
Abstract
Despite the great success that chimeric antigen receptor (CAR) T-cells have had in patients with B-cell malignancies and multiple myeloma, they continue to have limited efficacy against most solid tumors. Especially in the pediatric population, pre- and post-treatment biopsies are rarely performed due to ethical reasons, and thus, our understanding is still very limited regarding the mechanisms in the tumor microenvironment by which tumor cells exclude effectors and attract immune-suppressive cells. Nevertheless, based on the principles that are known, current T-cell engineering has leveraged some of these processes and created more potent CAR T-cells. The recent discovery of new oncofetal antigens and progress made in CAR design have expanded the potential pool of candidate antigens for therapeutic development. The most promising approaches to enhance CAR T-cells are novel CAR gating strategies, creative ways of cytokine delivery to the TME without enhancing systemic toxicity, and hijacking the chemokine axis of tumors for migratory purposes. With these new modifications, the next step in the era of CAR T-cell development will be the clinical validation of these promising preclinical findings.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Chimeric antigen receptor (CAR) T-cell therapy comprises genetically modified autologous T-cells that express a synthetic tumor-targeting CAR. Following lymphodepleting (LD) chemotherapy, this ex vivo manufactured “living drug” is reinfused into patients to eliminate cancer cells. The indications for CAR T-cells are quickly expanding and have changed clinical practice. To date, the U.S. Food and Drug Administration (FDA) has approved CD19-targeted CAR T-cells for patients with relapsed/refractory (R/R) leukemia and lymphoma, and B-cell maturation antigen (BCMA)-targeted CAR T-cells for patients with R/R multiple myeloma [1]. In a subset of these patients, CAR T-cells can induce long-term remissions and possibly cures alone or when combined with other consolidative therapies such as bone marrow transplantation [2,3,4]. In a study by the National Cancer Institute, close to two-thirds of the 43 patients with R/R B-cell lymphoma or chronic lymphocytic leukemia achieved a complete response (CR) after therapy with axicabtagene ciloleucel, a CD19-targeted CAR T-cell therapy. Additionally, 76% of these individuals remained in long-term remission without further therapy [5]. Comparable results were reported in a single-institution study of patients with lymphoma treated with tisagenlecleucel therapy, reporting a CR rate of 55% and 5-year event-free survival rates of 60% [6]. These studies and others have shown that durable responses were associated with a profound initial response, an absence of extramedullary disease, low tumor burden, receipt of LD chemotherapy, and high peak expansion of CAR T-cells—features that are likely shared in solid tumors as well. On the contrary, the development of CAR T-cells for solid tumors has not successfully transitioned beyond phase I/II trials due to the lack of robust activity despite promising preclinical work [7]. Many reviews have discussed potential key challenges and clinical determinants of success for CAR T-cells in solid tumors [8,9,10,11,12,13,14,15,16], some of which remain poorly understood, including (1) what characteristics of CAR T-cells enhance the trafficking to the tumor site, (2) what barriers in the tumor microenvironment can be therapeutically targeted to improve CAR T-cell performance, and (3) how CAR T-cells can be engineered to extend their function and activation without increasing toxicity in patients. The goal of this review is not to add another comprehensive review on this topic, but instead, to summarize new preclinical and clinical insights into factors that contribute to these challenges by highlighting key articles and concepts as well as providing suggestions on how to overcome them while balancing on-target off-tumor toxicity for the next generation of CARs.
1.1 CAR T-cell efficacy in solid tumors
The clinical landscape of CAR T-cell studies in pediatric solid tumors has been predominated by disialoganglioside (GD2)-CAR T-cells [17,18,19,20,21,22] (Table 1). However, across these studies, variable responses were reported. Among the most promising studies is a recent phase I/II trial (NCT03373097) of a 3rd generation (3G) GD2-CAR (GD2-CAR01) with a rimiducid-inducible caspase 9 suicide switch. In this study, researchers demonstrated feasibility and reported no safety concerns, testing three dose levels (3, 6, and 10 × 106 CAR+ T-cells/kg) following standard LD chemotherapy [17]. T-cells were transduced at high levels (> 70%) using a retroviral vector (iCasp9.2A.GD2.CD28.4-1BB.zeta) and expanded with IL-7 and IL-15. No dose-limiting toxicity (DLT) was reported in the 27 treated children with R/R neuroblastoma. Peak CAR T-cell expansion occurred in the second week after infusion, correlated with the dose level at which the patient was enrolled, and persisted for at least 3 months in 75% of participants. Interestingly, patients with low expansion (< 5% in the peripheral blood) had a significantly higher proportion of terminally differentiated CD45RA+ CCR7− CD4 T-cells in their infusion product. The trial reported a 63% response rate among which there were 9 CRs (33%) and 5 of 9 sustained CRs at a median follow-up of 1.7 years. Importantly though, the nine responders had SIOPEN MIBG scores of less than 4 with four patients having a score of 0, highlighting that responses in these patients and likely others with solid tumors occur in individuals with limited disease burden and that T-cell subsets may play an important role in anti-tumor activity of CAR T-cells.
In contrast to this trial, another recent study by the National Cancer Institute tested the 3G GD2-CAR, GD2.CD28.OX40.z and reported no objective responses in 12 patients with osteosarcoma and three with neuroblastoma [19]. These CAR T-cells were manufactured with retroviral vector (iC9-2A-14G2A.CD28.OX40Z), expanded in IL-2, and administered after LD chemotherapy at four different dose levels (0.1, 1, 3, and 10 × 106 CAR+ T-cells/kg). Again, no DLT was reported. Although the peak CAR T-cell expansion was decent (range, 0.01–3.4 × 105 copies/mcg of DNA), none of the 13 patients showed clinical responses (partial responses [PRs] or CRs), not even those 7 of 13 with low disease burden. However, despite the lack of activity, this study identified important factors that were associated with good CAR T-cell expansion. Patients with an abundance of naive CD45RA+ CCR7+ CD8 T-cells in their infusion product had good expansion, while patients with mainly CD45RA+ CCR7− CD38+ TBET+ CD11b+ CD122+ terminally differentiated effector TEMRA CD8 T-cells were poor expanders. Additionally, this study established a link between CXCR3+ or CXCR3hi classical monocytes and GD2-CAR T-cells and showed that their association can modulate CAR T-cell expansion and function.
In a third phase I clinical study (NCT04196413), pediatric and young adult patients with diffuse intrinsic pontine glioma (DIPG) and diffuse midline glioma (DMG) receive retrovirally transduced GD2-CAR T-cells (14G2A–4-1BB–CD3Ζ) in an ongoing study. These effectors were expanded in IL-7 and IL-15 with dasatinib on days 3 and 5 to improve T-cell fitness [23, 24]. Three out of four initial patients treated on the first dose level at 1 × 106 CAR+ T-cells/kg had clinical and radiographic improvement without reported on-target off-tumor toxicity. Further interim data from this trial corroborated these early promising findings in 13 patients [25]. Although GD2-CAR T-cells were tolerated on dose level 1, three patients subsequently developed grade 4 cytokine release syndrome (CRS) at the next dose level. All individuals developed tumor inflammation–associated neurotoxicity (TIAN), which was medically managed and resolved. These results prompted the study team to amend the protocol and administer intracerebroventricular GD2 CAR T-cells without LD chemotherapy preceded by a course of intravenous GD2-CAR T-cells with LD chemotherapy. On this modified arm, all patients have tolerated the intracerebroventricular injections without reported DLTs.
Even though CAR T-cell therapy trials in adult solid tumors target a larger variety of antigens in a more heterogeneous patient population, the anti-tumor efficacy of these treatments mirrors the response rates in pediatrics (Table 1). Given that to date, CAR T-cells are feasible and generally safe in clinical trials, the presented data raises the question of whether perhaps, there should be a shift in the focus of future clinical CAR T-cell studies in solid tumors. For example, one may want to target other antigens, combine CAR T-cells with immune-modulatory treatments, and expand on more in-depth correlative analyses to gain a more nuanced biological understanding of CAR T-cell biology.
1.2 What is an ideal CAR T-cell for solid tumors?
Based on the clinical responses from completed studies (Table 1), it is clear that CAR T-cell therapy in solid tumors has room for improvement. But what aspects of this treatment should be improved? Based on our mechanistic understanding of the challenges in the tumor microenvironment (TME) and the wealth of preclinical data on CAR engineering to enhance efficacy and T-cell fitness, we crafted an ideal CAR for solid tumors.
1.2.1 The ideal target
The problem: The “perfect” antigen is an oncogenic driver that is highly abundant and homogenously expressed on tumor cells while absent on healthy tissues and is widely shared among patients (“public epitope”) and across tumor types as a pan-marker [57, 58]. Most naturally occurring CAR targets are tumor-associated antigens (TAAs) and do not fulfill these criteria. While one can live with B-cell aplasia following CD19-CAR T-cell therapy, cross-reactivity against other TAAs can be detrimental to the patient [59, 60]. For example, 39% to 64% of patients receiving CD19-CAR T-cells develop immune effector cell–associated neurotoxicity syndrome (ICANS) that can range from mild neurologic symptoms to fatal encephalopathy [61, 62]. The occurrence of ICANS is mechanistically linked to the expression of CD19 in brain mural cells that form an integral part of the blood–brain barrier and when damaged, may allow CD19-CAR T-cells to invade the brain and cause neurotoxicity [63]. Interestingly, despite the exuberantly high expression of GD2 in mature neurons [64], patients treated with GD2-CAR T-cells do not develop neuronal cell death or as a result, overt encephalopathy, or severe peripheral neuropathy [17, 20,21,22, 65]. However, individuals with brain tumors but not those with extracranial solid tumors can manifest TIAN, which is thought to be caused by neuronal dysfunction due to local inflammation [66]. Clinical experience in humans is in stark contrast to preclinical findings in a study in which mice were treated with an affinity-enhanced GD2-CAR (GD2-E101K) and developed fatal encephalitis 19 to 27 days after GD2-CAR T-cell injection (though repeated studies by other groups were not able to reproduce these findings) [67, 68]. A pathologic review of the murine brains revealed T-cell infiltration and neuronal destruction. It is conceivable that the high antigen density threshold for most CARs is a possible reason why CAR T-cells do not cause more toxicity, considering that there are normal vital organs that share antigens with tumors such as the brain [22, 69]. Such toxicity may occur if this antigen threshold is lowered as in affinity-enhanced CARs. Other factors that mitigate toxicity remain to be discovered.
Strategies: The accurate identification of antigens for CAR therapy is essential. Especially in solid tumors, there is a scarcity of tumor-specific or tumor-associated targets not expressed in vital organs and tissues. Although tumor tissues are required for target discovery, we live in an era in medicine and science where there are large datasets publicly available that allow for in silico discovery work. Advances in bioinformatics and the broader availability of biocomputational resources have allowed for a comprehensive yet more nuanced re-analysis of these datasets. For example, recently, over 1500 RNA data sets from the St. Jude Cloud and the NCI TARGET cohort were analyzed for cancer-specific exons that present promising targets due to their exclusivity in cancer [70]. After cross-referencing potential targets with expression in normal tissues using the Genotype-Tissue Expression (GTEx) consortium data, the authors found 37 new gene-level or alternatively spliced exon targets that encode surface or matrix proteins and are absent in normal tissue. Approaches like this are particularly important in pediatric cancer where re-biopsies are rarely indicated for clinical reasons and are difficult to ethically justify for research purposes.
Antigens of recent interest are oncofetal proteins that are highly expressed during development but absent in most healthy tissues after birth, rendering them an attractive option for CAR development [71]. In addition, some antigens may have several isoforms, some of which are tumor-restricted, which may further minimize on-target off-tumor cross-reactivity and increase the therapeutic window of CAR T-cells [72, 73]. Ideally, target candidates also serve as oncogenic drivers for tumors. In this capacity, downregulation of the antigen and immune evasion are countered due to the antigen’s essential role in tumor growth [72, 73]. Glypican-2 (GPC2), a member of the glypican family, possesses most of these features. GPC2 is a neurodevelopmental heparan sulfate proteoglycan and is silenced after birth except for continued expression in the male testis [74]. The isoform GPC2-201 is expressed by several pediatric solid tumors including embryonal brain tumors and neuroblastoma [69, 72, 74,75,76,77]. In neuroblastoma, GPC2 has pro-tumorigenic effects by signaling through the WNT/β-catenin pathway. Preclinical efforts have identified efficacious CARs targeting GPC2 that are in clinical development across different centers in the U.S. Promising oncofetal antigens for CAR T-cells in adult cancers include the carcinoembryonic antigen (CEA) (NCT02349724), claudin 6 (CLDN6) (NCT04503278), or tumor-associated glycoprotein 72 (NCT05225363). Results from these clinical trials are pending.
1.2.2 The ideal CAR
The problem: The ideal CAR can sense specific antigens at a wide range of densities, potently activate T-cells to kill targets, and persist on the cell surface without causing tonic signaling. In comparison to TCRs, CARs typically require a much higher antigen density (> 1000 molecules/cell) due to factors like limited kinase recruitment, underdeveloped immune synapses, decreased co-receptor engagement, and increased activation of downstream regulators [9]. Though antigen specificity is a desired feature in CARs, we hypothesize that low level of antigen exposure may be beneficial for CAR persistence and long-term function. Thus, similar to the Goldilocks principle, CAR design has to enable “just the right amount” of signaling to activate CAR T-cells and trigger killing without causing exhaustion over time.
Strategies: In CARs that target highly expressed antigens, like GD2, CAR engineering becomes a central tool to fine-tune downstream signaling and CAR function. For example, the single-chain variable fragments of a 14G2a-based GD2-CAR exhibit antigen-independent clustering that causes tonic signaling and early exhaustion [78]. This phenomenon was not found with a CD19-CAR with structural similarities in the CAR construct. However, by using a CD28 instead of a 4-1BB costimulatory domain, CAR clustering and tonic signaling were dramatically reduced and CAR function improved. CARs that target low-density antigens like the GPC2-CAR also benefit from CAR engineering. Here, the choice of a CD28 transmembrane domain optimally enhances downstream signaling and CAR efficacy [69, 76].
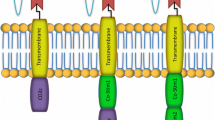
Other CAR engineering efforts have focused on Boolean logic–gating strategies to improve the specificity. Because combinatorial antigen sensing is required for CAR activation in this system, in theory, these receptors can better discriminate between tumor and healthy tissue, thereby expanding the repertoire of potential antigens (Fig. 1). The synNotch IF–THEN circuit incorporates a synthetic notch receptor, which upon antigen-specific engagement triggers the transcription of a conventional CAR against a TAA [79,80,81,82,83,84]. Thus, while both antigens are tumor-associated, their joint presence is required for CAR activation and cytotoxicity. However, despite effectively discriminating between single- and dual-antigen tumors, co-localization of TAA in healthy tissues remains a problem. Furthermore, this system can lead to sustained CAR expression and potential off-target effects as T-cells migrate into healthy tissue. Moreover, although the synNotch system has shown promising results in preclinical studies of neuroblastoma using GD2 and B7-H3 CARs, the scalability of this system poses a significant hurdle for clinical translation [81]. In the logic-gated intracellular network (“LINK”) AND gate platform, two proximal TCR signaling molecules, LAT and SLP76, are fused to membrane-bound scFvs and co-localize upon concurrent binding to induce T-cell activation [85]. Unlike in the synNotch system, this design enables reversible activation, contingent upon continuous and proximate interaction of both TAAs. In a preclinical ROR1 toxicity model, LINK CAR T-cells eradicated tumors presenting both antigens without causing toxicity. Mice treated with synNotch-engineered T-cells died from on-target off-tumor toxicity in this model [84]. CARs with “AND NOT” gating co-express a prototype CAR in trans with an inhibitory CAR (iCAR) [86]. The former triggers T-cell activation upon binding of a TAA, while the latter dampens activation when encountering an antigen on healthy tissue. The antigen-binding domain of iCARs is commonly fused to the signaling domains of negative checkpoints, such as PD-1 and CTLA-4 [87]. Temporary inhibition of cytokine secretion, cytotoxicity, and proliferation can provide rest to the T-cells and reinvigorate their function in the long term. These approaches demonstrate that re-purposing the TCR signaling machinery could open novel avenues for CAR engineering.

Summary of logic gates. OR gating (e.g., bispecific CAR): CAR is activated in response to a cell expressing antigen A or B. AND gating (e.g., SynNotch): SynNotch receptor releases a transcription factor upon activation that translocates into the nucleus and turns on the expression of a CAR for antigen B. AND/NOT (e.g., inhibitory (i) CAR): iCAR dampens the T-cell response when encountering an antigen on healthy tissue
Although combinatorial antigen-sensing may improve therapeutic safety, antigen escape remains an important mechanism of immune evasion and therapy failure that needs to be addressed. Bi-specific CARs target two antigens or two epitopes of one antigen (OR gate) [77, 88]. This can be achieved through the administration of a mixed cell product or a bicistronic or bivalent CAR [9]. As opposed to AND gates, this method allows for the recognition of two TAA but does not require concurrent antigen binding or simultaneous antigen expression. In the clinic, ciltacabtagene autoleucel, a bi-specific BCMA-targeting CAR, is highly effective in R/R multiple myeloma and received by the FDA based on the results from a phase II trial that demonstrated an overall response rate of 73% and a median progression-free survival of 8.8 months [89]. Preclinical work with a bicistronic GPC2 and B7-H3-targeted CAR shows promising results in preclinical models of neuroblastoma [77]. Future studies are needed to translate this and similar efforts into the clinic.
Antigen loss or downregulation is a well-established mechanism of immunotherapy resistance [90,91,92,93]. Multi-specific CAR T-cells could be a solution to overcome this therapy resistance mechanism. For example, in a glioblastoma model, bispecific CAR T-cells targeting both interleukin-13 receptor subunit alpha-2 (IL-13Rα2) and ephrin type-A receptor 2 (EphA2) demonstrated enhanced efficacy and reduced antigen escape compared to single CAR bearing T-cells [94]. However, increasing the therapeutic pressure in this manner can result in the downregulation of multiple antigens, as observed in pediatric ALL clinical trials where some patients relapsed with CD19lowCD22low tumors after treatment with bicistronic CD19/CD22 CAR T-cells [95]. In this instance, the development of a universal or adapter CAR offers greater flexibility. These CAR T-cells are engineered with a universal receptor that can be activated by a soluble adapter molecule containing an scFv region specific to a tumor antigen [96]. This design allows for the redirection of CAR T-cells to virtually any antigen target without needing further genetic modification or new T-cell production. When a tumor downregulates a specific antigen, a new adapter molecule can be generated to stimulate CAR T-cells in vivo. However, this strategy still relies on the identification of tumor-specific antigens. A potentially simpler approach involves creating T-cells that target synthetic markers. Recently, Vincent et al. developed an innovative system where tumor-colonizing probiotic bacteria are engineered to produce synthetic markers that label tumor tissue [97]. Probiotic CAR T-cells (ProCARs) are then designed to target these markers, leading to antigen-independent, tumor-specific cell lysis.
1.2.3 The ideal migratory and survival skills of a CAR T-cell
The problem: One of the greatest hurdles to effective CAR T-cell therapy against solid tumors is the presence of an immune-hostile TME. Unlike in hematological cancers, the solid tumor TME is a dynamic system composed of tumor cells, immune cells, stroma cells, abnormal vasculature, and dense disorganized extracellular matrices (ECMs) all designed to facilitate tumor survival, growth, and metastasis [98,99,100]. Tumor cells secrete chemokines that preferentially attract pro-tumor myeloid-derived suppressor cells (MDSCs), M2 tumor–associated macrophages (TAMs), and regulatory T-cells (Tregs) [101]. Once attracted, these immune cells secrete type II cytokines (e.g., IL-4, IL-10, and IL-13) that perpetuate further recruitment of immune-suppressive cells [101, 102], induce effector exhaustion through inhibitory checkpoint ligands, and force the development of a nutrient-deplete TME [102]. For example, MDSCs and M2 TAMs secrete reactive oxygen and nitrogen species and actively consume arginine. Tregs can act as cytokine “sinks,” depleting cytokines like IL-2 [103,104,105]. While immunosuppressive cells thrive in this environment, these features present physical and chemical barriers to the trafficking, infiltration, and persistence of CAR T-cells, limiting their therapeutic efficacy [106].
Strategy: CAR T-cells should possess the ability to extravasate and migrate into the tumor tissue going against a less favorable oxygen and metabolic gradient. As these cells arrive at the core of solid tumors, they must have the ability to survive, kill, and expand. This can be achieved by strong activation signals, for example, through the activation of the CAR, and by receiving additional signals through chemokines, cytokines, or other nurturing molecules or cues from the environment or the effectors themselves.
T-cells can transmigrate from the bloodstream when attracted by chemokines like CXCL9/10/11 and CCL5. After transversing across stromal cells and ECM, CAR T-cells then can seek direct tumor contact via intracellular adhesion molecule-1 (ICAM1) and execute anti-tumor cytotoxicity [107]. Importantly, although tumor cells secrete chemokines like CXCL1/2, CCL2, and IL-8 that attract myeloid cells to the tumor microenvironment (TME), they typically do not secrete chemokines for which CAR T-cells express receptors [108]. In addition, manufacturing procedures aim at generating CAR T-cells with a CCR7high CD62Lhigh CD45RO+ central memory effector phenotype because these cells exhibit greater cytotoxic capacity and lower propensity for functional exhaustion [109]. However, CCR7 and CD62L are known molecules that promote migration to secondary lymphoid tissues, contributing to a CAR T-cell-devoid TME [110,111,112]. Intraventricular injections in patients with brain tumors or intra-tumoral injections in breast cancers without preceding LD chemotherapy have shown to be feasible [113, 114]. Whether these alternate routes of administration improve CAR T-cell persistence in the TME and enhance cytotoxicity and clinical outcomes remains to be determined.
The persistence of CAR T-cells is measured in the peripheral blood in most clinical studies. CAR T-cell numbers are highest within the first 2 weeks after infusion and last for about a month before contracting [112]. The meaning of this information is unclear as the presence and persistence in the TME may be of greater biological importance. Although one may assume that the presence of peripheral blood CAR T-cells indirectly reflects CAR function, their presence is not a reliable biomarker for clinical responses. The best examples are the GD2-CAR T-cell studies where patients have high persistent levels of CAR (up to 3.4 × 105 copies/mcg of DNA) but do not show a clinical response after therapy. One major limitation of studying the migration and persistence of CAR T-cells in solid tumors is the lack of non-invasive detection methods. CAR T-cell imaging techniques are actively being developed for this purpose. Direct radiolabeling techniques employ nanoparticles or small molecules like 89Zr-oxine, 111In-oxine, or Technetium-99 m (99mTc)-hexamethylpropylene amine oxime and render T-cells detectable by single-photon emission computed tomography (SPECT) or positron emission tomography (PET). The half-lives of these molecules are relatively short, precluding longitudinal imaging [115, 116]. Reporter gene–based labeling with herpes simplex virus 1 thymidine kinase (HSV1-tk), norepinephrine transporter, and sodiumiodide symporter has become more popular as this approach allows for longer term tracking of T-cells [117, 118]. While promising, further refinement and validation of these methods are needed to reduce imaging background, increase resolution, and confirm their prognostic value.
All effectors, including therapeutic CAR T-cells, require a functional vascular network to enter the tumor [119, 120]. However, the TME of many solid tumors propagates the development of abnormal vessels, leading to poor CAR T-cell extravasation into tumors. This occurs in a forward propagating loop. The poor vasculature leads to hypoxia, which stimulates cells in the TME to secrete large amounts of angiogenic factors including vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), transforming growth factor-β (TGF-β), and platelet-derived growth factor (PDGF) [121,122,123]. This in turn leads to the rapid formation of dysfunctional and distorted blood vessels, ultimately leading to more hypoxia [121, 124, 125]. These aberrant vessels have reduced expression of adhesion molecules including vascular cell adhesion molecule-1 (VCAM1) and ICAM1, hindering extravasation, and leading to poor infiltration of CAR T-cells [126,127,128]. Several studies have found that blocking VEGF signaling can lead to the normalization of tumor vessels and improved tracking of adoptively transferred effectors cells into the TME [129,130,131]. The utility of VEGF-neutralizing antibodies could enhance the migration and improve the efficacy of CAR T-cells. As a proof of principle, in a recent syngeneic mouse model of glioblastoma, the co-administration of anti-VEGF and EGFR-vIII CAR T-cells therapy improved infiltration of CAR T-cells and prolonged survival of mice compared to CAR T-cells monotherapy [132].
1.3 What strategies will lead us to an ideal CAR T-cell therapy?
In the following, we will highlight and suggest different strategies to enhance CAR T-cell therapy in solid tumors (Fig. 2). Though each of these approaches is compelling, in the end, we suspect that a combination of several strategies is needed to optimize CAR T-cells and improve clinical outcomes.

Strategies to enhance CAR T-cell therapy against the immunosuppressive TME of solid tumors. Strategies are shown to enhance CAR T-cell function, persistence, and migration. Many of these advances offer overlapping benefits, optimizing multiple aspects of CAR T-cell functionality simultaneously
1.3.1 Combinatorial therapies
One-on-one, CAR T-cells are very potent killers of tumor cells, but they require help when facing additional challenges in the TME. As with conventional anti-cancer therapy, combinatorial therapies with CAR T-cells may have a synergistic effect and break down some of these barriers. However, since CAR T-cells are “living drugs,” one is limited by what agents to use concurrently. Lymphocytes, especially when activated, are exclusively sensitive to radiation and chemotherapy [133, 134]. In this context, LD chemotherapy is an important but undervalued aspect of treatment. Since LD chemotherapy and adoptive cell infusion occur in a timely staggered manner, CAR T-cells are typically not affected by LD chemotherapy, which is used to eradicate endogenous lymphocytes, create a niche for adoptive cells to engraft, and eliminate immune-suppressive cells in the TME. Fludarabine and cyclophosphamide (flu/cy) are the standard LD agents in leukemia and were adopted in solid tumor cell therapy. However, it is unclear whether this combination is the best for solid tumors or if patients could benefit from a third agent or a therapy modality that remodels the TME and skews the immune repertoire toward effectors. Tumor-targeted radiation could be one such modality. Radiation can eliminate tumor cells and activate the immune system in several ways such as through the cGAS/STING pathway via DNA damage or by inducing an abscopal effect or antigen spread [135,136,137]. Radiopharmaceutical therapy is an attractive approach to reach multiple tumor sites at the same time while administering low-dose radiation [138]. This approach was tested in immunologically “cold” tumors using 86Y-NM600, an alkylphosphocholine analog that accumulates in most tumor types, combined with immune checkpoint inhibitor (ICI) therapy [139]. Combination therapy led to CRs in 45 to 66% of mice compared to 0% with monotherapy and was dependent on STING expression in tumor cells. Interestingly, 86Y-NM600 promoted tumor infiltration by CD8 T-cells and enrichment of the T-cell memory compartment.
In another noteworthy paper, the deletion of the DNA methyltransferase 3 alpha (DNMT3A) in EphA2-CAR T-cells rendered these cells more potent against locoregional osteosarcoma [140]. The CAR T-cells had universally preserved proliferation and function despite being chronically exposed to antigen. These effects were mechanistically linked to an up-regulation of IL-10. Thus, 5′-azacytidine or decitabine could be promising combinatorial agents for CAR T-cell therapy while simultaneously epigenetically editing the tumor and immune landscape in the TME [141, 142].
1.3.2 Signal reprograming techniques
Based on promising preclinical results of CAR T-cell combination therapy with dual ICIs such as anti-PD1 and anti-T-cell immunoreceptor with Ig and ITIM domains (TIGIT), researchers have recently begun genetically engineering CAR T-cells with immunosuppressive countermeasures [143, 144]. One approach to this has been the development of chimeric immune–checkpoint switch receptors (CISR). CISR comprise a chimeric ectodomain that recognizes exhaustion signals such as PD-L1 and CD155 with an intracellular domain designed to stimulate cell activation and survival pathways such as CD28 [145]. In this way, these cells are co-opting immunosuppressive signals for the benefit of growth and survival. This model has shown success in preclinical syngeneic and xenograft mouse models exhibiting improved CAR T-cell tumor infiltration, proliferation, efficacy, and persistence [145]. Since the TME promotes high levels of these immunosuppressive factors, a strong CAR T-cell activation will preferentially occur in the tumor. However, given the robust presence of PD-L1 in the TME, these cells may get overactivation leading to systemic toxicities and premature CAR T-cell senescence. Similarly, inverted cytokine receptors (ICRs) link the extracellular domain of the IL-4 receptor to the transmembrane and intracellular domains of IL-10 [146]. In xenograft mouse models of pancreatic tumors, these CAR T-cells with ICRs demonstrated improved cytotoxicity, effector function, and cytokine release as well as a preserved less differentiated phenotype [146]. CISRs and ICRs demonstrate exciting initial results. Further studies are needed to validate these findings.
1.3.3 Cytokine/chemokine engineering
We have discussed that the chemokine repertoire in the TME attracts immune-suppressive cells rather than effector T-cells. To overcome this hurdle, CAR T-cells have been engineered to express receptors for such chemokines, engendering the ability for them to home to the TME. The list of potential candidates is long [108, 147, 148]. For example, in a xenograft model of lung adenocarcinoma, CAR T-cells with transgenic CCR6, the receptor for the chemokine CCL20, enhanced tumor infiltration and led to prolonged survival of treated mice [149]. Similar effects were noted with the transgenic expression of CCR2b, the receptor for the myeloid attractant CCL2, in a variety of tumor models [150, 151], including neuroblastoma [152]. This approach is in clinical testing in patients with R/R Hodgkin lymphoma and cutaneous T-cell lymphoma receiving CD30 CAR T-cells with transgenic CCR4 (NCT03602157), a chemokine receptor that is classically found in Tregs and type 2 T helper cells but not in effector T-cells. Preliminary results of this trial are promising with no DLT to date and CR rates of 75% [153].
CAR T-cells can also be used as a vehicle for cytokine delivery. Popular cytokines for this type of engineering are γc family cytokines, like IL-2, IL-15, and IL-21. The use of membrane-anchored IL-15 and IL-21 enhanced the cytotoxicity of GD2-CAR and E7-TCR T-cells in preclinical models of neuroblastoma and cervical cancer and limited the occurrence of NK-like T-cells after chronic antigen exposure [154]. Other cytokines that have been used for cytokine engineering are IL-12 and IL-18 [155,156,157]. Importantly, membrane-anchored cytokines can activate the carrier via cis presentation but may be able to influence bystander immune cells via transactivation. While soluble cytokines can achieve a similar effect, the obvious advantage of a cell-restricted delivery method is the local accumulation of the cytokines and potentially fewer side effects. Another elegant method of TME-targeted cytokine delivery is the use IL-12 genetically engineered myeloid cells [158]. After homing to the tumor, they remodel the immune-suppressive TME and decrease metastasis while augmenting T and NK cell activity. Critically, IL12-engineered myeloid cells had limited persistence over time, albeit acutely elevated levels in the lungs. Elevated IL-12 levels were also found in tumors. These engineered cells would be a very attractive approach as a combinatorial strategy with CAR T-cells, given that IL-12 is a potent T-cell pro-inflammatory activator [159,160,161,162]. It remains to be determined if this cell product will cause respiratory toxicity and whether it can synergize with CAR T-cells to fight cancer cells.
1.3.4 ECM remodeling approaches
As one of the hallmarks of cancer, cancer-associated fibrosis results from an excess in collagen and ECM and is a controversial topic since this process can be both pro and anti-cancer [163]. The main cells that contribute to fibrosis are fibroblasts, fibrocytes, stellate cells, and mesenchymal stem cells [164,165,166]. These cells exist in the body for physiologic processes like wound healing and inflammation, but in cancer, they are hijacked and reprogrammed to support the TME [163]. Several studies explored strategies to degrade the ECM in solid tumors and allow for CAR T-cells to migrate into the TME [167]. For example, hyaluronidase-producing CAR T-cells enzymatically degrade hyaluronic acid (HA), a component of the ECM implicated in tumor rigidity and density. When co-administered with ICIs, CAR T-cells with hyaluronidase enriched in the TME and exhibited better antitumor activity than without hyaluronidase in syngeneic models of lymphoma and colon cancer [168]. Generally, each solid tumor has a different composition of the ECM; thus, strategic efforts should aim at broadly characterizing these components to attempt a concerted effort to target them. Attempts to deplete ECM-producing cells like FAP-expressing fibroblasts can be successfully undertaken with antibody–drug conjugates or CAR T-cells [169,170,171]. These and related strategies are still in preclinical development.
Lastly, the area of tumor-mediated post-translational modifications has gained more attention in the past years. Altered glycosylation as a characteristic of carcinogenesis frequently manifests as incomplete synthesis of O-glycans and increased branching of N-glycans [128, 172]. Whereas the former provides neoantigens for cellular therapies, N-glycans may directly impact tumor cell recognition or conceal epitopes. In this role, N-glycans protect solid tumors from synapse formation and ultimately, CAR T-cell killing. Inhibition of N-glycan synthesis with the glucose/mannose analog, 2DG, could offset this shield and restore susceptibility to CAR T-cells [173].
1.3.5 Logistical approaches
The manufacturing time for CAR T-cells presents an additional challenge to delivering cell therapy timely, particularly in patients with rapidly progressing cancer. The manufacturing process typically involves leukapheresis, T-cell isolation and activation, CAR transduction, and ex vivo expansion [174]. The average vein-to-vein time, including transportation, ranges from 3 to 5 weeks, often necessitating bridging therapy [175]. It is therefore critical to accelerate CAR T-cell production if possible. Tackling this challenge, next-generation manufacturing protocols try to accelerate CAR T-cell production through in vivo or in situ transduction methods. For example, in vivo generation of CAR T-cell uses lentiviruses, adeno-associated viruses, lipid nanoparticles, and polymeric nanocarriers, with targeting achieved via single-chain variable fragments for CD4, CD8, or CD3 [176,177,178,179]. Pandit et al. developed the Drydux technology to produce CAR T-cells in situ within 3 days post T-cell isolation using a macroporous biomaterial scaffold [180]. Preclinically, this approach successfully generated CAR T-cells and induced long-term remissions in preclinical models of ovarian, lung, and pancreatic cancer. Although further biomechanical studies are needed to fully delineate the properties of Drydux as an in situ transduction vehicle, early results demonstrate a promising technology for accelerating turnaround times. With all the above-mentioned approaches, risks of off-target gene delivery and concerns for genotoxicity necessitate further research to ensure human safety and efficacy. Another process of accelerating CAR T-cell manufacturing is the generation of CAR T-cells without ex vivo activation and expansion. This process can generate CAR T-cells ready for infusion within 24 h of T-cell isolation [181, 182], bypassing the expansion stage and preserving T-cell stemness, which is linked to prolonged therapeutic benefit [183]. In a phase I trial for multiple myeloma, patients treated with 24-h manufactured anti-CD19 CAR T-cells tolerated the infusion and showed a 98% overall response rate, with CAR T-cells detectable in 71% of patients at 12 months [181]. Potential concerns regarding CAR detection and meeting the regulatory requirements on product release remain a challenge with this approach. Ongoing trials are needed to further clarify the safety and efficacy of these products, emphasizing the importance of understanding biological differences across manufacturing platforms.
2 Conclusions
In reviewing the latest literature on CAR T-cell development, we think that there is momentum in the field to improve this treatment for patients with solid tumors. Though still in the early stages of development, recent years have shown that new creative approaches, advances in technology, and an increasing number of clinical trials have allowed us to understand and tackle the most challenging aspects of CAR T-cell therapy in solid tumors. While these scientific advances are ongoing, the mindset in clinical trials must change with regard to correlative biospecimens. They are the most valuable source for researchers and clinicians to learn what CAR, host, and tumor-related factors prohibit or support CAR T-cell therapy and will allow us to determine in what direction we need to go from here on.
Data availability
No datasets were generated or analysed during the current study.
References
Verma, M., Obergfell, K., Topp, S., Panier, V., & Wu, J. (2023). The next-generation CAR-T therapy landscape. Nature Reviews. Drug Discovery, 22(10), 776–777. https://doi.org/10.1038/d41573-023-00140-7
Maude, S. L., Teachey, D. T., Porter, D. L., & Grupp, S. A. (2015). CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood, 125(26), 4017–4023. https://doi.org/10.1182/blood-2014-12-580068
Maude, S. L., Frey, N., Shaw, P. A., Aplenc, R., Barrett, D. M., Bunin, N. J., et al. (2014). Chimeric antigen receptor T cells for sustained remissions in leukemia. New England Journal of Medicine, 371(16), 1507–1517. https://doi.org/10.1056/NEJMoa1407222
Maude, S. L., Laetsch, T. W., Buechner, J., Rives, S., Boyer, M., Bittencourt, H., et al. (2018). Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. New England Journal of Medicine, 378(5), 439–448. https://doi.org/10.1056/NEJMoa1709866
Cappell, K. M., & Kochenderfer, J. N. (2023). Long-term outcomes following CAR T cell therapy: What we know so far. Nature Reviews Clinical Oncology, 20(6), 359–371. https://doi.org/10.1038/s41571-023-00754-1
Chong, E. A., Ruella, M., Schuster, S. J., Lymphoma Program Investigators at the University of, P. (2021). Five-year outcomes for refractory B-cell lymphomas with CAR T-cell therapy. New England Journal of Medicine, 384(7), 673–674. https://doi.org/10.1056/NEJMc2030164
Morotti, M., Albukhari, A., Alsaadi, A., Artibani, M., Brenton, J. D., Curbishley, S. M., et al. (2021). Promises and challenges of adoptive T-cell therapies for solid tumours. British Journal of Cancer, 124(11), 1759–1776. https://doi.org/10.1038/s41416-021-01353-6
June, C. H., & Sadelain, M. (2018). Chimeric antigen receptor therapy. New England Journal of Medicine, 379(1), 64–73. https://doi.org/10.1056/NEJMra1706169
Labanieh, L., & Mackall, C. L. (2023). CAR immune cells: Design principles, resistance and the next generation. Nature, 614(7949), 635–648. https://doi.org/10.1038/s41586-023-05707-3
Majzner, R. G., & Mackall, C. L. (2019). Clinical lessons learned from the first leg of the CAR T cell journey. Nature Medicine, 25(9), 1341–1355. https://doi.org/10.1038/s41591-019-0564-6
Sadelain, M., Riviere, I., & Riddell, S. (2017). Therapeutic T cell engineering. Nature, 545(7655), 423–431. https://doi.org/10.1038/nature22395
Rafiq, S., Hackett, C. S., & Brentjens, R. J. (2020). Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nature Reviews Clinical Oncology, 17(3), 147–167. https://doi.org/10.1038/s41571-019-0297-y
Wagner, J., Wickman, E., DeRenzo, C., & Gottschalk, S. (2020). CAR T cell therapy for solid tumors: Bright future or dark reality? Molecular Therapy, 28(11), 2320–2339. https://doi.org/10.1016/j.ymthe.2020.09.015
Tantalo, D. G., Oliver, A. J., von Scheidt, B., Harrison, A. J., Mueller, S. N., Kershaw, M. H., et al. (2021). Understanding T cell phenotype for the design of effective chimeric antigen receptor T cell therapies. Journal for ImmunoTherapy of Cancer. https://doi.org/10.1136/jitc-2021-002555
Maher, J., & Davies, D. M. (2023). CAR-based immunotherapy of solid tumours-A survey of the emerging targets. Cancers (Basel). https://doi.org/10.3390/cancers15041171
Gumber, D., & Wang, L. D. (2022). Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. eBioMedicine, 77, 103941. https://doi.org/10.1016/j.ebiom.2022.103941
Del Bufalo, F., De Angelis, B., Caruana, I., Del Baldo, G., De Ioris, M. A., Serra, A., et al. (2023). GD2-CART01 for relapsed or refractory high-risk neuroblastoma. New England Journal of Medicine, 388(14), 1284–1295. https://doi.org/10.1056/NEJMoa2210859
Heczey, A., Louis, C. U., Savoldo, B., Dakhova, O., Durett, A., Grilley, B., et al. (2017). CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Molecular Therapy, 25(9), 2214–2224. https://doi.org/10.1016/j.ymthe.2017.05.012
Kaczanowska, S., Murty, T., Alimadadi, A., Contreras, C. F., Duault, C., Subrahmanyam, P. B., et al. (2024). Immune determinants of CAR-T cell expansion in solid tumor patients receiving GD2 CAR-T cell therapy. Cancer Cell, 42(1), 35–51. https://doi.org/10.1016/j.ccell.2023.11.011. e38.
Louis, C. U., Savoldo, B., Dotti, G., Pule, M., Yvon, E., Myers, G. D., et al. (2011). Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood, 118(23), 6050–6056. https://doi.org/10.1182/blood-2011-05-354449
Pule, M. A., Savoldo, B., Myers, G. D., Rossig, C., Russell, H. V., Dotti, G., et al. (2008). Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nature Medicine, 14(11), 1264–1270. https://doi.org/10.1038/nm.1882
Straathof, K., Flutter, B., Wallace, R., Jain, N., Loka, T., Depani, S., et al. (2020). Antitumor activity without on-target off-tumor toxicity of GD2-chimeric antigen receptor T cells in patients with neuroblastoma. Science Translational Medicine. https://doi.org/10.1126/scitranslmed.abd6169
Majzner, R. G., Ramakrishna, S., Yeom, K. W., Patel, S., Chinnasamy, H., Schultz, L. M., et al. (2022). GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. https://doi.org/10.1038/s41586-022-04489-4
Weber, E. W., Lynn, R. C., Sotillo, E., Lattin, J., Xu, P., & Mackall, C. L. (2019). Pharmacologic control of CAR-T cell function using dasatinib. Blood Advances, 3(5), 711–717. https://doi.org/10.1182/bloodadvances.2018028720
Majzner, R. G., Mahdi, J., Ramakrishna, S., Patel, S., Chinnasamy, H., Yeom, K., et al. (2022). Abstract CT001: Major tumor regressions in H3K27M-mutated diffuse midline glioma (DMG) following sequential intravenous (IV) and intracerebroventricular (ICV) delivery of GD2-CAR T cells. Cancer Research, 82(12_Supplement), CT001–CT001. https://doi.org/10.1158/1538-7445.Am2022-ct001
Rooney, C. M., Smith, C. A., Ng, C. Y. C., Loftin, S. K., Sixbey, J. W., Gan, Y., et al. (1998). Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus–induced lymphoma in allogeneic transplant recipients. Blood, 92(5), 1549–1555. https://doi.org/10.1182/blood.V92.5.1549
Thomas, S., Straathof, K., Himoudi, N., Anderson, J., & Pule, M. (2016). An optimized GD2-targeting retroviral cassette for more potent and safer cellular therapy of neuroblastoma and other cancers. PLoS One1, 11(3), e0152196. https://doi.org/10.1371/journal.pone.0152196
Yu, L., Huang, L., Lin, D., Lai, X., Wu, L., Liao, X., et al. (2022). GD2-specific chimeric antigen receptor-modified T cells for the treatment of refractory and/or recurrent neuroblastoma in pediatric patients. Journal of Cancer Research and Clinical Oncology, 148(10), 2643–2652. https://doi.org/10.1007/s00432-021-03839-5
Quintarelli, C., Orlando, D., Boffa, I., Guercio, M., Polito, V. A., Petretto, A., et al. (2018). Choice of costimulatory domains and of cytokines determines CAR T-cell activity in neuroblastoma. Oncoimmunology, 7(6), e1433518. https://doi.org/10.1080/2162402x.2018.1433518
Navai, S. A., Derenzo, C., Joseph, S., Sanber, K., Byrd, T., Zhang, H., et al. (2019). Abstract LB-147: Administration of HER2-CAR T cells after lymphodepletion safely improves T cell expansion and induces clinical responses in patients with advanced sarcomas. Cancer Research, 79(13_Supplement), LB-147-LB-147. https://doi.org/10.1158/1538-7445.Am2019-lb-147
Rainusso, N., Brawley, V. S., Ghazi, A., Hicks, M. J., Gottschalk, S., Rosen, J. M., et al. (2012). Immunotherapy targeting HER2 with genetically modified T cells eliminates tumor-initiating cells in osteosarcoma. Cancer Gene Therapy, 19(3), 212–217. https://doi.org/10.1038/cgt.2011.83
Sabina, K., Sneha, R., Tara, M., Cristina, C., Ahmad, A., Norma, G., et al. (2022). 397 Deep myeloid cell profiling provides new insights into modulators of CAR T cell expansion in patients with solid tumor malignancies. Journal for ImmunoTherapy of Cancer, 10(Suppl 2), A418. https://doi.org/10.1136/jitc-2022-SITC2022.0397
Albert, C. M., Pinto, N. R., Taylor, M., Wilson, A., Rawlings-Rhea, S., Mgebroff, S., et al. (2022). STRIvE-01: Phase I study of EGFR806 CAR T-cell immunotherapy for recurrent/refractory solid tumors in children and young adults. Journal of Clinical Oncology, 40(16_suppl), 2541–2541. https://doi.org/10.1200/JCO.2022.40.16_suppl.2541
Pinto, N. R., Albert, C. M., Taylor, M., Wilson, A., Rawlings-Rhea, S., Huang, W., et al. (2022). STRIVE-02: A first-in-human phase 1 trial of systemic B7H3 CAR T cells for children and young adults with relapsed/refractory solid tumors. Journal of Clinical Oncology, 40(16_suppl), 10011–10011. https://doi.org/10.1200/JCO.2022.40.16_suppl.10011
Majzner, R. G., Ramakrishna, S., Yeom, K. W., Patel, S., Chinnasamy, H., Schultz, L. M., et al. (2022). GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature, 603(7903), 934–941. https://doi.org/10.1038/s41586-022-04489-4
Monje, M., Majzner, R., Mahdi, J., Ramakrishna, S., Patel, S., Chinnasamy, H., et al. (2022). DIPG-15. Major tumor regressions in H3K27M-mutated diffuse midline glioma (DMG) following sequential intravenous (IV) and intracerebroventricular (ICV) delivery of GD2-CAR T-cells. Neuro-Oncology, 24(Suppl 1), i20-21. https://doi.org/10.1093/neuonc/noac079.072
Mount, C. W., Majzner, R. G., Sundaresh, S., Arnold, E. P., Kadapakkam, M., Haile, S., et al. (2018). Potent antitumor efficacy of anti-GD2 CAR T cells in H3–K27M(+) diffuse midline gliomas. Nature Medicine, 24(5), 572–579. https://doi.org/10.1038/s41591-018-0006-x
Fu, Q., Zheng, Y., Fang, W., Zhao, Q., Zhao, P., Liu, L., et al. (2023). RUNX-3-expressing CAR T cells targeting glypican-3 in patients with heavily pretreated advanced hepatocellular carcinoma: A phase I trial. EClinicalMedicine, 63, 102175. https://doi.org/10.1016/j.eclinm.2023.102175
Tang, J., Sheng, J., Zhang, Q., Ji, Y., Wang, X., Zhang, J., et al. (2023). Runx3-overexpression cooperates with ex vivo AKT inhibition to generate receptor-engineered T cells with better persistence, tumor-residency, and antitumor ability. Journal for Immunotherapy of Cancer. https://doi.org/10.1136/jitc-2022-006119
Li, D., Guo, X., Yang, K., Yang, Y., Zhou, W., Huang, Y., et al. (2023). EpCAM-targeting CAR-T cell immunotherapy is safe and efficacious for epithelial tumors. Science Advances, 9(48), eadg9721. https://doi.org/10.1126/sciadv.adg9721
Zhang, C., Wang, Z., Yang, Z., Wang, M., Li, S., Li, Y., et al. (2017). Phase I escalating-dose trial of CAR-T therapy targeting CEA(+) metastatic colorectal cancers. Molecular Therapy, 25(5), 1248–1258. https://doi.org/10.1016/j.ymthe.2017.03.010
Wang, Y., Chen, M., Wu, Z., Tong, C., Dai, H., Guo, Y., et al. (2018). CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology, 7(7), e1440169. https://doi.org/10.1080/2162402x.2018.1440169
Brown, C. E., Alizadeh, D., Starr, R., Weng, L., Wagner, J. R., Naranjo, A., et al. (2016). Regression of glioblastoma after chimeric antigen receptor T-cell therapy. New England Journal of Medicine, 375(26), 2561–2569. https://doi.org/10.1056/NEJMoa1610497
Brown, C. E., Hibbard, J. C., Alizadeh, D., Blanchard, M. S., Natri, H. M., Wang, D., et al. (2024). Locoregional delivery of IL-13Rα2-targeting CAR-T cells in recurrent high-grade glioma: A phase 1 trial. Nature Medicine. https://doi.org/10.1038/s41591-024-02875-1
Qi, C., Gong, J., Li, J., Liu, D., Qin, Y., Ge, S., et al. (2022). Claudin18.2-specific CAR T cells in gastrointestinal cancers: Phase 1 trial interim results. Nature Medicine, 28(6), 1189–1198. https://doi.org/10.1038/s41591-022-01800-8
Jiang, H., Shi, Z., Wang, P., Wang, C., Yang, L., Du, G., et al. (2019). Claudin18.2-Specific chimeric antigen receptor engineered T cells for the treatment of gastric cancer. Journal of the National Cancer Institute, 111(4), 409–418. https://doi.org/10.1093/jnci/djy134
Qi, C., Xie, T., Zhou, J., Wang, X., Gong, J., Zhang, X., et al. (2023). CT041 CAR T cell therapy for Claudin18.2-positive metastatic pancreatic cancer. Journal of Hematology Oncology, 16(1), 102. https://doi.org/10.1186/s13045-023-01491-9
Qi, C., Chang, L., Li, J., Gong, J., Wang, X., Wang, Z., et al. (2023). 1018O Phase I study of GCC CAR-T therapy IM96 in patients with advanced colorectal cancer. Annals of Oncology, 34, S620. https://doi.org/10.1016/j.annonc.2023.09.2157
Specht, J., Lee, S., Turtle, C., Berger, C., Balakrishnan, A., Srivastava, S., et al. (2019). Abstract P2–09–13: A phase I study of adoptive immunotherapy for ROR1+ advanced triple negative breast cancer (TNBC) with defined subsets of autologous T cells expressing a ROR1-specific chimeric antigen receptor (ROR1-CAR). Cancer Research, 79(4_Supplement), P2-09-13-P02-09–13. https://doi.org/10.1158/1538-7445.Sabcs18-p2-09-13
Srivastava, S., Furlan, S. N., Jaeger-Ruckstuhl, C. A., Sarvothama, M., Berger, C., Smythe, K. S., et al. (2021). Immunogenic chemotherapy enhances recruitment of CAR-T cells to lung tumors and improves antitumor efficacy when combined with checkpoint blockade. Cancer Cell, 39(2), 193-208.e110. https://doi.org/10.1016/j.ccell.2020.11.005
Guo, Y., Feng, K., Liu, Y., Wu, Z., Dai, H., Yang, Q., et al. (2018). Phase I study of chimeric antigen receptor-modified T cells in patients with EGFR-positive advanced biliary tract cancers. Clinical Cancer Research, 24(6), 1277–1286. https://doi.org/10.1158/1078-0432.Ccr-17-0432
Feng, K., Guo, Y., Dai, H., Wang, Y., Li, X., Jia, H., et al. (2016). Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Science China Life sciences, 59(5), 468–479. https://doi.org/10.1007/s11427-016-5023-8
Liu, Y., Guo, Y., Wu, Z., Feng, K., Tong, C., Wang, Y., et al. (2020). Anti-EGFR chimeric antigen receptor-modified T cells in metastatic pancreatic carcinoma: A phase I clinical trial. Cytotherapy, 22(10), 573–580. https://doi.org/10.1016/j.jcyt.2020.04.088
Grosser, R., Cherkassky, L., Chintala, N., & Adusumilli, P. S. (2019). Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell, 36(5), 471–482. https://doi.org/10.1016/j.ccell.2019.09.006
Adusumilli, P. S., Zauderer, M. G., Rivière, I., Solomon, S. B., Rusch, V. W., O’Cearbhaill, R. E., et al. (2021). A phase I trial of regional mesothelin-targeted CAR T-cell therapy in patients with malignant pleural disease, in combination with the anti-PD-1 agent pembrolizumab. Cancer Discovery, 11(11), 2748–2763. https://doi.org/10.1158/2159-8290.Cd-21-0407
Narayan, V., Barber-Rotenberg, J. S., Jung, I. Y., Lacey, S. F., Rech, A. J., Davis, M. M., et al. (2022). PSMA-targeting TGFβ-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: A phase 1 trial. Nature Medicine, 28(4), 724–734. https://doi.org/10.1038/s41591-022-01726-1
Brochier, W., Bricard, O., & Coulie, P. G. (2023). Facts and hopes in cancer antigens recognized by T cells. Clinical Cancer Research, 29(2), 309–315. https://doi.org/10.1158/1078-0432.Ccr-21-3798
Leko, V., & Rosenberg, S. A. (2020). Identifying and targeting human tumor antigens for T cell-based immunotherapy of solid tumors. Cancer Cell, 38(4), 454–472. https://doi.org/10.1016/j.ccell.2020.07.013
DeRenzo, C., Krenciute, G., & Gottschalk, S. (2018). The landscape of CAR T cells beyond acute lymphoblastic leukemia for pediatric solid tumors. American Society of Clinical Oncology Educational Book, 38, 830–837. https://doi.org/10.1200/edbk_200773
Newick, K., Moon, E., & Albelda, S. M. (2016). Chimeric antigen receptor T-cell therapy for solid tumors. Molecular Therapy Oncolytics, 3, 16006. https://doi.org/10.1038/mto.2016.6
Neelapu, S. S., Locke, F. L., Bartlett, N. L., Lekakis, L. J., Miklos, D. B., Jacobson, C. A., et al. (2017). Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. New England Journal of Medicine, 377(26), 2531–2544. https://doi.org/10.1056/NEJMoa1707447
Schuster, S. J., Svoboda, J., Chong, E. A., Nasta, S. D., Mato, A. R., Anak, O., et al. (2017). Chimeric antigen receptor T cells in refractory B-cell lymphomas. New England Journal of Medicine, 377(26), 2545–2554. https://doi.org/10.1056/NEJMoa1708566
Parker, K. R., Migliorini, D., Perkey, E., Yost, K. E., Bhaduri, A., Bagga, P., et al. (2020). Single-cell analyses identify brain mural cells expressing CD19 as potential off-tumor targets for CAR-T immunotherapies. Cell, 183(1), 126–142. https://doi.org/10.1016/j.cell.2020.08.022. e117.
Lammie, G., Cheung, N., Gerald, W., Rosenblum, M., & Cordoncardo, C. (1993). Ganglioside gd(2) expression in the human nervous-system and in neuroblastomas - An immunohistochemical study. International Journal of Oncology, 3(5), 909–915. https://doi.org/10.3892/ijo.3.5.909
Heczey, A., Courtney, A. N., Montalbano, A., Robinson, S., Liu, K., Li, M., et al. (2020). Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: An interim analysis. Nature Medicine, 26(11), 1686–1690. https://doi.org/10.1038/s41591-020-1074-2
Mahdi, J., Dietrich, J., Straathof, K., Roddie, C., Scott, B. J., Davidson, T. B., et al. (2023). Tumor inflammation-associated neurotoxicity. Nature Medicine, 29(4), 803–810. https://doi.org/10.1038/s41591-023-02276-w
Richman, S. A., Nunez-Cruz, S., Moghimi, B., Li, L. Z., Gershenson, Z. T., Mourelatos, Z., et al. (2018). High-affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunology Research, 6(1), 36–46. https://doi.org/10.1158/2326-6066.CIR-17-0211
Majzner, R. G., Weber, E. W., Lynn, R. C., Xu, P., & Mackall, C. L. (2018). Neurotoxicity associated with a high-affinity GD2 CAR-letter. Cancer Immunology Research, 6(4), 494–495. https://doi.org/10.1158/2326-6066.CIR-18-0089
Heitzeneder, S., Bosse, K. R., Zhu, Z., Zhelev, D., Majzner, R. G., Radosevich, M. T., et al. (2022). GPC2-CAR T cells tuned for low antigen density mediate potent activity against neuroblastoma without toxicity. Cancer Cell, 40(1), 53-69.e59. https://doi.org/10.1016/j.ccell.2021.12.005
Shaw, T. I., Wagner, J., Tian, L., Wickman, E., Poudel, S., Wang, J., et al. (2024). Discovery of immunotherapy targets for pediatric solid and brain tumors by exon-level expression. Nature Communications, 15(1), 3732. https://doi.org/10.1038/s41467-024-47649-y
Yan, Q., Fang, X., Li, C., Lan, P., & Guan, X. (2022). Oncofetal proteins and cancer stem cells. Essays in Biochemistry, 66(4), 423–433. https://doi.org/10.1042/EBC20220025
Bosse, K. R., Raman, P., Zhu, Z., Lane, M., Martinez, D., Heitzeneder, S., et al. (2017). Identification of GPC2 as an oncoprotein and candidate immunotherapeutic target in high-risk neuroblastoma. Cancer Cell, 32(3), 295–309. https://doi.org/10.1016/j.ccell.2017.08.003. e212.
Li, N., Fu, H., Hewitt, S. M., Dimitrov, D. S., & Ho, M. (2017). Therapeutically targeting glypican-2 via single-domain antibody-based chimeric antigen receptors and immunotoxins in neuroblastoma. Proceedings of the National Academy of Sciences USA, 114(32), E6623–E6631. https://doi.org/10.1073/pnas.1706055114
Li, N., Torres, M. B., Spetz, M. R., Wang, R., Peng, L., Tian, M., et al. (2021). CAR T cells targeting tumor-associated exons of glypican 2 regress neuroblastoma in mice. Cell Rep Med, 2(6), 100297. https://doi.org/10.1016/j.xcrm.2021.100297
Foster, J. B., Griffin, C., Rokita, J. L., Stern, A., Brimley, C., Rathi, K., et al. (2022). Development of GPC2-directed chimeric antigen receptors using mRNA for pediatric brain tumors. Journal for ImmunoTherapy of Cancer. https://doi.org/10.1136/jitc-2021-004450
Sun, M., Cao, Y., Okada, R., Reyes-Gonzalez, J. M., Stack, H. G., Qin, H., et al. (2023). Preclinical optimization of a GPC2-targeting CAR T-cell therapy for neuroblastoma. Journal for ImmunoTherapy of Cancer. https://doi.org/10.1136/jitc-2022-005881
Tian, M., Cheuk, A. T., Wei, J. S., Abdelmaksoud, A., Chou, H. C., Milewski, D., et al. (2022). An optimized bicistronic chimeric antigen receptor against GPC2 or CD276 overcomes heterogeneous expression in neuroblastoma. The Journal of Clinical Investigation. https://doi.org/10.1172/JCI155621
Long, A. H., Haso, W. M., Shern, J. F., Wanhainen, K. M., Murgai, M., Ingaramo, M., et al. (2015). 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nature Medicine, 21(6), 581–590. https://doi.org/10.1038/nm.3838
Allen, G. M., Frankel, N. W., Reddy, N. R., Bhargava, H. K., Yoshida, M. A., Stark, S. R., et al. (2022). Synthetic cytokine circuits that drive T cells into immune-excluded tumors. Science, 378(6625), 1624. https://doi.org/10.1126/science.aba1624. eaba1624.
Hyrenius-Wittsten, A., Su, Y., Park, M., Garcia, J. M., Alavi, J., Perry, N., et al. (2021). SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Science Translational Medicine, 13(591), 8836.
Moghimi, B., Muthugounder, S., Jambon, S., Tibbetts, R., Hung, L., Bassiri, H., et al. (2021). Preclinical assessment of the efficacy and specificity of GD2-B7H3 SynNotch CAR-T in metastatic neuroblastoma. Nature Communications, 12(1), 511. https://doi.org/10.1038/s41467-020-20785-x
Morsut, L., Roybal, K. T., Xiong, X., Gordley, R. M., Coyle, S. M., Thomson, M., et al. (2016). Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell, 164(4), 780–791. https://doi.org/10.1016/j.cell.2016.01.012
Roybal, K. T., Williams, J. Z., Morsut, L., Rupp, L. J., Kolinko, I., Choe, J. H., et al. (2016). Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell, 167(2), 419–432. https://doi.org/10.1016/j.cell.2016.09.011. e416.
Srivastava, S., Salter, A. I., Liggitt, D., Yechan-Gunja, S., Sarvothama, M., Cooper, K., et al. (2019). Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell, 35(3), 489–503. https://doi.org/10.1016/j.ccell.2019.02.003. e488.
Tousley, A. M., Rotiroti, M. C., Labanieh, L., Rysavy, L. W., Kim, W. J., Lareau, C., et al. (2023). Co-opting signalling molecules enables logic-gated control of CAR T cells. Nature, 615(7952), 507–516. https://doi.org/10.1038/s41586-023-05778-2
Fedorov, V. D., Themeli, M., & Sadelain, M. (2013). PD-1–and CTLA-4–based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Science Translational Medicine, 5(215), 215ra172-215ra172.
Fedorov, V. D., Themeli, M., & Sadelain, M. (2013). PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Science Translational Medicine, 5(215), 215ra172. https://doi.org/10.1126/scitranslmed.3006597
Shalabi, H., Qin, H., Su, A., Yates, B., Wolters, P. L., Steinberg, S. M., et al. (2022). CD19/22 CAR T-cells in children and young adults with B-ALL: Phase I results and development of a novel bicistronic CAR. Blood. https://doi.org/10.1182/blood.2022015795
Munshi, N. C., Anderson, L. D., Jr, Shah, N., Madduri, D., Berdeja, J., Lonial, S., et al. (2021). Idecabtagene vicleucel in relapsed and refractory multiple myeloma. New England Journal of Medicine, 384(8), 705–716. https://doi.org/10.1056/NEJMoa2024850
Foran, J. M., Norton, A. J., Micallef, I. N., Taussig, D. C., Amess, J. A., Rohatiner, A. Z., et al. (2001). Loss of CD20 expression following treatment with rituximab (chimaeric monoclonal anti-CD20): A retrospective cohort analysis. British Journal of Haematology, 114(4), 881–883. https://doi.org/10.1046/j.1365-2141.2001.03019.x
Jones, J. D., Hamilton, B. J., & Rigby, W. F. (2012). Rituximab mediates loss of CD19 on B cells in the absence of cell death. Arthritis and Rheumatism, 64(10), 3111–3118. https://doi.org/10.1002/art.34560
Kramer, K., Gerald, W. L., Kushner, B. H., Larson, S. M., Hameed, M., & Cheung, N. K. (2001). Disaloganglioside GD2 loss following monoclonal antibody therapy is rare in neuroblastoma. Medical and Pediatric Oncology, 36(1), 194–196. https://doi.org/10.1002/1096-911X(20010101)36:1<194::AID-MPO1046>3.0.CO;2-B
Majzner, R. G., & Mackall, C. L. (2018). Tumor antigen escape from CAR T-cell therapy. Cancer Discovery, 8(10), 1219–1226. https://doi.org/10.1158/2159-8290.CD-18-0442
Muhammad, N., Wang, R., Li, W., Zhang, Z., Chang, Y., Hu, Y., et al. (2022). A novel TanCAR targeting IL13Ralpha2 and EphA2 for enhanced glioblastoma therapy. Molecular Therapy Oncolytics, 24, 729–741. https://doi.org/10.1016/j.omto.2022.02.012
Roddie, C., Lekakis, L. J., Marzolini, M. A. V., Ramakrishnan, A., Zhang, Y., Hu, Y., et al. (2023). Dual targeting of CD19 and CD22 with bicistronic CAR-T cells in patients with relapsed/refractory large B-cell lymphoma. Blood, 141(20), 2470–2482. https://doi.org/10.1182/blood.2022018598
McCue, A. C., Yao, Z., & Kuhlman, B. (2022). Advances in modular control of CAR-T therapy with adapter-mediated CARs. Advanced Drug Delivery Reviews, 187, 114358. https://doi.org/10.1016/j.addr.2022.114358
Vincent, R. L., Gurbatri, C. R., Li, F., Vardoshvili, A., Coker, C., Im, J., et al. (2023). Probiotic-guided CAR-T cells for solid tumor targeting. Science, 382(6667), 211–218. https://doi.org/10.1126/science.add7034
Mayer, S., Milo, T., Isaacson, A., Halperin, C., Miyara, S., Stein, Y., et al. (2023). The tumor microenvironment shows a hierarchy of cell-cell interactions dominated by fibroblasts. Nature Communications, 14(1), 5810. https://doi.org/10.1038/s41467-023-41518-w
Anderson, N. M., & Simon, M. C. (2020). The tumor microenvironment. Current Biology, 30(16), R921–R925. https://doi.org/10.1016/j.cub.2020.06.081
Liu, G., Rui, W., Zhao, X., & Lin, X. (2021). Enhancing CAR-T cell efficacy in solid tumors by targeting the tumor microenvironment. Cellular & Molecular Immunology, 18(5), 1085–1095. https://doi.org/10.1038/s41423-021-00655-2
Boutilier, A. J., & Elsawa, S. F. (2021). Macrophage polarization states in the tumor microenvironment. International Journal of Molecular Sciences. https://doi.org/10.3390/ijms22136995
Wynn, T. A. (2008). Cellular and molecular mechanisms of fibrosis. The Journal of Pathology, 214(2), 199–210. https://doi.org/10.1002/path.2277
Quail, D. F., & Joyce, J. A. (2013). Microenvironmental regulation of tumor progression and metastasis. Nature Medicine, 19(11), 1423–1437. https://doi.org/10.1038/nm.3394
Togashi, Y., Shitara, K., & Nishikawa, H. (2019). Regulatory T cells in cancer immunosuppression - Implications for anticancer therapy. Nature Reviews Clinical Oncology, 16(6), 356–371. https://doi.org/10.1038/s41571-019-0175-7
Molon, B., Ugel, S., Del Pozzo, F., Soldani, C., Zilio, S., Avella, D., et al. (2011). Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. Journal of Experimental Medicine, 208(10), 1949–1962. https://doi.org/10.1084/jem.20101956
Peng, J. J., Wang, L., Li, Z., Ku, C. L., & Ho, P. C. (2023). Metabolic challenges and interventions in CAR T cell therapy. Science Immunology, 8(82), eabq3016. https://doi.org/10.1126/sciimmunol.abq3016
Kantari-Mimoun, C., Barrin, S., Vimeux, L., Haghiri, S., Gervais, C., Joaquina, S., et al. (2021). CAR T-cell entry into tumor islets is a two-step process dependent on IFNgamma and ICAM-1. Cancer Immunology Research, 9(12), 1425–1438. https://doi.org/10.1158/2326-6066.CIR-20-0837
Sun, R., Sun, Y., Wu, C., Liu, Y., Zhou, M., Dong, Y., et al. (2023). CXCR4-modified CAR-T cells suppresses MDSCs recruitment via STAT3/NF-kappaB/SDF-1alpha axis to enhance efficacy against pancreatic cancer. Molecular Therapy, 31(11), 3193–3209. https://doi.org/10.1016/j.ymthe.2023.09.010
Turtle, C. J., Hanafi, L. A., Berger, C., Gooley, T. A., Cherian, S., Hudecek, M., et al. (2016). CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. The Journal of Clinical Investigation, 126(6), 2123–2138. https://doi.org/10.1172/JCI85309
Weninger, W., Crowley, M. A., Manjunath, N., & von Andrian, U. H. (2001). Migratory properties of naive, effector, and memory CD8(+) T cells. Journal of Experimental Medicine, 194(7), 953–966. https://doi.org/10.1084/jem.194.7.953
Choi, H., Song, H., & Jung, Y. W. (2020). The roles of CCR7 for the homing of memory CD8+ T cells into their survival niches. Immune Network, 20(3), e20. https://doi.org/10.4110/in.2020.20.e20
Albelda, S. M. (2024). CAR T cell therapy for patients with solid tumours: Key lessons to learn and unlearn. Nature Reviews Clinical Oncology, 21(1), 47–66. https://doi.org/10.1038/s41571-023-00832-4
Tchou, J., Zhao, Y., Levine, B. L., Zhang, P. J., Davis, M. M., Melenhorst, J. J., et al. (2017). Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunology Research, 5(12), 1152–1161. https://doi.org/10.1158/2326-6066.CIR-17-0189
Vitanza, N. A., Johnson, A. J., Wilson, A. L., Brown, C., Yokoyama, J. K., Künkele, A., et al. (2021). Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: An interim analysis. Nature Medicine, 27(9), 1544–1552. https://doi.org/10.1038/s41591-021-01404-8
Fisher, B., Packard, B. S., Read, E. J., Carrasquillo, J. A., Carter, C. S., Topalian, S. L., et al. (1989). Tumor localization of adoptively transferred indium-111 labeled tumor infiltrating lymphocytes in patients with metastatic melanoma. Journal of Clinical Oncology, 7(2), 250–261. https://doi.org/10.1200/JCO.1989.7.2.250
Griffith, K. D., Read, E. J., Carrasquillo, J. A., Carter, C. S., Yang, J. C., Fisher, B., et al. (1989). In vivo distribution of adoptively transferred indium-111-labeled tumor infiltrating lymphocytes and peripheral blood lymphocytes in patients with metastatic melanoma. Journal of the National Cancer Institute, 81(22), 1709–1717. https://doi.org/10.1093/jnci/81.22.1709
Yaghoubi, S. S., Jensen, M. C., Satyamurthy, N., Budhiraja, S., Paik, D., Czernin, J., et al. (2009). Noninvasive detection of therapeutic cytolytic T cells with 18F-FHBG PET in a patient with glioma. Nature Clinical Practice Oncology, 6(1), 53–58. https://doi.org/10.1038/ncponc1278
Keu, K. V., Witney, T. H., Yaghoubi, S., Rosenberg, J., Kurien, A., Magnusson, R., et al. (2017). Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Science Translational Medicine. https://doi.org/10.1126/scitranslmed.aag2196
Huang, Y., Kim, B. Y. S., Chan, C. K., Hahn, S. M., Weissman, I. L., & Jiang, W. (2018). Improving immune-vascular crosstalk for cancer immunotherapy. Nature Reviews Immunology, 18(3), 195–203. https://doi.org/10.1038/nri.2017.145
Lanitis, E., Irving, M., & Coukos, G. (2015). Targeting the tumor vasculature to enhance T cell activity. Current Opinion in Immunology, 33, 55–63. https://doi.org/10.1016/j.coi.2015.01.011
Carmeliet, P., & Jain, R. K. (2000). Angiogenesis in cancer and other diseases. Nature, 407(6801), 249–257. https://doi.org/10.1038/35025220
Shweiki, D., Itin, A., Soffer, D., & Keshet, E. (1992). Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature, 359(6398), 843–845. https://doi.org/10.1038/359843a0
Kourembanas, S., Hannan, R. L., & Faller, D. V. (1990). Oxygen tension regulates the expression of the platelet-derived growth factor-B chain gene in human endothelial cells. The Journal of Clinical Investigation, 86(2), 670–674. https://doi.org/10.1172/jci114759
Jain, R. K. (2005). Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science, 307(5706), 58–62. https://doi.org/10.1126/science.1104819
Jain, R. K. (2014). Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell, 26(5), 605–622. https://doi.org/10.1016/j.ccell.2014.10.006
Buckanovich, R. J., Facciabene, A., Kim, S., Benencia, F., Sasaroli, D., Balint, K., et al. (2008). Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nature Medicine, 14(1), 28–36. https://doi.org/10.1038/nm1699
Wu, N. Z., Klitzman, B., Dodge, R., & Dewhirst, M. W. (1992). Diminished leukocyte-endothelium interaction in tumor microvessels. Cancer Research, 52(15), 4265–4268.
Granovsky, M., Fata, J., Pawling, J., Muller, W. J., Khokha, R., & Dennis, J. W. (2000). Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nature Medicine, 6(3), 306–312. https://doi.org/10.1038/73163
Huang, Y., Yuan, J., Righi, E., Kamoun, W. S., Ancukiewicz, M., Nezivar, J., et al. (2012). Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proceedings of the National Academy of Sciences USA, 109(43), 17561–17566. https://doi.org/10.1073/pnas.1215397109
Fukumura, D., Kloepper, J., Amoozgar, Z., Duda, D. G., & Jain, R. K. (2018). Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nature Reviews. Clinical Oncology, 15(5), 325–340. https://doi.org/10.1038/nrclinonc.2018.29
Chinnasamy, D., Yu, Z., Theoret, M. R., Zhao, Y., Shrimali, R. K., Morgan, R. A., et al. (2010). Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. The Journal of Clinical Investigation, 120(11), 3953–3968. https://doi.org/10.1172/jci43490
Dong, X., Ren, J., Amoozgar, Z., Lee, S., Datta, M., Roberge, S., et al. (2023). Anti-VEGF therapy improves EGFR-vIII-CAR-T cell delivery and efficacy in syngeneic glioblastoma models in mice. Journal for ImmunoTherapy of Cancer. https://doi.org/10.1136/jitc-2022-005583
Trowell, O. A. (1952). The sensitivity of lymphocytes to ionising radiation. The Journal of Pathology and Bacteriology, 64(4), 687–704. https://doi.org/10.1002/path.1700640403
Louagie, H., Van Eijkeren, M., Philippe, J., Thierens, H., & de Ridder, L. (1999). Changes in peripheral blood lymphocyte subsets in patients undergoing radiotherapy. International Journal of Radiation Biology, 75(6), 767–771. https://doi.org/10.1080/095530099140113
Burnette, B. C., Liang, H., Lee, Y., Chlewicki, L., Khodarev, N. N., Weichselbaum, R. R., et al. (2011). The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Research, 71(7), 2488–2496. https://doi.org/10.1158/0008-5472.CAN-10-2820
Postow, M. A., Callahan, M. K., Barker, C. A., Yamada, Y., Yuan, J., Kitano, S., et al. (2012). Immunologic correlates of the abscopal effect in a patient with melanoma. New England Journal of Medicine, 366(10), 925–931. https://doi.org/10.1056/NEJMoa1112824
Lussier, D. M., Alspach, E., Ward, J. P., Miceli, A. P., Runci, D., White, J. M., et al. (2021). Radiation-induced neoantigens broaden the immunotherapeutic window of cancers with low mutational loads. Proceedings of the National Academy of Sciences USA. https://doi.org/10.1073/pnas.2102611118
Salerno, K. E., Roy, S., Ribaudo, C., Fisher, T., Patel, R. B., Mena, E., et al. (2023). A primer on radiopharmaceutical therapy. International Journal of Radiation Oncology Biology Physics, 115(1), 48–59. https://doi.org/10.1016/j.ijrobp.2022.08.010
Patel, R. B., Hernandez, R., Carlson, P., Grudzinski, J., Bates, A. M., Jagodinsky, J. C., et al. (2021). Low-dose targeted radionuclide therapy renders immunologically cold tumors responsive to immune checkpoint blockade. Science Translational Medicine. https://doi.org/10.1126/scitranslmed.abb3631
Prinzing, B., Zebley, C. C., Petersen, C. T., Fan, Y., Anido, A. A., Yi, Z., et al. (2021). Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Science Translational Medicine, 13(620), 0272. https://doi.org/10.1126/scitranslmed.abh0272
Kim, K., Skora, A. D., Li, Z., Liu, Q., Tam, A. J., Blosser, R. L., et al. (2014). Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proceedings of the National Academy of Sciences, 111(32), 11774–11779. https://doi.org/10.1073/pnas.1410626111
Lu, Z., Zou, J., Li, S., Topper, M. J., Tao, Y., Zhang, H., et al. (2020). Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature, 579(7798), 284–290. https://doi.org/10.1038/s41586-020-2054-x
Hung, A. L., Maxwell, R., Theodros, D., Belcaid, Z., Mathios, D., Luksik, A. S., et al. (2018). TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology, 7(8), e1466769. https://doi.org/10.1080/2162402x.2018.1466769
Banta, K. L., Xu, X., Chitre, A. S., Au-Yeung, A., Takahashi, C., O’Gorman, W. E., et al. (2022). Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8(+) T cell responses. Immunity, 55(3), 512-526.e519. https://doi.org/10.1016/j.immuni.2022.02.005
Zhao, J., Dong, J., Deng, C., Zhang, Q., Sun, S., Li, H., et al. (2023). Enhancing T cell anti-tumor efficacy with a PD1-TIGIT chimeric immune-checkpoint switch receptor. Oncoimmunology, 12(1), 2265703. https://doi.org/10.1080/2162402X.2023.2265703
Zhou, Y., Farooq, M. A., Ajmal, I., He, C., Gao, Y., Guo, D., et al. (2023). Co-expression of IL-4/IL-15-based inverted cytokine receptor in CAR-T cells overcomes IL-4 signaling in immunosuppressive pancreatic tumor microenvironment. Biomedicine & Pharmacotherapy, 168, 115740. https://doi.org/10.1016/j.biopha.2023.115740
Adachi, K., Kano, Y., Nagai, T., Okuyama, N., Sakoda, Y., & Tamada, K. (2018). IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nature Biotechnology, 36(4), 346–351. https://doi.org/10.1038/nbt.4086
Luo, H., Su, J., Sun, R., Sun, Y., Wang, Y., Dong, Y., et al. (2020). Coexpression of IL7 and CCL21 increases efficacy of CAR-T cells in solid tumors without requiring preconditioned lymphodepletion. Clinical Cancer Research, 26(20), 5494–5505. https://doi.org/10.1158/1078-0432.Ccr-20-0777
Jin, L., Cao, L., Zhu, Y., Cao, J., Li, X., Zhou, J., et al. (2021). Enhance anti-lung tumor efficacy of chimeric antigen receptor-T cells by ectopic expression of C-C motif chemokine receptor 6. Science Bulletin, 66(8), 803–812. https://doi.org/10.1016/j.scib.2020.12.027
Moon, E. K., Carpenito, C., Sun, J., Wang, L. C., Kapoor, V., Predina, J., et al. (2011). Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clinical Cancer Research, 17(14), 4719–4730. https://doi.org/10.1158/1078-0432.Ccr-11-0351
Wang, Y., Wang, J., Yang, X., Yang, J., Lu, P., Zhao, L., et al. (2021). Chemokine receptor CCR2b enhanced anti-tumor function of chimeric antigen receptor T cells targeting mesothelin in a non-small-cell lung carcinoma model. Frontiers in Immunology, 12, 628906. https://doi.org/10.3389/fimmu.2021.628906
Craddock, J. A., Lu, A., Bear, A., Pule, M., Brenner, M. K., Rooney, C. M., et al. (2010). Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. Journal of Immunotherapy, 33(8), 780–788. https://doi.org/10.1097/CJI.0b013e3181ee6675
Meier, J. A., Savoldo, B., & Grover, N. S. (2022). The emerging role of CAR T cell therapy in relapsed/refractory Hodgkin lymphoma. Journal of Personalized Medicine. https://doi.org/10.3390/jpm12020197
Nguyen, R., Doubrovina, E., Mousset, C. M., Jin, B. Y., Okada, R., Zhang, X., et al. (2023). Cooperative armoring of CAR and TCR T-cells by T cell-restricted IL-15 and IL-21 universally enhances solid tumor efficacy. Clinical Cancer Research. https://doi.org/10.1158/1078-0432.CCR-23-1872
Zhang, L., Davies, J. S., Serna, C., Yu, Z., Restifo, N. P., Rosenberg, S. A., et al. (2020). Enhanced efficacy and limited systemic cytokine exposure with membrane-anchored interleukin-12 T-cell therapy in murine tumor models. Journal for ImmunoTherapy of Cancer. https://doi.org/10.1136/jitc-2019-000210
Hu, B., Ren, J., Luo, Y., Keith, B., Young, R. M., Scholler, J., et al. (2017). Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Reports, 20(13), 3025–3033. https://doi.org/10.1016/j.celrep.2017.09.002
Jaspers, J. E., Khan, J. F., Godfrey, W. D., Lopez, A. V., Ciampricotti, M., Rudin, C. M., et al. (2023). IL-18-secreting CAR T cells targeting DLL3 are highly effective in small cell lung cancer models. The Journal of Clinical Investigation. https://doi.org/10.1172/JCI166028
Kaczanowska, S., Beury, D. W., Gopalan, V., Tycko, A. K., Qin, H., Clements, M. E., et al. (2021). Genetically engineered myeloid cells rebalance the core immune suppression program in metastasis. Cell, 184(8), 2033–2052. https://doi.org/10.1016/j.cell.2021.02.048. e2021.
Agliardi, G., Liuzzi, A. R., Hotblack, A., De Feo, D., Nunez, N., Stowe, C. L., et al. (2021). Intratumoral IL-12 delivery empowers CAR-T cell immunotherapy in a pre-clinical model of glioblastoma. Nature Communications, 12(1), 444. https://doi.org/10.1038/s41467-020-20599-x
Chmielewski, M., Kopecky, C., Hombach, A. A., & Abken, H. (2011). IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Research, 71(17), 5697–5706. https://doi.org/10.1158/0008-5472.CAN-11-0103
Yang, Y., Yang, H., Alcaina, Y., Puc, J., Birt, A., Vedvyas, Y., et al. (2023). Inducible expression of interleukin-12 augments the efficacy of affinity-tuned chimeric antigen receptors in murine solid tumor models. Nature Communications, 14(1), 2068. https://doi.org/10.1038/s41467-023-37646-y
Zhang, L., Kerkar, S. P., Yu, Z., Zheng, Z., Yang, S., Restifo, N. P., et al. (2011). Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Molecular Therapy, 19(4), 751–759. https://doi.org/10.1038/mt.2010.313
Chandler, C., Liu, T., Buckanovich, R., & Coffman, L. G. (2019). The double edge sword of fibrosis in cancer. Translational Research, 209, 55–67. https://doi.org/10.1016/j.trsl.2019.02.006
McDonald, L. T., Russell, D. L., Kelly, R. R., Xiong, Y., Motamarry, A., Patel, R. K., et al. (2015). Hematopoietic stem cell–derived cancer–associated fibroblasts are novel contributors to the pro-tumorigenic microenvironment. Neoplasia, 17(5), 434–448. https://doi.org/10.1016/j.neo.2015.04.004
Albrengues, J., Bertero, T., Grasset, E., Bonan, S., Maiel, M., Bourget, I., et al. (2015). Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nature Communications, 6(1), 10204. https://doi.org/10.1038/ncomms10204
Quante, M., Tu, S. P., Tomita, H., Gonda, T., Wang, S. S. W., Takashi, S., et al. (2011). Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell, 19(2), 257–272. https://doi.org/10.1016/j.ccr.2011.01.020
Hong, M., Talluri, S., & Chen, Y. Y. (2023). Advances in promoting chimeric antigen receptor T cell trafficking and infiltration of solid tumors. Current Opinion in Biotechnology, 84, 103020. https://doi.org/10.1016/j.copbio.2023.103020
Ni, Y., Zhou, X., Yang, J., Shi, H., Li, H., Zhao, X., et al. (2021). The role of tumor-stroma interactions in drug resistance within tumor microenvironment. Front Cell Dev Biol, 9, 637675. https://doi.org/10.3389/fcell.2021.637675
Ostermann, E., Garin-Chesa, P., Heider, K. H., Kalat, M., Lamche, H., Puri, C., et al. (2008). Effective immunoconjugate therapy in cancer models targeting a serine protease of tumor fibroblasts. Clinical Cancer Research, 14(14), 4584–4592. https://doi.org/10.1158/1078-0432.CCR-07-5211
Lo, A., Wang, L. S., Scholler, J., Monslow, J., Avery, D., Newick, K., et al. (2015). Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Research, 75(14), 2800–2810. https://doi.org/10.1158/0008-5472.CAN-14-3041
Wang, L. C., Lo, A., Scholler, J., Sun, J., Majumdar, R. S., Kapoor, V., et al. (2014). Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunology Research, 2(2), 154–166. https://doi.org/10.1158/2326-6066.CIR-13-0027
Dennis, J. W., Laferté, S., Waghorne, C., Breitman, M. L., & Kerbel, R. S. (1987). Beta 1–6 branching of Asn-linked oligosaccharides is directly associated with metastasis. Science, 236(4801), 582–585. https://doi.org/10.1126/science.2953071
Greco, B., Malacarne, V., De Girardi, F., Scotti, G. M., Manfredi, F., Angelino, E., et al. (2022). Disrupting N-glycan expression on tumor cells boosts chimeric antigen receptor T cell efficacy against solid malignancies. Science Translational Medicine, 14(628), eabg3072. https://doi.org/10.1126/scitranslmed.abg3072
Ayala Ceja, M., Khericha, M., Harris, C. M., Puig-Saus, C., & Chen, Y. Y. (2024). CAR-T cell manufacturing: Major process parameters and next-generation strategies. Journal of Experimental Medicine. https://doi.org/10.1084/jem.20230903
Mikhael, J., Fowler, J., & Shah, N. (2022). Chimeric antigen receptor T-cell therapies: Barriers and solutions to access. JCO Oncology Practice, 18(12), 800–807. https://doi.org/10.1200/OP.22.00315
Agarwal, S., Hanauer, J. D. S., Frank, A. M., Riechert, V., Thalheimer, F. B., & Buchholz, C. J. (2020). In vivo generation of CAR T cells selectively in human CD4(+) lymphocytes. Molecular Therapy, 28(8), 1783–1794. https://doi.org/10.1016/j.ymthe.2020.05.005
Frank, A. M., Braun, A. H., Scheib, L., Agarwal, S., Schneider, I. C., Fusil, F., et al. (2020). Combining T-cell-specific activation and in vivo gene delivery through CD3-targeted lentiviral vectors. Blood Advances, 4(22), 5702–5715. https://doi.org/10.1182/bloodadvances.2020002229
Rurik, J. G., Tombacz, I., Yadegari, A., Mendez Fernandez, P. O., Shewale, S. V., Li, L., et al. (2022). CAR T cells produced in vivo to treat cardiac injury. Science, 375(6576), 91–96. https://doi.org/10.1126/science.abm0594
Smith, T. T., Stephan, S. B., Moffett, H. F., McKnight, L. E., Ji, W., Reiman, D., et al. (2017). In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nature Nanotechnology, 12(8), 813–820. https://doi.org/10.1038/nnano.2017.57
Pandit, S., Agarwalla, P., Song, F., Jansson, A., Dotti, G., & Brudno, Y. (2024). Implantable CAR T cell factories enhance solid tumor treatment. Biomaterials, 308, 122580. https://doi.org/10.1016/j.biomaterials.2024.122580
Yang, J., He, J., Zhang, X., Li, J., Wang, Z., Zhang, Y., et al. (2022). Next-day manufacture of a novel anti-CD19 CAR-T therapy for B-cell acute lymphoblastic leukemia: First-in-human clinical study. Blood Cancer Journal, 12(7), 104. https://doi.org/10.1038/s41408-022-00694-6
Ghassemi, S., Durgin, J. S., Nunez-Cruz, S., Patel, J., Leferovich, J., Pinzone, M., et al. (2022). Rapid manufacturing of non-activated potent CAR T cells. Nature Biomedical Engineering, 6(2), 118–128. https://doi.org/10.1038/s41551-021-00842-6
Barba, P., Kwon, M., Briones, J., Jaeger, U., Bachy, E., Blaise, D., et al. (2022). YTB323 (rapcabtagene autoleucel) demonstrates durable efficacy and a manageable safety profile in patients with relapsed/refractory diffuse large B-cell lymphoma: Phase I study update. Blood, 140(Supplement 1), 1056–1059. https://doi.org/10.1182/blood-2022-162520
Acknowledgements
We thank Dr. Ira Phadke for her input on the design of the figures.
Funding
Open access funding provided by the National Institutes of Health This work was supported by the Center for Cancer Research (CCR; ZIA BC 012066 to R. Nguyen), the Children’s Cancer Foundation (R. Nguyen), and the Deutsche Forschungsgemeinschaft (T. Trautmann). Figures were created with Biorender.
Author information
Authors and Affiliations
Contributions
All three authors conceptualized the review, wrote the main manuscript text, and prepared the figures.
Corresponding author
Ethics declarations
Ethical approval
N/A.
Informed consent
N/A
Competing interests
R.N. filed a patent (WO2023158986A1) for GPC2-CARs.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Trautmann, T., Yakobian, N. & Nguyen, R. CAR T-cells for pediatric solid tumors: where to go from here?. Cancer Metastasis Rev (2024). https://doi.org/10.1007/s10555-024-10214-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10555-024-10214-6