
Abstract
Purpose
Acute myocardial infarction (AMI) is a leading cause of mortality. Neutrophils penetrate injured heart tissue during AMI or ischemia–reperfusion (I/R) injury and produce inflammatory factors, chemokines, and extracellular traps that exacerbate heart injury. Inhibition of the TRAIL-DR5 pathway has been demonstrated to alleviate cardiac ischemia–reperfusion injury in a leukocyte-dependent manner. However, it remains unknown whether TRAIL-DR5 signaling is involved in regulating neutrophil extracellular traps (NETs) release.
Methods
This study used various models to examine the effects of activating the TRAIL-DR5 pathway with soluble mouse TRAIL protein and inhibiting the TRAIL-DR5 signaling pathway using DR5 knockout mice or mDR5-Fc fusion protein on NETs formation and cardiac injury. The models used included a co-culture model involving bone marrow-derived neutrophils and primary cardiomyocytes and a model of myocardial I/R in mice.
Results
NETs formation is suppressed by TRAIL-DR5 signaling pathway inhibition, which can lessen cardiac I/R injury. This intervention reduces the release of adhesion molecules and chemokines, resulting in decreased neutrophil infiltration and inhibiting NETs production by downregulating PAD4 in neutrophils.
Conclusion
This work clarifies how the TRAIL-DR5 signaling pathway regulates the neutrophil response during myocardial I/R damage, thereby providing a scientific basis for therapeutic intervention targeting the TRAIL-DR5 signaling pathway in myocardial infarction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute myocardial infarction is now the primary cause of death from cardiovascular illness, which poses a serious hazard to human health due to its high prevalence [1, 2]. Current treatment options for acute myocardial infarction include thrombolysis, bypass grafting, and percutaneous coronary intervention (PCI) [3, 4]. However, these interventions may induce cardiac I/R injury [5]. Therefore, the development of effective therapeutic procedures that may overcome the shortcomings of present treatment modalities is crucial.
Cardiac I/R injury is a complex process wherein neutrophils recruit more inflammatory cells and release pathogenic proteases, including matrix metalloproteinases, chemotactic factors, and myeloperoxidase (MPO), which exacerbate cardiac injury in the infarcted myocardium [6, 7]. Furthermore, it has been demonstrated that NETs formation, characterized by the release of web-like structures during neutrophil cell death, also contributes to the inflammatory damage process in myocardial infarction [8, 9]. NETs are complex fibrous structures consisting of chromatin DNA derived from neutrophils and a diverse range of intracellular granular proteins, which play a crucial role in capturing and eliminating pathogens, thereby exerting potent anti-infective effects [10]. However, NETs can promote platelet adhesion and aggregation, providing a supportive structure for thrombus formation [11]. They also provoke acute myocardial infarction by increasing fibrin deposition and enhancing red blood cell agglutination, which causes thrombus development [12, 13]. These results imply that NETs are essential to the pathophysiology of myocardial I/R injury and that treatments that specifically target NETs may be effective.
TRAIL and its receptor DR5 are essential components of the pathophysiology of myocardial infarction, including the induction of cardiomyocyte apoptosis and the coordination of immune cell recruitment and activation, both of which lead to cardiac damage [14,15,16]. However, it remains unclear whether TRAIL can induce the release of NETs to exacerbate cardiac I/R injury. We used mDR5-Fc protein and DR5 knockout mice to further understand the regulatory function and mechanism of the TRAIL-DR5 signaling pathway in NET production following cardiac IR injury.
Materials and Methods
Animals
The C57BL/6N male mice, obtained from Vital River Company, were acclimated to the environment for one week prior to the study. The DR5 Knockout mice were generously provided by Professor Yinming Liang (Xinxiang Medical University), and bred in the animal facility of Joint National Laboratory for Antibody Drug Engineering. Mice were housed in a specific pathogen-free (SPF) environment, with up to five animals per cage, following a 12-h light/dark cycle, maintaining relative humidity between 55–70%, temperature conditions at 22–25 °C, and provided ad libitum access to food and water.
For the mice cardiac I/R injury model, anesthesia was induced using 2% isoflurane while ensuring proper disinfection with iodine solution and alcohol. A thoracic incision between the ribs allowed for exposure of the heart. Ligation of the left anterior descending coronary artery (LAD) was accomplished utilizing a 6–0 suture for a period lasting 40 min, after which reperfusion commenced upon release of this ligation. Subsequently, muscle and skin were sutured using a 4–0 suture for closure.
For Evan’s blue and TTC double staining, mice were killed 24 h after I/R injury. The blood vessels at the ligation site were ligated using a 4–0 suture and followed by Evans blue injection through the aorta. The hearts were excised and cryopreserved at -80 °C. The hearts under the ligation site were cut into five slices for TTC staining, and quantification of the infarct (white), risk (red), and non-infarct (blue) areas were analyzed using ImageJ software.
Preparation of Mouse DR5-Fc Fusion Protein
The extracellular segment of mouse DR5 and the Fc region of mouse IgG1 were constructed into the pcDNA3.1 vector and subsequently transfected into 293F cells. The mDR5-Fc protein was purified from 293F culture supernatant using protein A affinity chromatography, followed by buffer exchange into PBS. Subsequently, the purified protein was filtered through a 0.22 μm membrane and quantified using the BCA assay kit. Purity assessment was performed via SDS-PAGE Coomassie Brilliant Blue staining.
Neutrophil Depletion
To deplete neutrophils in mice, an anti-Ly6G antibody (Biolegend, 127,649) or isotype antibody (Biolegend, 400,565) was administered i.v. 0.2 mg per mouse, and the depletion efficiency was assessed using flow cytometry.
Isolation and Culture of Primary Cells
For isolation of neutrophils from mouse bone marrow, mice were euthanized by cervical dislocation. The bone marrow was flushed into 1640 medium and filtered through a 40 μm sieve. Subsequently, cells were centrifuged at room temperature for 5 min at 500 g and resuspended in 2 mL of supplemented 1640 medium containing 10% FBS. The cell suspension was then transferred to a new 15 mL centrifuge tube containing Percoll solutions with concentrations of 78% and 65%. Centrifugation was performed at a speed of 2500 rpm for 30 min. The cell layer located at the interface between the layers of Percoll solutions was carefully collected into another clean 15 mL centrifuge tube, followed by washing with PBS. Finally, purified neutrophils were obtained and cultured short-term in a complete growth medium containing 10% FBS supplemented with 1640 medium.
The isolation of cardiomyocytes was performed according to a previously described protocol [14]. Briefly, neonatal rat ventricular myocytes (NRVMs) were isolated from 2 d old Wistar rats by mincing the hearts and digesting in a C-tube containing enzymatic solution (Worthington Biochemical) using gentleMACS™ (Miltenyi Biotec). Cardiomyocytes were then collected through percoll centrifugation and cultured in a medium supplemented with 20% FBS (Gibco).
Co-Culture of Neutrophil Derived NETs and NRVM
Mouse bone marrow-derived neutrophils were stimulated with mTRAIL alone or in combination with mDR5-Fc, washed twice with culture medium, resuspended in 1640 medium supplemented with 10% FBS, and subsequently co-cultured with NRVM cells. After a duration of 24 h, the supernatant of the NRVM cell culture was collected to quantify LDH levels, while NRVM apoptosis was assessed using a TUNEL assay.
Immunohistochemical
The left and right atria appendages were incised, and the left ventricle and right ventricle were flushed with 15–20 mL of heparinized PBS thoroughly to remove any residual blood. Subsequently, the hearts were fixed in a solution containing 4% paraformaldehyde.
After embedding, sectioning, dewaxing, and hydration of the tissue, endogenous peroxidase activity was blocked using hydrogen peroxide. The tissue slices were incubated 1 h with 5% BSA at room temperature and then overnight for primary antibody binding at 4 °C. Secondary antibodies were then incubated at room temperature for 1 h. Finally, the slices were analyzed using a microscope scanning system after chromogenic staining, hematoxylin counterstaining of nuclei, and sealing with a neutral resin.
Immunofluorescence
The tissue sections underwent deparaffinization, hydration, and antigen retrieval and were subsequently incubated in 1% Triton X-100 PBS. Following blocking with 5% BSA, the slices were then incubated overnight at 4 °C with the primary antibody. Subsequently, the secondary antibody was applied, and cellular nuclei were stained with DAPI. Finally, fluorescence microscopy was employed to capture images of the slides.
For TUNEL staining, the heart tissue sections were subjected to a TUNEL staining kit according to the manufacturer’s protocol and subsequently examined using fluorescence microscopy.
Western Blot
The mouse heart was lysed using RIPA lysis buffer supplemented with protease inhibitors and PMSF, followed by tissue homogenization. Subsequently, the lysate was incubated on ice for 30 min. After centrifugation, the supernatant was collected and subjected to BCA protein quantification. SDS-PAGE analysis was conducted, and the proteins were transferred onto a PVDF membrane. The membrane was then incubated with 5% skim milk and primary antibody overnight at 4 °C. The membrane was incubated with a secondary antibody and substrate for detection.
qPCR
RNA was extracted from mouse cardiac tissue using the TRIZOL method, followed by reverse transcription into cDNA. qPCR was performed and subsequent detection was carried out using QuantStudio 3. The primer sequences used for qPCR were as follows.
Tnfa AGGCGGTGCCTATGTCTCA, GGGAGGCCATTTGGGAACTTCT; Il1b TGCCACCTTTTGACAGTGATG, TGATGTGCTGCTGCGAGATT; Mpo AGTTGTGCTGAGCTGTATGGA, CGGCTGCTTGAAGTAAAACAGG; Cxcl15 TGGGTGAAGGCTACTGTTGG, AGCTTCATTGCCGGTGGAAA; Icam1 CCGCTACCATCACCGTGTATT, GGTGAGGTCCTTGCCTACTT; Itgam AAAGAACAACACACGCAGGC, CAGAACTGGTCGGAGGTTCC; Pad4 GGCTCATTCCCCTCACCATC, TTGTCAGAAACCCTGCACAC; Gapdh GGTGAAGGTCGGTGTGAACG, CTCGCTCCTGGAAGATGGTG.
Detection of Peripheral Blood and Plasma Samples
The quantification of plasma lactate dehydrogenase (LDH) and creatine kinase (CK) levels was conducted using biochemical instrumentation (IDEXX). For dsDNA detection, ELISA was performed according to the manufacturer’s protocol (Solarbio, P9740).
Flow Cytometry
After perfusion with PBS, the mouse heart was dissected to remove adherent tissues and the aorta [17]. The mouse heart was then weighed, minced, and digested in a digestion solution containing type II collagenase (Worthington Biochemical), neutral protease (Roche), and DNase I (D4513, Sigma-Aldrich). Subsequently, the digestion was neutralized with 2%FBS, and the digested cells were passed through a 70 μm sieve and lysed red blood cells. Anti-mouse CD16/32 antibody (eBioscience, 14–0161-85) was used for blocking at 4 °C followed by incubation with antibodies (CD45, eBioscience, 47–0451-82; CD11b, eBioscience, 63–0112-82; Ly6G, eBioscience, 46–9668-82) at 4 °C in the dark for 30 min. Finally, flow cytometry analysis was performed using Bio-Rad ZE5.
Quantification and Statistical Analysis
The data analysis in this study was performed using GraphPad Prism9. The results were presented as means ± SEM and subjected to statistical analysis using the unpaired t test or one-way ANOVA followed by Tukey’s post hoc test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.001).
Results
Cardiac I/R Induced NETs Formation and Release
We looked at the release of NETs in the hearts of mice after I/R injury to determine whether NETs are involved in cardiac I/R injury. Citrullinated histone H3 (citH3), an essential component of NET formation reflecting NET production, exhibited increased expression at 3 and 6 h post-reperfusion, which remained elevated even at 24 h post-reperfusion as determined by Western blotting analysis (Fig. 1a). ELISA analysis revealed elevated levels of double-stranded DNA (dsDNA) content in the plasma following reperfusion compared to the sham group (Fig. 1b). MPO is one of the components present on the DNA scaffold of NETs. Therefore, we assessed the number of MPO-positive cells in the heart tissues at various time points after reperfusion and observed a significant increase in MPO-positive cells 24 h post-reperfusion compared to the sham group (Fig. 1c, d). Neutrophils undergo an inflammatory cell death mechanism called NETosis. To explore whether early-stage NETs contribute to impaired cardiac recovery after I/R injury, we employed Ly6G antibody for neutrophil depletion. Flow cytometry analysis confirmed the effective depletion of neutrophils (Fig. 1e, f), while TTC/Evans blue dual staining showed a reduction by 35% in myocardial infarct area upon neutralizing neutrophils compared to control group mice subjected to cardiac I/R injury (Fig. 1g, h). On the basis of these data, neutrophils may migrate into injured tissues during the initial phases of cardiac I/R injury, releasing extracellular traps that harm the heart.

NETs were increased and involved in cardiac I/R injury. a The expression of citH3 in the hearts of mice was assessed by western blot analysis following sham surgery or 40 min of ischemia with subsequent reperfusion for the indicated periods. b Detection of dsDNA content in mouse plasma using ELISA assay following sham surgery or 40 min of ischemia with subsequent reperfusion for the indicated periods. c–d Representative IHC results (c) and statistical analysis (d) of MPO expression in mouse heart tissue following sham surgery or I40min/R24h. e–f Flow cytometry detection (e) and statistical analysis (f) of neutrophils clearance efficiency in the mice peripheral blood. g–h Representative results (g) and statistical (h) of TTC-Evans blue double stain heart sections after I40min/R24h treated with isotype or anti-Ly6G antibody for neutrophils depletion. AAR, area at risk (white, red); IS, infarction size (white); LV, left ventricle. Quantitative data are shown as means ± SEM. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001 as determined by a two-tailed t test (b, d, f, and h)
The Beneficial Effect of DR5 Knockout on Cardiac I/R Injury is Dependent on Neutrophil
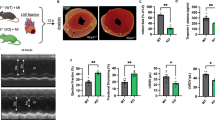
Our previous studies have demonstrated that blocking the TRAIL-DR5 signaling pathway with sDR5-Fc can attenuate cardiac I/R injury in monkeys, pigs, and rats [14]. To further elucidate the underlying mechanisms, we validated this effect in DR5 knockout mice using the I40min/R24h model. DR5 knockout mice exhibited a reduced infarct area (Fig. 2a, b), decreased levels of CK (Fig. 2c) and LDH (Fig. 2d) in plasma, fewer TUNEL-positive cells in cardiac tissue (Fig. 2e, f), as well as lower transcription levels of Il1b and Tnfa in cardiac tissue detected by qPCR compared to the wild-type group (Fig. 2g, h). These results suggest that DR5 knockout can lessen cardiac IR injury in rodents.

The protective effect against cardiac I/R injury in DR5 knockout mice relies on neutrophils. a–b Representative results (a) and statistical analysis (b) of TTC-Evans blue double stain heart sections following I40min/R24h in wild type (WT, n = 5) or DR5 knockout (DR5-KO, n = 5) mice. c–d Plasma concentrations of CK (c) and LDH (d) of WT or DR5-KO mice following sham or I 40 min/R 24 h were analyzed using the Catalyst Dx Chemistry Analyzer. e–f Immunofluorescence detection (e) and statistical analysis (f) of TUNEL in heart tissues of WT or DR5-KO mice following sham or I40min/R24h. g–h mRNA expression of Tnfa and Il1b in the hearts of sham or I/R mice as detected by quantitative PCR (n = 3 for each group). i–j Representative results (i) and statistical analysis (j) of TTC-Evans blue double stain heart sections following I40min/R24h in WT mice treated with isotype antibody (n = 6), DR5-KO mice treated with isotype antibody (n = 6) and DR5-KO mice treated with anti-Ly6G antibody for neutrophils depletion (n = 5). Quantitative data are shown as means ± SEM. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001 as determined by two-tailed t test (b–d, f–h, and j)
We employed a well-established paradigm of anti-Ly6G-induced neutrophil depletion to investigate whether neutrophil-derived NETs have a detrimental role during the early inflammatory phase of cardiac electrical resistance injury. Neutrophil depletion resulted in a reduced infarct area compared to the control group. However, no significant differences in infarction area were found between the WT or DR5-KO groups after neutrophil depletion (Fig. 2i, j), suggesting that knocking out DR5 can reduce cardiac I/R injury, which is neutrophil dependent.
Knockout of DR5 Reduces NETs Release During Cardiac IR Injury
NETs derived from neutrophils play an important role in the process of inflammation [18]. To investigate the impact of the TRAIL-DR5 pathway on NETs release in myocardial tissue and peripheral blood following cardiac IR injury, we collected the heart tissue and peripheral blood plasma. Western blot analysis revealed citH3 expression in all groups after I/R injury, with a higher level observed in the WT group compared to the DR5-KO group (Fig. 3a). ELISA analysis showed elevated levels of dsDNA content in plasma in the WT group and decreased dsDNA in DR5-KO mice after cardiac IR injury (Fig. 3b). Immunofluorescence results demonstrated a reduced presence of MPO-positive cells and citH3-positive cells in cardiac tissues (Fig. 3c, d). qRT-PCR analysis showed elevated Mpo mRNA levels in mouse heart tissue after I/R injury, with higher levels detected in the WT group compared to the DR5-KO group (Fig. 3e). These findings collectively imply that DR5 deletion can reduce the release of NETs from infiltrating neutrophils and peripheral blood.

Knockout of DR5 decreases NETs after cardiac I/R injury. a The expression of citH3 in the hearts of WT or DR5-KO mice was assessed by western blot analysis following sham surgery or I40min/R24h. b Detection of dsDNA content in WT or DR5-KO mice plasma using ELISA assay following sham surgery or I40min/R24h. c–d Representative IF results (c) and statistical analysis (d) of MPO expression in WT or DR5-KO mice heart tissue following sham surgery or I40min/R24h. e mRNA expression of Mpo in WT or DR5-KO mice hearts following sham surgery or I40min/R24h as detected by quantitative PCR (n = 3 for each group). Quantitative data are shown as means ± SEM. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001 as determined by a two-tailed t test (b, d, and e)
Blocking TRAIL-DR5 Pathway Through DR5-Fc Fusion Protein Reduces Heart NETs Release During Cardiac IR Injury
The efficacious blocking of the TRAIL-DR5 signaling pathway and the cardioprotective impact of the human sDR5-Fc antibody fusion protein in rats, pigs, and monkeys have been verified by our earlier investigation. However, given the modest homology of approximately 63% between mouse and human TRAIL, we developed a mouse-derived mDR5-Fc antibody fusion protein using the 293F protein eukaryotic expression system and purified it with affinity chromatography. mDR5-Fc was identified through Coomassie brilliant blue staining (Fig. 4a). Twenty-four hours after cardiac I/R injury, we observed a significant decrease in the area of myocardial infarction (Fig. 4b, c), less TUNEL positive in mouse heart tissue (Fig. 4d, e), reduced levels of plasma CK (Fig. 4f) and LDH (Fig. 4g), as well as lower transcription levels of Tnfa (Fig. 4h) and Il1b (Fig. 4i) in cardiac tissue detected by qPCR compared to the wild-type group in mice treated with mDR5-Fc compared to the PBS group. According to these findings, mice’s myocardial I/R damage is well mitigated by the mDR5-Fc fusion protein.

Administration of mDR5-Fc ameliorated mice’s cardiac I/R injury and attenuated the presence of NETs. a SDS-PAGE Coomassie brilliant blue staining for 293F culture media (sample), flow-through solution for sample affinity chromatography (flow-through), and purified mDR5-Fc (elution). b–c Representative results (b) and statistical analysis (c) of TTC-Evans blue double stain heart sections following sham or I40min/R24h in mice treated with PBS or mDR5-Fc. d–e Immunofluorescence detection (d) and statistical analysis (e) of TUNEL in heart tissues of mice following sham or I40min/R24h treated with PBS or mDR5-Fc. f–g Plasma concentrations of CK (f) and LDH (g) of mice following sham or I40minR/24h treated with PBS or mDR5-Fc were analyzed using the Catalyst Dx Chemistry Analyzer. h–i mRNA expression of Tnfa and Il1b in the hearts of sham or I40min/R24h mice treated with PBS or mDR5-Fc, as detected by quantitative PCR. j Detection of dsDNA content in mice plasma using ELISA assay following sham surgery or I40min/R24h treated with PBS or mDR5-Fc. k Immunofluorescence detection of MPO in mice heart tissue following sham surgery or I40min/R24h treated with PBS or mDR5-Fc. l mRNA expression of Mpo in the hearts of sham or I40min/R24h mice treated with PBS or mDR5-Fc, as detected by quantitative PCR. m The expression of citH3 in mice hearts was assessed by western blot analysis following sham or I40min/R24h treated with PBS or mDR5-Fc. Quantitative data are shown as means ± SEM. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001 as determined by a two-tailed t test (c) or one-way ANOVA followed by Tukey’s post hoc test (e–j, and l)
Considering the reduced release of NETs observed in DR5 knockout mice following I/R injury, we investigate the potential impact of mDR5-Fc on NETs release. Higher levels of dsDNA in plasma were observed in mice treated with PBS than those treated with mDR5-Fc (Fig. 4j). Moreover, immunofluorescence results revealed a notable decrease of MPO-positive cells within cardiac tissues subsequent to mDR5-Fc therapy (Fig. 4k). qRT-PCR analysis revealed significantly increased mRNA levels of Mpo in mouse cardiac tissue of the PBS group following I/R injury compared to the mDR5-Fc group (Fig. 4l). Western blot analysis of mouse cardiac tissue revealed an upregulation in the expression of citH3 following I/R injury compared to the sham group, with significantly higher levels observed in mice treated with PBS as opposed to those treated with mDR5-Fc (Fig. 4m). These results imply that mDR5-Fc treatment confers protection against myocardial I/R injury by efficiently reducing the release of NETs from cardiac tissues.
Blocking TRAIL-DR5 Pathway Reduces Neutrophil Infiltration by Diminishing Chemokines and Adhesion Molecules
Neutrophils respond to adhesion and chemotaxis, infiltrating the damaged tissue and releasing NETs, cytokines, and chemokines to exacerbate the inflammatory [19,20,21] response further. To investigate whether the reduction of NETs in the cardiac tissue after I/R injury in DR5 knockout mice and mDR5-Fc treated mice is attributed to a decline in neutrophil population, we quantified the neutrophil count in both peripheral blood and cardiac tissue while also evaluating the expression levels of chemotactic, adhesion, and integrin molecules within the cardiac tissue. The blood routine tests revealed a significant reduction in the neutrophil count in the peripheral blood of both DR5-KO and mDR5-Fc treated groups following cardiac I/R (Fig. 5a, e). Additionally, qPCR analysis revealed a significant downregulation of key adhesion molecule Icam1, chemokine Cxcl15, and integrin Itgam expression levels in DR5 knockout mouse hearts compared to WT controls (Fig. 5b–d). Similarly, treatment with mDR5-Fc resulted in reduced expression levels of Icam1, Cxcl15, and Itgam compared to mouse hearts treated with PBS (Fig. 5f–h). Flow cytometry results revealed a significant reduction in infiltrating neutrophils within the cardiac tissue of mDR5-Fc-treated mice 24 h post-I/R (Fig. 5i, j). According to these results, blocking the TRAIL-DR5 signaling pathway may be a useful strategy to lessen neutrophil infiltration into I/R hearts and help lower the number of NETs produced.

Blocking the TRAIL-DR5 pathway reduces NEU infiltration. a Quantification of neutrophils in the peripheral blood of WT or DR5-KO mice following sham surgery or I40min/R24h, as detected by Auto Hematology Analyzer. b–d mRNA expression of Cxcl15, Icam1 and Itgam in the WT or DR5-KO mice hearts following sham or I40min/R24h treatment, as detected by quantitative PCR. e Quantification of neutrophils in the peripheral blood of mice following sham surgery or I40min/R24h treated with PBS or mDR5-Fc, as detected by Auto Hematology Analyzer. f–h mRNA expression of Cxcl15, Icam1, and Itgam in mice hearts following sham surgery or I40min/R24h treated with PBS or mDR5-Fc, as detected by quantitative PCR. i Gating strategy for neutrophils analysis in mice cardiac tissue using flow cytometry. j Quantitative analysis of neutrophil infiltration in mice heart tissue, the data was quantified by neutrophils numbers per milligram of cardiac tissue. Quantitative data are shown as means ± SEM. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001 as determined by a two-tailed t test (a–d) or one-way ANOVA followed by Tukey’s post hoc test (e–h and j)
Blocking TRAIL-DR5 Pathway Through mDR5-Fc Administration Directly Attenuates NETs Release
We used Percoll density gradient centrifugation to separate bone marrow-derived neutrophils to analyze the direct effect of blocking the TRAIL-DR5 signaling pathway on NETs release (Fig. 6a). The isolated neutrophils were exposed to mouse TRAIL recombinant protein (mTRAIL) and/or mDR5-Fc, resulting in a significant upregulation of citH3 expression as detected by immunofluorescence (Fig. 6b–c). The administration of mDR5-Fc resulted in a significant reduction in Mpo compared to the mTRAIL-treated group, as detected by qPCR and immunofluorescence staining (Fig. 6d–f). Accordingly, these findings suggest that the activation of the DR5 signaling pathway directly triggers neutrophil formation of NETs, while mDR5-Fc inhibits NETs induced by mTRAIL.

Administration of mDR5-Fc directly attenuates NETs release of bone marrow-derived neutrophil. a Percoll density gradient centrifugation was used for the isolation of neutrophils derived from bone marrow. b–c Immunofluorescence detection (b) and statistical analysis (c) of citH3 in neutrophils treated with PBS (control), mTRAIL alone, or co-administration of mTRAIL and mDR5-Fc. d mRNA expression of Mpo in neutrophils treated with or without mTRAIL or co-administration of mTRAIL and mDR5-Fc, as detected by quantitative PCR. e–f Immunofluorescence detection (e) and statistical analysis (f) of MPO in neutrophils treated with PBS (control), mTRAIL alone, or co-administration of mTRAIL and mDR5-Fc. Quantitative data are shown as means ± SEM. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001 as determined by one-way ANOVA followed by Tukey’s post hoc test (c, d, and f)
Administration of mDR5-Fc Inhibits the Formation of NETs, Thereby Attenuating Apoptosis in Cardiomyocytes
NETs are essential in the pathophysiology of cardiovascular disorders because they can cause harm to cardiac cells. To investigate the potential protective effect of mDR5-Fc against myocardial tissue damage induced by NET formation, we conducted a co-culture experiment to assess the impact of neutrophil-derived NETs on NRVM apoptosis. After co-culturing with pre-treated neutrophils-derived NETs, NRVM was tested for TUNEL staining. The supernatant was collected for LDH assay (Fig. 7a). Increased LDH was observed in NRVMs culture media, wherein the mTRAIL-induced NETs stimulated group exhibited significantly higher levels compared to the mDR5-Fc blockade group (Fig. 7b). Similarly, immunofluorescence-based TUNEL detection revealed enhanced apoptosis in NRVM cells induced by mTRAIL-induced NETs. In contrast, less apoptosis was detected in NRVM treated with mTRAIL/mDR5-Fc induced NETs (Fig. 7c–d). These results imply that myocardial damage, which can be effectively mitigated by mDR5-Fc, may be exacerbated by the induction of NETs by mTRAIL.

The administration of mDR5-Fc effectively attenuates the detrimental effects of NETs induced by sTRAIL, thus protecting NRVM. a A diagram for the induction of NETs and treatment of NRVM (by Figdraw). b Detection of LDH expression in the culture supernatant of NRVMs induced by NETs obtained from various treatments. c–d Immunofluorescence detection (c) and statistical analysis (d) of TUNEL in NRVM treated with NETs obtained from various treatments. Quantitative data are shown as means ± SEM. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001 as determined by one-way ANOVA followed by Tukey’s post hoc test (b and d)
Blocking the TRAIL-DR5 Signaling Pathway Can Effectively Downregulate PAD4 Expression
Peptidyl arginine deiminase 4 (PAD4) is a pivotal histone-modifying enzyme implicated in the mediation of NETs formation following ischemia/reperfusion injury [22, 23]. To elucidate the inhibitory mechanism of NETs release by blocking the TRAIL-DR5 pathway, we evaluated the impact of TRAIL-DR5 on PAD4 expression. Immunohistochemistry analysis revealed an upregulation of PAD4 expression in the hearts of mice subjected to IR injury, whereas knockout of DR5 (Fig. 8a–b) or administration of mDR5-Fc (Fig. 8c–d) resulted in a substantial reduction in PAD4 expression. qPCR analyses revealed an upregulation of Pad4 expression in the mouse heart following ischemia/reperfusion (I/R) injury, which could be effectively attenuated by administration of mDR5-Fc (Fig. 8e). Additionally, western blot analyses revealed an upregulation of PAD4 expression in the mouse heart following ischemia/reperfusion (I/R) injury, which could be effectively attenuated by knocking out DR5 or administration of mDR5-Fc (Fig. 8f–g). Moreover, qPCR and western blot analyses demonstrated an upregulation of PAD4 expression in bone marrow-derived neutrophils treated with mTRAIL. However, this effect was attenuated upon treatment with mDR5-Fc (Fig. 8h–i). These results imply that blocking the TRAIL-DR5 pathway reduces the expression of PAD4, which in turn reduces the release of NETs.

Blocking the TRAIL-DR5 pathway could suppress PAD4 expression. a–b Representative IHC results (a) and statistical analysis (b) of PAD4 expression in WT or DR5-KO mice heart tissue following sham surgery or I40min/R24h. c–d Representative IHC results (c) and statistical analysis (d) of PAD4 expression in mouse heart tissue following sham surgery or I40min/R24h treated with PBS or mDR5-Fc. e The expression of Pad4 in mouse heart tissue following sham surgery or I40min/R24h treated with PBS or mDR5-Fc, as detected by quantitative PCR. f The expression of PAD4 in WT or DR5-KO mice hearts was assessed by western blot analysis following sham or I40min/R24h. g The expression of PAD4 in mouse heart tissue following sham surgery or I40min/R24h treated with PBS or mDR5-Fc, as detected by western blot. h–i The expression of PAD4 in bone marrow-derived neutrophils treated with PBS, mTRAIL alone, or co-administration of mTRAIL and mDR5-Fc, as detected by quantitative PCR (h) and western blot (i). Quantitative data are shown as means ± SEM. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001 as determined by a two-tailed t test (b) or one-way ANOVA followed by Tukey’s post hoc test (d, g, and i)
Discussion
Heart I/R injury is a serious clinical issue that needs to be addressed right away since it involves complex pathogenic mechanisms such as an abrupt increase in reactive oxygen species, calcium overload, and an inflammatory response during vascular reperfusion. Currently, there is a lack of specific drug prevention and treatment strategies targeting heart I/R injury and therapeutic targets. Owing to the complexity of the underlying process causing this illness, several studies have been conducted looking at different ways to reduce reperfusion injury from different angles. Inflammatory response plays a pivotal role in acute myocardial infarction and I/R injury, leading researchers to explore multiple endeavors aimed at mitigating the inflammatory response [24,25,26,27]. For instance, studies have revealed that S100a8/a9 levels rapidly increase during the early stages of cardiac I/R injury [28]. Knocking out S100A9 or employing neutralizing antibodies against S100A9 alleviates cardiac I/R injury in mice [29]. Dectin-1 expressed by macrophages exacerbates myocardial I/R injury by regulating macrophage polarization and recruiting neutrophil infiltration, thus making it a potential target for cardiac therapy [30]. The clinical trials have utilized Anakinra, an Il-1β receptor antagonist [31,32,33], and etanercept, a TNF-α antibody [34], to inhibit inflammatory mediators that exacerbate myocardial I/R injury.
In our previous study, we discovered that inhibition of the TRAIL-DR5 signaling pathway could attenuate myocardial cell death and mitigate inflammatory responses, thereby reducing infarct size and ameliorating heart I/R injury. We found that monocytes/macrophages and granulocytes are required for the protective effects of sDR5-Fc [14]. Blocking the TRAIL-DR5 pathway modifies neutrophil involvement in cardiac I/R damage, although the mechanism behind this modulation remains unknown.
Through NETosis, neutrophils can release NETs that aggregate platelets and encourage thrombus formation, aggravating inflammation and inducing an innate immune response [35,36,37] that can lead to heart damage. However, the role of neutrophils in cardiac injury remains multifaceted. Some studies have demonstrated that inhibiting neutrophils can ameliorate acute inflammation in a mouse model of viral myocarditis [38]. Previous studies have demonstrated that depletion of neutrophils can attenuate infarct size in a mouse model of myocardial I/R, which is consistent with our research findings. However, it has been observed that neutrophil depletion does not impact infarct size within 24 h after myocardial infarction but progressively impairs cardiac function in a mouse model of permanent LAD occlusion. Neutrophils are capable of promoting macrophage polarization towards the M2 phenotype during cardiac repair and remodeling processes, highlighting their beneficial contribution [39]. These differences could be the result of differences in treatment regimens, animal models, and detection intervals. Our findings reveal that employing Ly6G antibodies for neutrophil removal leads to a reduction in I/R-induced myocardial infarct area at 24 h post-injury, indicating that early neutrophil suppression may be advantageous for cardioprotection in cases of I/R damage.
The activation of the TRAIL-DR5 pathway can induce apoptosis through death-inducing signaling complex (DISC) formation and subsequent activation of caspase 3, or trigger an inflammatory response via activation of the NF-κB transcription factor [40, 41]. In the context of acute myocardial infarction and cardiac I/R injury, cardiomyocytes undergo cell death and release damage-associated molecular patterns (DAMPs), which activate inflammation and upregulate chemokines and adhesion molecules, thereby promoting neutrophil infiltration. Therefore, we looked into how blocking the TRAIL-DR5 pathway affected the invasion of neutrophils. By deleting DR5 or administering mDR5-Fc, we successfully ameliorated cardiac reperfusion injury, suppressed the gene expression of pro-inflammatory factors Il1b and Tnfa, chemokine Cxcl15, as well as adhesion molecule Vcam1, additionally reducing neutrophil infiltration in the heart. Previous studies have demonstrated that NETosis primarily requires PAD4 in response to calcium ionophores and immune complexes involving chromatin decondensation and nuclear envelope rupture [11, 42]. We observed that inhibition of the TRAIL-DR5 pathway could suppress PAD4 expression in NEU cells and cardiac tissue. As a result, DR5 deletion or mDR5-Fc administration efficiently prevented the creation of NETs via a variety of methods.
We studied the pharmacodynamics of sDR5-Fc in rhesus monkeys, pigs, and rats as animal models. However, owing to the unavailability of antibodies for mechanistic research purposes and the high cost involved in conducting such studies using large animal models, we opted to perform mechanistic research using mice instead. Initially, we validated our findings using DR5 knockout mice and observed that knocking out DR5 can alleviate cardiac I/R injury. On the other hand, in a mouse cardiac I/R model, the protective impact of human-derived sDR5-Fc was insufficient. The subsequent investigation revealed a species disparity in mouse DR5, with a 38.61% homology to humans, and in mouse TRAIL, exhibiting a 63.35% homology to humans (as predicted by UniProt). Additionally, it was found that while the extracellular domain of mouse DR5 possesses multiple N-glycosylation sites, the human-derived DR5 extracellular domain lacks any N-glycosylation sites [43, 44]. This discrepancy partly explains the low effectiveness of human-derived sDR5-Fc in the mouse cardiac IR model. As a result, we created mouse-derived mDR5-Fc for additional mechanistic studies.
In this study, we show that by lowering the development of NETs in the heart, suppression of the TRAIL-DR5 signaling pathway attenuates myocardial I/R injury. Neutrophils play a pivotal role in this process, and blockade of TRAIL-DR5 not only diminishes adhesion and chemotactic factors to decrease neutrophil infiltration but also directly suppresses PAD4 expression to inhibit NETs formation. Moreover, our findings suggest that TRAIL-induced NETs release promotes primary cardiomyocyte apoptosis, which can be blocked by mDR5-Fc (Fig. 9). Although neutrophils are detrimental during the acute phase of myocardial infarction, careful consideration is necessary due to their beneficial role in the healing phase. These dual effects, like a “double-edged sword,” may present challenges in clinical translation. Therefore, further comprehensive research is urgently needed to investigate aspects such as optimal timing and dosage of administration, as well as underlying mechanisms. On the basis of essential mechanistic studies, sDR5-Fc may be explored in clinical settings as a potential treatment drug for heart IR injury.

A summary diagram illustrates the impact of blocking the TRAIL-DR5 pathway on neutrophil infiltration and NETs formation following cardiac I/R injury (by Figdraw). In a mouse model of cardiac ischemia for 40 min followed by reperfusion for 24 h, inhibition of the TRAIL-DR5 signaling pathway resulted in decreased expression of the chemokine CXCL15, adhesion molecule ICAM1, and integrin ITGAM, thereby attenuating neutrophil infiltration. Simultaneously, blockade of the TRAIL-DR5 signaling pathway directly suppressed PAD4 expression in neutrophils, leading to reduced formation of NETs. These effects collectively contribute to diminished cardiomyocyte death, alleviation of the cardiac inflammatory response, and ultimately result in a reduced myocardial infarct size
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Code Availability
Not applicable.
References
Crea F. The burden of cardiovascular risk factors: a global perspective. Eur Heart J. 2022;43(30):2817–20. https://doi.org/10.1093/eurheartj/ehac430
Crea F, Badimon L, Berry C, et al. The European Heart Journal: leading the fight to reduce the global burden of cardiovascular disease. Eur Heart J. 2020;41(33):3113–6. https://doi.org/10.1093/eurheartj/ehaa674
Lopes RD, Hong H, Harskamp RE, et al. Safety and efficacy of antithrombotic strategies in patients with atrial fibrillation undergoing percutaneous coronary intervention: a network meta-analysis of randomized controlled trials. JAMA Cardiol. 2019;4(8):747–55. https://doi.org/10.1001/jamacardio.2019.1880
Yang S, Kang J, Park KW, et al. Comparison of antiplatelet monotherapies after percutaneous coronary intervention according to clinical, ischemic, and bleeding risks. J Am Coll Cardiol. 2023;82(16):1565–78. https://doi.org/10.1016/j.jacc.2023.07.031
Heusch G. Myocardial ischemia/reperfusion: translational pathophysiology of ischemic heart disease. Med. 2024;5(1):10–31. https://doi.org/10.1016/j.medj.2023.12.007
Wu X, Liu L, Zheng Q, et al. Dihydrotanshinone I preconditions myocardium against ischemic injury via PKM2 glutathionylation sensitive to ROS. Acta Pharm Sin B. 2023;13(1):113–27. https://doi.org/10.1016/j.apsb.2022.07.006
Heusch G, Andreadou I, Bell R, et al. Health position paper and redox perspectives on reactive oxygen species as signals and targets of cardioprotection. Redox Biol. 2023;67:102894. https://doi.org/10.1016/j.redox.2023.102894
Chen C, Zhang H, Xie R, Wang Y, Ma Y. Gut microbiota aggravate cardiac ischemia-reperfusion injury via regulating the formation of neutrophils extracellular traps. Life Sci. 2022;303:120670. https://doi.org/10.1016/j.lfs.2022.120670
Ge L, Zhou X, Ji WJ, et al. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy. Am J Physiol Heart Circ Physiol. 2015;308(5):H500–9. https://doi.org/10.1152/ajpheart.00381.2014
Sharma S, Hofbauer TM, Ondracek AS, et al. Neutrophil extracellular traps promote fibrous vascular occlusions in chronic thrombosis. Blood. 2021;137(8):1104–16. https://doi.org/10.1182/blood.2020005861
Thiam HR, Wong SL, Wagner DD, Waterman CM. Cellular mechanisms of NETosis. Annu Rev Cell Dev Biol. 2020;36:191–218. https://doi.org/10.1146/annurev-cellbio-020520-111016
Jankowski J, Floege J, Fliser D, Böhm M, Marx N. Cardiovascular disease in chronic kidney disease: pathophysiological insights and therapeutic options. Circulation. 2021;143(11):1157–72. https://doi.org/10.1161/circulationaha.120.050686
He L, Liu R, Yue H, et al. Interaction between neutrophil extracellular traps and cardiomyocytes contributes to atrial fibrillation progression. Signal Transduct Target Ther. 2023;8(1):279. https://doi.org/10.1038/s41392-023-01497-2
Wang Y, Zhang H, Wang Z et al. Blocking the death checkpoint protein TRAIL improves cardiac function after myocardial infarction in monkeys, pigs, and rats. Sci Transl Med. 2020;12(540). https://doi.org/10.1126/scitranslmed.aaw3172
Nash M, McGrath JP, Cartland SP, Patel S, Kavurma MM. Tumour necrosis factor superfamily members in ischaemic vascular diseases. Cardiovasc Res. 2019;115(4):713–20. https://doi.org/10.1093/cvr/cvz042
Crunkhorn S. TRAIL blockade improves heart function. Nat Rev Drug Discov. 2020;19(6):388. https://doi.org/10.1038/d41573-020-00087-z
Aronoff L, Epelman S, Clemente-Casares X. Isolation and identification of extravascular immune cells of the heart. J Vis Exp. 2018(138). https://doi.org/10.3791/58114
Van Bruggen S, Sheehy CE, Kraisin S et al. Neutrophil peptidylarginine deiminase 4 plays a systemic role in obesity-induced chronic inflammation in mice. J Thromb Haemost. 2024. https://doi.org/10.1016/j.jtha.2024.01.022
Dong Y, Kang Z, Zhang Z, et al. Single-cell profile reveals the landscape of cardiac immunity and identifies a cardio-protective Ym-1(hi) neutrophil in myocardial ischemia-reperfusion injury. Sci Bull (Beijing). 2024. https://doi.org/10.1016/j.scib.2024.02.003
Wong R, Lénárt N, Hill L, et al. Interleukin-1 mediates ischaemic brain injury via distinct actions on endothelial cells and cholinergic neurons. Brain Behav Immun. 2019;76:126–38. https://doi.org/10.1016/j.bbi.2018.11.012
Korbecki J, Kojder K, Kapczuk P et al. The effect of hypoxia on the expression of CXC chemokines and CXC chemokine receptors-a review of literature. Int J Mol Sci. 2021;22(2). https://doi.org/10.3390/ijms22020843
Du M, Yang W, Schmull S, Gu J, Xue S. Inhibition of peptidyl arginine deiminase-4 protects against myocardial infarction induced cardiac dysfunction. Int Immunopharmacol. 2020;78:106055. https://doi.org/10.1016/j.intimp.2019.106055
Thiam HR, Wong SL, Qiu R, et al. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proc Natl Acad Sci U S A. 2020;117(13):7326–37. https://doi.org/10.1073/pnas.1909546117
Zuurbier CJ, Abbate A, Cabrera-Fuentes HA, et al. Innate immunity as a target for acute cardioprotection. Cardiovasc Res. 2019;115(7):1131–42. https://doi.org/10.1093/cvr/cvy304
Andreadou I, Cabrera-Fuentes HA, Devaux Y, et al. Immune cells as targets for cardioprotection: new players and novel therapeutic opportunities. Cardiovasc Res. 2019;115(7):1117–30. https://doi.org/10.1093/cvr/cvz050
Hausenloy DJ, Bøtker HE, Ferdinandy P, et al. Cardiac innervation in acute myocardial ischaemia/reperfusion injury and cardioprotection. Cardiovasc Res. 2019;115(7):1167–77. https://doi.org/10.1093/cvr/cvz053
Swirski FK, Nahrendorf M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol. 2018;18(12):733–44. https://doi.org/10.1038/s41577-018-0065-8
Sreejit G, Abdel Latif A, Murphy AJ, Nagareddy PR. Emerging roles of neutrophil-borne S100A8/A9 in cardiovascular inflammation. Pharmacol Res. 2020;161:105212. https://doi.org/10.1016/j.phrs.2020.105212
Li Y, Chen B, Yang X, et al. S100a8/a9 signaling causes mitochondrial dysfunction and cardiomyocyte death in response to ischemic/reperfusion injury. Circulation. 2019;140(9):751–64. https://doi.org/10.1161/circulationaha.118.039262
Fan Q, Tao R, Zhang H, et al. Dectin-1 contributes to myocardial ischemia/reperfusion injury by regulating macrophage polarization and neutrophil infiltration. Circulation. 2019;139(5):663–78. https://doi.org/10.1161/circulationaha.118.036044
Abbate A, Trankle CR, Buckley LF, et al. Interleukin-1 blockade inhibits the acute inflammatory response in patients with ST-segment-elevation myocardial infarction. J Am Heart Assoc. 2020;9(5):e014941. https://doi.org/10.1161/jaha.119.014941
Smith CJ, Hulme S, Vail A, et al. SCIL-STROKE (subcutaneous interleukin-1 receptor antagonist in ischemic stroke): a randomized controlled phase 2 trial. Stroke. 2018;49(5):1210–6. https://doi.org/10.1161/strokeaha.118.020750
Morton AC, Rothman AM, Greenwood JP, et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: the MRC-ILA heart study. Eur Heart J. 2015;36(6):377–84. https://doi.org/10.1093/eurheartj/ehu272
Padfield GJ, Din JN, Koushiappi E, et al. Cardiovascular effects of tumour necrosis factor α antagonism in patients with acute myocardial infarction: a first in human study. Heart. 2013;99(18):1330–5. https://doi.org/10.1136/heartjnl-2013-303648
Dou H, Kotini A, Liu W, et al. Oxidized phospholipids promote NETosis and arterial thrombosis in LNK(SH2B3) deficiency. Circulation. 2021;144(24):1940–54. https://doi.org/10.1161/circulationaha.121.056414
Yang M, Jiang H, Ding C, et al. STING activation in platelets aggravates septic thrombosis by enhancing platelet activation and granule secretion. Immunity. 2023;56(5):1013-26.e6. https://doi.org/10.1016/j.immuni.2023.02.015
Holm S, Kared H, Michelsen AE, et al. Immune complexes, innate immunity, and NETosis in ChAdOx1 vaccine-induced thrombocytopenia. Eur Heart J. 2021;42(39):4064–72. https://doi.org/10.1093/eurheartj/ehab506
Carai P, González LF, Van Bruggen S, et al. Neutrophil inhibition improves acute inflammation in a murine model of viral myocarditis. Cardiovasc Res. 2023;118(17):3331–45. https://doi.org/10.1093/cvr/cvac052
Horckmans M, Ring L, Duchene J, et al. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J. 2017;38(3):187–97. https://doi.org/10.1093/eurheartj/ehw002
Sullivan GP, O’Connor H, Henry CM, et al. TRAIL receptors serve as stress-associated molecular patterns to promote ER-stress-induced inflammation. Dev Cell. 2020;52(6):714-30.e5. https://doi.org/10.1016/j.devcel.2020.01.031
Yang J, LeBlanc FR, Dighe SA, et al. TRAIL mediates and sustains constitutive NF-κB activation in LGL leukemia. Blood. 2018;131(25):2803–15. https://doi.org/10.1182/blood-2017-09-808816
Castanheira FVS, Kubes P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood. 2019;133(20):2178–85. https://doi.org/10.1182/blood-2018-11-844530
Dufour F, Rattier T, Shirley S, et al. N-glycosylation of mouse TRAIL-R and human TRAIL-R1 enhances TRAIL-induced death. Cell Death Differ. 2017;24(3):500–10. https://doi.org/10.1038/cdd.2016.150
Estornes Y, Dondelinger Y, Weber K, et al. N-glycosylation of mouse TRAIL-R restrains TRAIL-induced apoptosis. Cell Death Dis. 2018;9(5):494. https://doi.org/10.1038/s41419-018-0544-7
Funding
This work was supported by the Natural Science Foundation of Henan (No.242300421021), Program for Science & Technology Innovation Talents in Universities of Henan Province (No.22HASTIT046), National Natural Science Foundation of China (No.U22A20382), Young Scientists Fund of the National Natural Science Foundation of China (No. 82101913), and Medical Science and Technique Foundation of Henan Province (No.SBGJ202102195).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. XW and RX acquired the data and performed statistical analysis. GW, RX, WS, YD, and WW performed the mice cardiac I/R surgery. YXW, LZ, TM, and XT prepared the mDR5-Fc protein. YW, DZ, and YC developed the method for NRVM isolation. RC, YXW, and YW conceived and designed the research. XW drafted the manuscript. YW and YM handled funding and supervision. RC, YW, YXW, DZ, LZ, and YM made critical revisions to the manuscript for key intellectual content. All co-authors edited and reviewed the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
All animal experiments conducted in this study received approval from the Institutional Animal Ethics Committee of Henan University Joint National Laboratory for Antibody Drug Engineering, and the experimental designs and animal care were conducted in accordance with the ARRIVE guidelines. All applicable international, national and/or institutional guidelines for the care and use of animals were followed.
Consent to Participate
Not Applicable.
Consent for Publication
Not applicable.
Conflicts of Interest
The authors have no conflicts of interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, X., Xie, R., Zhao, D. et al. Blocking the TRAIL-DR5 Pathway Reduces Cardiac Ischemia–Reperfusion Injury by Decreasing Neutrophil Infiltration and Neutrophil Extracellular Traps Formation. Cardiovasc Drugs Ther (2024). https://doi.org/10.1007/s10557-024-07591-z
Accepted:
Published:
DOI: https://doi.org/10.1007/s10557-024-07591-z