au deposition. In the spectrum of SG, absorptions at 2,962, 2,934 and 2,872 cm−1 are present, which correlate well with C–H stretching vibrations of Titanium (IV) isopropoxide. Absorptions at 1,465 and 1,372 cm−1 are the corresponding C–H bending modes. Clearly the spectrum reveals that not all the Titanium (IV) isopropoxide is hydrolyzed in the preparation procedure. For the Au–SG material the isopropoxide bands are no longer present. Upon Au deposition, the SG material was exposed to a pH of 7.5 in a Na2CO3 solution at 70 °C, which is favorable for the hydrolysis of isopropoxide. By comparing the intensity of the spectra in the range of 3,300–3,800 cm−1, where OH-groups are expected [28], clearly the intensity after Au deposition on SG is higher, confirming the hydrolysis of isopropoxide groups, also in agreement with the NH3–TPD data (Table 1). Completing hydrolysis of isopropoxide also makes SG more hydophylic, as is evident from the intensity differences in the broad stretching modes of adsorbed water (remaining at 120 °C), between 3,600 and 2,500 cm−1, and the corresponding bending modes centered at 1,630 cm−1.

IR spectra of the samples studied: a SG and Au–SG, b H and Au–H
For the H and Au–H materials, the DRIFT spectra are shown in Fig. 2b. The OH- and water associated bands are less intense after Au deposition, in agreement with data presented in Table 1. The lower amount of water and Ti–OH–groups found after Au deposition, is the result of the pH of the applied solution, which was at 6: a condition known to lower the amount of acidic OH sites [29].
TEM pictures of the samples Au–SG and Au–H are shown in Fig. 3. The dark spots in the micrographs were confirmed to be gold by means of EDX analysis. Figure 3a shows some Au particles of large dimensions (20–60 nm), and a large fraction of about 70% of particles in the size range of 4–6 nm. For the Au–SG sample, Fig. 3b, big Au particles as in the case of the Au–H were not observed, and around 90% of the particle sizes were in the range of 5–10 nm.

TEM pictures of the Au catalysts used in this study. a Au–H b Au–SG
The metal loading of the catalysts was determined by XRF. Although a loading of 1 wt% was aimed for, for the Au–SG sample the obtained metal loading was 0.5 wt% and for Au–H, 2.6 wt%. Apparently the different surface composition of Hombikat relative to P25, for which the synthesis procedure was optimized [25], resulted in an excess of Au to be deposited. The relatively small Au loading of Au–SG is most likely a consequence of the large degree of propoxylation of the SG-Anatase, limiting the initially present number of OH-anchoring sites for the Au precursor species.
Absorbance spectra for the two Au catalysts are shown in Fig. 4. The absorbance fraction, F A, is the relative amount of photons actually attenuated by the catalyst layer. The absorption edge of Au–H shows a slight blue shift from 365 to 360 nm. This is related to the crystalline particle size, which is lower in Hombikat, as compared to TiO2 prepared by the sol–gel method. This was confirmed by XRD data, from which by application of the Scherrer equation, crystal sizes of ~8 nm for Au–SG and ~6 nm for Au–H were determined. Around 550 nm an absorption band can be observed which is due to the plasmon absorption of metallic Gold. The absorption in this area showed to be more accentuated in the Au–H catalyst, as compared to the Au–SG catalyst, in particular considering the higher loading of the Au–H catalyst. This again indicates a difference in particle size, and possibly also in the reduction degree of the Au present in the catalytic materials, which requires further analyses.

UV/Vis spectra of the Au samples studied, as measured with an integrating sphere under ambient conditions
3.2 Photocatalytic Oxidation of Cyclohexane
Previous work in our group [4] showed that the main products of the cyclohexane selective photocatalytic oxidation are cyclohexanone and cyclohexanol, with a ketone to alcohol selectivity higher than 98%. Cyclohexane conversion is very low, <2% and CO2 contributions, due to complete oxidation, are not significant (cyclohexanone and cyclohexanol are reported to be produced with selectivities >95% relative to CO2) [4]. While radical chemistry in solution can provide for other products, in view of the applied wavelengths (>275 nm) this is not expected to occur, as confirmed by our GC analyses.
Cyclohexanone formation upon photo-oxidation of cyclohexane is shown for the 4 applied catalysts in Fig. 5. The shape of the profiles is the result of catalyst deactivation, induced by carbonate and carboxylate formation on the catalyst surface [30]. The amount of cyclohexanone formed over the different catalysts follows the order H > Au–SG > Au–H > SG. After the Au deposition procedure, the activity of the SG catalyst is apparently largely enhanced, whereas an opposite effect is observed for the H catalyst.

a Cyclohexanone production profiles for H, Au–H, Au–SG and SG. The cyclohexanone value reaches a plateau after prolonged time of reaction, as a result of loss of activity of the catalyst. The inset shows the final cyclohexanol amounts obtained (at t = 420 min). b Cylohexanone profiles for H, HT HCl, and Au–H. The inset shows the final cyclohexanol amounts obtained (at t = 120 min)
In addition to cyclohexanone, the inset of Fig. 5a shows the amount of cyclohexanol produced for the materials under study. Albeit small, a different trend in catalyst behaviour for cyclohexanol production as compared to cyclohexanone was found, the production order being Au–H > Au–SG > H > SG. Cyclohexanol formation was below the GC detection limit for the SG material. Figure 5b shows a comparison in cyclohexanone production of the H and AuH with the reference catalyst HT HCl, prepared in the exact same way as Au–H, but substituting HAuCl4 for HCl. Please note that the time scales of (Fig. 5a, b) are different. Figure 5b clearly shows that the preparation conditions largely affect the performance of H, irrespective of the presence of Au: the performance of Au–H and HT HCl is comparable. The inset of Fig. 5b shows the one order of magnitude smaller cyclohexanol production after 120 min of reaction: in agreement with Fig. 5a, Au has a positive affect on cyclohexanol production.
Figure 6 shows a plot of the estimated initial cycohexanone production rate using the amount produced at t = 40, Fig. 5 as a function of the amount of OH-groups present on the TiO2 surface, as determined by the TPD experiment. An exponential correlation is obtained.

Dependency of the initial total product formation rate on the initial amount of surface hydroxyl groups present on the catalyst surface
4 Discussion
4.1 Effect of the Preparation Procedure on Catalyst Constitution
Various research groups have reported a positive effect on photocatalytic performance of noble metal (Pt, Pd, Au) deposition on the surface of TiO2 catalysts. Generally the enhancement in activity is attributed to an increased rate of charge separation and/or decreasing charge carrier recombination velocity [19, 21, 23]. The effect of the deposition of noble metals on the surface chemical properties of the catalyst (semiconductor oxide) is typically not taken into consideration. In the present study, it is apparent that the Au deposition procedure can dramatically affect the amount of surface OH-groups as is shown in Table 1 and evident from the DRIFT spectra in Fig. 2. The amount of Au is not enough to account for the large differences in the number of hydroxyl groups on the TiO2 surfaces. Alternatively, during the Au deposition procedure the TiO2 surface is exposed to conditions which induce the significant surface changes. Two surface chemical processes have been identified in the present study: (a) the hydrolysis of remaining Ti-isopropoxide groups, explaining the significant increase in the amount surface OH-groups for the SG catalyst upon Au deposition and (b) solution induced removal of acid sites in the case of the H catalyst. In the latter case, the control experiments, shown in Fig. 5b, clearly indicate that the acid treatment of the catalyst upon Au deposition is the cause of the reduced activity of Au–H compared to H. The nature of the OH-groups (Acid sites) on the catalyst surface will be discussed on the basis of the NH3–TPD spectra (Fig. 1) in the following.
It is believed that desorbed ammonia originates from ammonium ions (NH4 +) and NH3 species molecularly coordinated to Lewis acid sites [4, 31]. If the NH3–TPD profiles shown in Fig. 1 are deconvoluted they consist of two overlapping peaks which correspond to two types of adsorption strengths. The low-temperature peak (363–543K) characterizes desorption states of ammonium ions and weakly coordinated ammonia (Fig. 1). The high temperature peak (598–793 K) is related to strongly coordinated ammonia at Lewis acid sites. All sites appear affected by the Au deposition procedure, leading to a non-selective change in the amount of acid sites [31].
In the following we will discuss why the hydroxyl groups are so important in determining the catalyst performance in photo-oxidation of cyclohexane, as is evident from the trend shown in Fig. 6.
4.2 The Role of OH-groups in Photocatalytic Reactions in Organic Media
Surface OH-groups (\( {\text{OH}}_{{_{\text{ads}} }}^{ - } \)) have been reported in the literature as being of great importance for the studied reaction [32] and in the degradation of pollutants [33, 34]. In Fig. 7, the initial steps of the selective photo-oxidation of cyclohexane are depicted, including cyclohexyl radical formation. In the beginning of the photo-process an electron-hole (e−, h+) pair is formed by light excitation of the semiconductor, TiO2. Once the electron and hole reach the surface of the catalyst, the hole is trapped by either a surface hydroxyl group (\( {\text{OH}}_{{_{\text{ads}} }}^{ - } \)) or adsorbed water (H2Oads), to form surface hydroxyl radicals. These surface hydroxyl radicals react with surface adsorbed cyclohexane, yielding a cyclohexyl radical. The electron interacts with the Ti(IV) center, that is reduced and further reacts with molecular oxygen to superoxide anions, O2 −. The cyclohexyl radical combines with the superoxide anion, forming oxygenated products (i.e., cyclohexanone) [30]. Reactions involving free OH radicals are not considered here, since desorption of OH radicals into the organic solution is unlikely [35].

Simplified scheme of TiO2 induced photocatalytic cyclohexane oxidation
The message of Fig. 7 is that an important step in the formation of cyclohexanone is the reaction of cyclohexane with a OH-radical originating from a surface OH-group. Since the reaction is performed in a (dried) cyclohexane medium, the initial reaction rate is thus a strong function of the amount of hydroxyl groups initially present on the catalyst surface. The importance of surface OH-groups is also illustrated by the rapid deactivation of the catalyst systems. Products are accumulating on the surface and interact with surface OH-groups, thus limiting the reaction rate. It should be mentioned that since the samples are taken from the liquid, the amount of product measured is a lower limit of the amount actually formed [12].
4.3 A Role of Gold?
Clearly, for the initial reaction rate in cyclohexane oxidation the initial concentration of surface OH-groups is much more important than the presence or absence of Au. However, a positive effect of gold has been observed in the literature for aqueous phase reactions. It is thus important to distinguish photo-catalysis performed in an aqueous medium (e.g., wastewater treatment) from that in organic medium (e.g., cyclohexane photo-oxidation) in evaluating the role of Au. Considering a wastewater degradation type reaction, water and hydroxyl groups are present at high concentration on the catalyst surface, and less likely to be limiting the reaction. Photo-generated holes are constantly being trapped by the abundant presence of surface water and OH-groups, and thus the amount of photo-generated holes might be limiting the reaction. Certainly, improving charge separation is needed in this case for a better catalyst performance. This justifies changing the opto-electronic properties of the catalyst, and most likely explains the observed effects of Au on photo-catalytic performance in water phase reactions.
In the present study, Au deposition enhances the amount of cyclohexanol formed, the amount being still significantly smaller compared to cyclohexanone (see inset in Fig 5a, b). This phenomenon requires further investigation, but might indicate that Au affects the decomposition chemistry of cyclohexyl hydroperoxide, which is often considered the intermediate in the photon induced formation of cyclohexanone [12]. The electron trapping effect of Au might also induce a higher amount of superoxide anion (O2 −, Fig. 7), leading to the observed selectivity changes. The opto-electronic changes that Au induces in a semiconductor like TiO2 (anatase and/or rutile phase), are currently further analyzed in our laboratory, to validate the claims made in the literature and to support the above hypothesis for enhanced cyclohexanol formation.
In order to improve catalytic performance in selective oxidation reactions such as the one studied, increasing the amount of surface OH-groups on the semiconductor surface should be established. This could be achieved, e.g., by pre-treatment of the catalysts by (mild) steaming.
5 Conclusions
Gold deposition on Hombikat negatively affects the activity of this Anatase catalyst in selective photo-oxidation of cyclohexane, explained by a significant modification of the surface OH-population of the catalyst induced by the Au preparation procedure as determined by ammonia TPD and DRIFT spectroscopy. When a photoreaction is performed in an organic medium, the initial amount of surface hydroxyl groups is apparently largely affecting the initial rate. This was confirmed by the experiments performed with a surface propoxylated Sol–Gel catalyst, before and after Au deposition. Surface modifications of the TiO2 as a result of the preparation procedure, and the deposition recipe of noble metals should thus be taken into account in evaluating and explaining photocatalytic performance of TiO2, in particular in non-aqueous phase reactions.
References
Almquist CB, Biswas P (2001) Appl Catal A-Gen 214(2):259
Boarini P, Carassiti V, Maldotti A, Amadelli R (1998) Langmuir 14(8):2080
Brusa MA, Di Iorio Y, Churio MS, Grela MA (2007) J Mol Catal A-Chem 268(1–2):29
Du P, Moulijn JA, Mul G (2006) J Catal 238(2):342
Giannotti C, Legreneur S, Watts O (1983) Tetrahedron Lett 24(46):5071
Herrmann JM, Mu W, Pichat P (1991) Stud Surface Sci Catal 59:405
Li XY, Chen GH, Po-Lock Y, Kutal C (2003) J Chem Technol Biotechnol 78(12):1246
Li XY, Quan X, Kutal C (2004) Scr Mater 50(4):499
Lu GX, Gao HX, Suo JH, Li SB (1994) J Chem Soc-Chem Commun 21:2423
Mu W, Herrmann JM, Pichat P (1989) Catal Lett 3(1):73
Su BT, He YF, Li XY, Lin EH, Li QL (1997) Indian J Chem Sect A-Inorg Bio-Inorg Phys Theor & Anal Chem 36(9):785
Du P, Cameiro JT, Moulijn JA, Mul G (2008) Appl Catal A-Gen 334(1–2):119
Van Gerven T, Mul G, Moulijn J, Stankiewicz A (2008) Chem Eng Process 46(9):781
Augugliaro V, Litter M, Palmisano L, Soria J (2006) J Photochem Photobiol C-Photochem Rev 7(4):127
Bahnemann D (2004) Sol Energy 77(5):445
Dillert R, Cassano AE, Goslich R, Bahnemann D (1999) Catal Today 54(2–3):267
Arana J, Dona-Rodriguez JM, Cabo CGI, Gonzalez-Diaz O, Herrera-Melian JA, Perez-Pena J (2004) Appl Catal B-Environ 53(4):221
Peral J, Domenech X, Ollis DF (1997) J Chem Technol Biotechnol 70(2):117
Li HX, Bian ZF, Zhu J, Huo YN, Li H, Lu YF (2007) J Am Chem Soc 129(15):4538
Orlov A, Jefferson DA, Macleod N, Lambert RM (2004) Catal Lett 92(1–2):41
Sonawane RS, Dongare MK (2006) J Mol Catal A-Chem 243(1):68
Chan SC, Barteau MA (2005) Langmuir 21(12):5588
Sakthivel S, Shankar MV, Palanichamy M, Arabindoo B, Bahnemann DW, Murugesan V (2004) Water Res 38(13):3001
Sreethawong T, Yoshikawa S (2005) Catal Commun 6(10):661
Yu WY, Lee WS, Yang CP, Wan BZ (2007) J Chin Inst Chem Eng 38(2):151
Mohapatra P, Moma J, Parida KM, Jordaan WA, Scurrell MS (2007) Chem Commun 10:1044
Haruta M (1997) Catal Today 36(1):153
Panayotov DA, Yates JT (2005) Chem Phys Lett 410(1–3):11
Kozlov D, Bavykin D, Savinov E (2003) Catal Lett 86(4):169
Almeida AR, Moulijn JA, Mul G (2008) J Phys Chem C 112(5):1552
Popova NM, Sokolova LA, Marchenko EA, Bobrova LN (1998) React Kinet Catal Lett 65(2):371
Gonzalez MA, Howell SG, Sikdar SK (1999) J Catal 183(1):159
Turchi CS, Ollis DF (1990) J Catal 122(1):178
Okamoto K, Yamamoto Y, Tanaka H, Tanaka M, Itaya A (1985) Bull Chem Soc Jpn 58(7):2015
Lawless D, Serpone N, Meisel D (1991) J Phys Chem 95(13):5166
Acknowledgments
We would like to acknowledge the X-ray facilities of the 3ME Department, Faculty of Applied Sciences, of the Delft University of Technology for the X-Ray analyses. Dr. ing. V. C. L. Butselaar of DCT/NCHREM, Delft University of Technology, is acknowledged for performing the electron microscopy investigations, and Sander Brouwer for performing the NH3–TPD and N2 physisorption measurements. STW (VIDI Project DPC.7065), is gratefully acknowledged for financial support.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Carneiro, J.T., Yang, CC., Moma, J.A. et al. How Gold Deposition Affects Anatase Performance in the Photo-catalytic Oxidation of Cyclohexane. Catal Lett 129, 12–19 (2009). https://doi.org/10.1007/s10562-008-9801-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-008-9801-1