
Abstract
Tumor microenvironment (TME) has been demonstrated to play a significant role in tumor initiation, progression, and metastasis. Cancer-associated fibroblasts (CAFs) are the major component of TME and exhibit heterogeneous properties in their communication with tumor cells. This heterogeneity of CAFs can be attributed to various origins, including quiescent fibroblasts, mesenchymal stem cells (MSCs), adipocytes, pericytes, endothelial cells, and mesothelial cells. Moreover, single-cell RNA sequencing has identified diverse phenotypes of CAFs, with myofibroblastic CAFs (myCAFs) and inflammatory CAFs (iCAFs) being the most acknowledged, alongside newly discovered subtypes like antigen-presenting CAFs (apCAFs). Due to these heterogeneities, CAFs exert multiple functions in tumorigenesis, cancer stemness, angiogenesis, immunosuppression, metabolism, and metastasis. As a result, targeted therapies aimed at the TME, particularly focusing on CAFs, are rapidly developing, fueling the promising future of advanced tumor-targeted therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The tumor microenvironment is a complex ecosystem consisting of a heterogeneous population of cells, including tumor cells and recruited stromal cells [1]. These stromal cells, along with tumor cells, form a solid “castle” both physically and chemically. This abnormal and stiffened structure of tumor vasculature and lymphatic vessels results in interstitial hypoxia, acidic interstitial pH, and drug retention, creating an immunosuppressive and anti-drug barrier in the TME [2]. Furthermore, the heterogenous composition of the TME leads to the release of high levels of chemokines and cytokines, such as transforming growth factor β (TGF-β), interleukin 6 (IL-6), and tumor necrosis factor-α (TNFα). These molecules promote chronic inflammation and extracellular matrix (ECM) remodeling [3].
Cancer-associated fibroblasts (CAFs) are a significant component of the TME. However, their characterization remains imprecise due to their heterogeneity in origin, phenotype, and function. CAFs have been reported to derive from various sources, including quiescent fibroblasts, mesenchymal stem cells (MSCs), epithelial cells, adipocytes, and pericytes, through different but interconnected signaling pathways [4]. Given the diversity of origins and the specific TME of different tumors, CAFs can exhibit multiple phenotypes. In 2018, Bartoschek et al. employed single-cell RNA sequencing to identify several distinct subclasses of breast CAFs, which they classified as vascular CAFs (vCAFs), matrix CAFs (mCAFs), developmental CAFs (dCAFs), and circulating CAFs (cCAFs). These subclasses originate from perivascular cells, resident fibroblasts, malignant cells that have undergone epithelial-to-mesenchymal transition (EMT), and proliferating vCAFs, respectively [5]. As detection technology evolves and interest in CAF research grows, two major CAF subtypes have been widely recognized: myofibroblastic CAFs (myCAFs) involved in ECM remodeling, and inflammatory CAFs (iCAFs) regulating tumor immunity [6]. Several markers have been identified for the identification of CAFs, such as α-SMA, FAP, and COL1A1, but none of these markers is highly specific [7]. Therefore, it is urgently needed to develop a well-established system for accurately distinguishing the heterogenous CAFs.
Tumor-targeted therapy, which has been under development since the early 2000s, has emerged as a viable and remarkable option for cancer patients. These therapies include various approaches such as drugs, viruses and gene therapy [8, 9]. Among the significant components in solid tumors, CAFs play an indispensable role. Consequently, targeted therapies aimed at CAFs have been in prosperous development, ranging from direct CAF depletion to molecular CAF reprograming. However, the majority of these CAF-targeted therapies have faced challenges during clinical trials, likely due to the lack of a specific CAF marker or the occurrence of severe adverse effect.
In this review, we provide a comprehensive definition of CAFs, taking into consideration their peculiar heterogeneity in origin, phenotype, and markers. Furthermore, we make an effort to examine and discuss the diverse regulatory functions of CAFs in tumorigenesis, progression, and metastasis, with a particular focus on the unique molecular pathways involved. Additionally, we endeavor to outline the latest advancements in CAF-targeted therapies, with the ultimate goal of establishing an efficient TME-targeted therapy to overcome the challenges faced in cancer treatment.
2 CAF and its heterogeneity
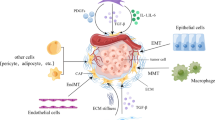
Fibroblasts are universal and fundamental cells that play a crucial role in building connective tissue. They are spindle-shaped cells with ability of adhesion, signifying their diverse functions in synthesis, construction, and wound healing [10]. In response to tissue injury, quiescent fibroblasts undergo reversible activation to facilitate tissue repair and regeneration. During this process, the activated fibroblasts express α-SMA and vimentin, undergo a transformation into a stellate shape, and exhibit enhanced secretory and migratory function [11]. However, in the context of tumorigenesis, various stimuli drive the injury-activated fibroblasts towards a state of increased secretion and proliferation but reduced contractility. This state is known as CAF [12]. Consequently, CAFs play a role in promoting cancer progression. Unlike the highly preserved resident fibroblasts, CAFs are composed of heterogenous subsets with specific division of duties (Fig. 1).

Heterogeneity of CAFs in origin and phenotype. CAFs can be derived from various origins, including quiescent fibroblasts, MSCs, epithelial cells, endothelial cells, adipocytes, pericytes, macrophages, and mesothelial cells, stimulated by respective factors. Moreover, CAFs also exhibit diverse phenotypes, such as myofibroblastic CAFs, inflammatory CAFs, and antigen-presenting CAFs. These distinct phenotypes share several collective markers while also express particular function-associated markers. MSC, mesenchymal stem cell; IL-1, interleukin 1; IL-17A, interleukin 17A; TNF, tumor necrosis factor; TGF-β, transforming growth factor β; LIF, leukemia inhibitory factor; ROS, reactive oxygen species; EMT, epithelial-to-mesenchymal transition; PAI-1, plasminogen activator inhibitor 1; EndMT, endothelial-mesenchymal transition; NF-κB, nuclear factor kappa-B; PFT, pericyte-fibroblast transition; MMT, macrophage-myofibroblast transition; iCAF, inflammatory CAF; myCAF, myofibroblastic CAF; apCAF, antigen-presenting CAF; CXCL12, C-X-C motif chemokine ligand 12; PDPN, podoplanin; α-SMA, α-smooth muscle actin; LRRC15, leucine-rich repeat-containing protein 15; TnC, tenascin-C; CNN1, calponin 1; ANTXR1, anthrax toxin receptor 1; ITGA11, integrin α11; SPARC, secreted protein acidic and rich in cysteine; CDH11, cadherin-11
2.1 The heterogeneity of CAFs in origin
Accumulating evidences have shown that CAFs derive from a heterogenous population of cells. Quiescent tissue fibroblasts and MSCs can be induced to transform into stellate-shaped myofibroblasts when activated by several cytokines, chemokines, and exosomes. For example, TGF-β has been confirmed to activate fibroblasts through both SMAD and non-canonical signaling pathways [13]. Leukemia inhibitory factor (LIF), a member of the IL-6 pro-inflammatory cytokine family, promotes and sustains the pro-invasive conversion of fibroblasts through crosstalk between the JAK1/STAT3 and RhoA/ROCK/MLC2 signaling pathways [14]. Li et al. demonstrated that aggressive cancer cells from both lung cancer and melanoma can produce methylmalonic acid, an oncometabolite increased with aging, which activates fibroblasts through reactive oxygen species (ROS)-activated nuclear factor kappa-B (NF-κB) and TGF-β signaling [15]. Furthermore, Fang et al. reported that hepatocellular carcinoma cells-derived exosomal miR-1247-3p target β-1,4-galactosyltransferases III (B4GALT3), a protein involved in glycosylation, leading to the activation of integrin β1/NF-κB signaling in fibroblasts in lung metastatic niche [16]. In addition to soluble factors, hypoxia has been reported to activate hypoxia-inducible factor 1 (HIF-1) and induce autocrine TGF-β signaling to promote prostate cancer-associated myofibroblast activation [17]. Apart from quiescent fibroblasts and MSCs, there are various non-fibroblastic sources that have the potential to be induced to transdifferentiate into CAFs.
EMT is a pivotal process present universally in both physiological and pathological scenarios. During EMT, a polarized epithelial cell undergoes multiple biochemical changes, leading to the loss of epithelial phenotype and the acquisition of mesenchymal phenotype. The mesenchymal phenotype exhibits enhanced migratory and invading capacity, elevated resistance to apoptosis, and increased productivity of ECM components [18]. EMT is classified into three subtypes. Type-1 EMT is involved in embryo implantation and development, causing neither fibrosis nor an invasive fibroblastic phenotype. Type-2 EMT is associated with wound healing, tissue regeneration, and organ fibrosis in response to stimuli like trauma or inflammatory damage, and the transformed fibroblasts are typically marked by the expression of fibroblast-specific protein 1 (FSP1, also known as S100A4), α-SMA, and collagen I [19]. On the other hand, type-3 EMT usually occurs in epithelial neoplastic cells, playing a critical role in the acquisition of malignant phenotypes with invasion and disseminating capacities [20]. EMT can be initiated and promoted by multiple cytokines, microRNAs, exosomes, and hypoxia. These factors activate various transcriptional regulators, such as Snail, Slug, Twist, and ZEB1/ZEB2, leading to the downregulation of epithelial markers (e.g., E-cadherin, occludin, claudin-1, β-catenin) and the acquisition of mesenchymal markers (e.g., N-cadherin, vimentin, fibronectin) through signaling cascades, including Wnt/β‐catenin, Notch, Sonic hedgehog, NF-κB, receptor tyrosine kinases, PI3K/AKT/mTOR, Hippo, and TGF-β pathways, among others [21]. While it is widely accepted that cancer induces type-2 EMT for “wound” healing, it remains uncertain whether type-3 EMT is involved in the origination of CAFs from resident epithelial cells or if it modulates crosstalk with type-2 EMT through genomic alterations.
Endothelial cells also serve as major sources of CAFs through a process known as endothelial-mesenchymal transition (EndMT) [22]. EndMT is initiated by specific inductors secreted by cancer cells, such as TGF-β, IL-1β, tumor necrosis factor-α (TNF-α), NF-kB transcription factor, and endotoxin [23,24,25]. TGF-β, among the most common EndMT inducers, exists in three isoforms (TGF-β1, TGF-β2, and TGF-β3). EndMT is primarily induced by the first two isoforms, which bind to the corresponding TGF-β transmembrane receptor type II (TGFβR2) and phosphorylate TGF-β receptor type I (TGFβR1 or ALK5). This activation leads to the phosphorylation of Smad2/3, which forms a complex with Smad4, translocates to the nucleus, and triggers the expression of multiple genes specific for EndMT, including NOTCH1, TWIST1, SLUG, and SNAI1/2. EndMT can also be induced through alternative signaling pathways, such as TGFβ/ALK5/PI3K/Akt pathway.
Adipocytes in TME are also recognized as a source of CAFs, known as cancer-associated adipocytes (CAAs). CAAs undergo dedifferentiation and transition into fibroblast-like cells termed adipocyte-derived fibroblasts (ADFs), with an increased expression of CAF markers such as fibroblast-specific protein-1 (FSP1), but not α-SMA [26]. This process is often initiated by Wnt3a secreted by tumor cells through the Wnt/β-catenin pathway, as discovered in breast cancer by Bochet et al. [27]. Consistent with Bochet’s finding, Iyoshi et al. identified omental adipocyte-derived fibroblast dissected from the metastatic lesions of ovarian cancer and found it exhibiting both mesenchymal stem cell and myofibroblast-like features, according to the expression of CD73 and α-SMA. This pro-tumoral phenotype of omental adipocyte-derived fibroblast is also found to be induced by Wnt3a instead of Wnt5a [28]. The incompatible expression of α-SMA in ADFs may indicate different subpopulations of CAFs originating from adipocytes in different tumor context. Hence, deciphering the heterogeneity of adipocytes-derived CAFs might be instructive for the therapy of fat-rich cancers.
Pericyte, known for promoting angiogenesis and vessel maturation in cancers, can also undergo a transition into CAF-like phenotypes, termed pericyte-fibroblast transition (PFT) [29]. PFT is mainly induced by platelet-derived growth factor type BB (PDGF-BB) and the relevant PDGF-BB-PDGFRβ axis. In the acute phase of benign inflammation, microvascular endothelial cells contribute to most circulating PDGF-BB, while during the chronic inflammation of cancer, the tumor bulk can produce a high level of PDGF-BB, promoting angiogenesis and initiating PFT [30]. Hosaka et al. induced mouse pericytes with PDGF-BB, and whereafter found it losing Ng2 expression while gaining of Fsp1 and α-Sma in pericytes, additionally discovering the function of stimulating tumor growth and metastasis in PDGF-BB-primed pericytes [31].
Furthermore, there are several other origins of CAFs, such as a subset derived from the macrophage lineage found in non-small-cell lung carcinoma, regulated by Smad3-induced macrophage-myofibroblast transition (MMT) [32]. Mesothelial cells can also transform into a typical antigen-presenting CAFs (apCAFs) when induced by IL-1 and TGF-β, discovered in pancreatic cancer [33]. The exact origin of CAFs still lacks precise elaboration, and further approaches like lineage tracing, fate-mapping, pseudotime analysis, and RNA velocity may help uncover new primogenitors of CAFs.
2.2 The heterogeneity of CAFs in phenotype
Previous studies have indicated that α-SMA, FAP, vimentin, FSP1 (also known as S100A4), PDGFRα, and PDGFRβ could serve as markers to define CAFs. These studies have also shown that CAFs play a tumor-promoting function in various ways. However, the lack of a specific CAF marker and the discovery of tumor-restraining CAFs have driven research on the functional phenotypes of CAFs [11, 34]. Due to the various origins of CAFs, it is evident that CAF subpopulations exhibit significant diversity. The proximity of CAFs to cancer cells and the presence of different soluble factors in the TME may also contribute to this heterogeneity [35]. With the advancement of single-cell RNA sequencing, researchers have identified multiple and distinct populations of CAFs with characteristic markers. Table 1 shows several markers found in CAFs and its subpopulations; however, none of these markers have been strictly proven to be specific.
myCAFs are a subpopulation of CAFs that exhibit similarities with myofibroblasts involved in the wound healing process. Due to these similarities, myCAFs have been reported to play a major role in producing ECM components and remodeling ECM [36]. Based on the heterogenous expression of SDC1, LAMP5, and CD9, Kieffer et al. further divided ANTXR1+ myCAFs into different subtypes: SDC1+LAMP5− ecm-myCAF, which expresses genes coding for ECM proteins; LAMP5+SDC1+/− TGFβ-myCAF, expressing the TGFβ pathway; SDC1−LAMP5−CD9+ wound-myCAF, expressing wound healing proteins; and acto-myCAF, programming acto-myosin pathway. This highlights the high heterogeneity in the myCAF phenotype [37]. However, it has been shown that the phenotype of myCAF is flexible rather than fixed. Mosa et al. demonstrated that high and low levels of Wnt induce myCAF and iCAFs subtype, respectively, indicating the regulation of tumor growth and malignancy through CAF subtypes transition under the influence of the Wnt/β-catenin pathway [38].
iCAFs have been identified in multiple cancers, indicating their universal presence within CAF clusters. It has been demonstrated that iCAFs share similar transcriptional profiles and signaling pathway activation with senescent fibroblasts [34]. In pancreatic cancer, iCAFs derived from pancreatic stellate cells (PSCs) exhibit significantly lower α-SMA expression but intensely high expression of inflammatory cytokines, such as IL-6 and IL-11, compared to the homologous myCAFs [39]. Similarly, in bladder carcinoma, single-cell sequencing in COL1A1+ fibroblasts has revealed a PDGFRA+ iCAF cluster that exhibit strong expression of cytokines and chemokines, including CXCL12, IL6, CXCL14, CXCL1, and CXCL2, compared to RSG5+ myCAF cluster. The pro-proliferation effect of this cluster has been confirmed [40]. On the other hand, in cholangiocarcinoma, iCAFs identified in COL1A1+ fibroblasts show high expression of RSG5, Lrat, and Reln, with enrichment of inflammatory, growth factor, and antigen-presentation genes as well as receptor-ligand, growth factor, and cytokine activity pathways [41]. These findings collectively confirm the inflammatory cytokine-secreting property of iCAFs while also indicating the heterogeneity of iCAFs in different organs.
The generation of the inflammatory phenotype in iCAFs has been demonstrated to be induced by various factors, including cytokines, circRNAs, T cells, and hypoxia. Biffi et al. illustrated that IL-1 induces leukemia inhibitory factor (LIF) expression to activate JAK/STAT signaling pathways, generating an iCAF phenotype, and TGF-β antagonizes this process by downregulating IL1R1 expression, promoting differentiation into myCAFs [42]. Wnt activity in CAFs is also associated with distinct subtypes, where low and high levels induce an iCAF subtype or contractile myCAFs, respectively [38]. Besides cytokines, Zheng et al. found that the upregulation of circCUL2 expression in normal fibroblasts (NFs) induces the iCAF phenotype and promotes tumorigenesis and metastasis of pancreatic ductal adenocarcinoma (PDAC) cells via miR-203a-3p/MyD88/NF-κB/IL6 axis [43]. Tc17, a novel protumorigenic CD8+ T cell subtype in PDAC, has been demonstrated to induce iCAF differentiation via synergism of IL-17A and TNF [44]. Furthermore, Schwörer et al. revealed that hypoxia drives fibroblasts to gain an inflammatory gene expression signature and synergizes with cancer cell-derived cytokines to promote an iCAF phenotype in an HIF-1α dependent fashion, and experimental evidence supporting this finding [45, 46].
In 2019, Elyada et al. unveiled a new population of CAFs expressing MHC class II and CD74 but lacking classical co-stimulatory molecules, leading to their designation as antigen-presenting CAFs (apCAFs) [47]. These apCAFs were found to originate from mesothelial cells through mesothelial-mesenchymal transition, which is induced via IL-1/NF-kB and TGF-β/Smad signaling pathway [33, 48]. The presence of MHC II molecules on apCAFs enables them to present antigens to CD4 + T cells. However, due to the absence of co-stimulation molecules (such as CD40, CD80, and CD86) on apCAFs, they promote the formation of regulatory T cells, which may suppress the immune response [49]. Although it has been observed that patients with a high abundance of apCAFs in PDAC have a decreased survival probability, the specific mechanism by which apCAFs promote cancer progression is still not fully understood [50]. Future research and evidence are required to elucidate the exact role and impact of apCAFs in cancer development and progression.
In addition to the well-known subtypes of CAFs, such as myCAF, iCAF, and apCAF, there are other rare subtypes of CAFs that have been identified through single-cell RNA sequencing, highlighting the high diversity of CAF population and the different criteria used for their classification. For example, there are vascular CAFs (vCAFs) characterized by their high expression of angiogenic or microvasculature-associated genes, like CAD146. Another subtype is the PLA2G2A+ metabolic CAFs (meCAFs), which have been found to be correlated with the presence of immune cells [51]. As CAF-detecting techniques continue to advance, it becomes increasingly important to establish a uniform classification system for CAFs to facilitate more precise and targeted therapies. The heterogeneity of CAFs underscores the need for a comprehensive understanding of their distinct subtypes and functional roles in the TME, which may ultimately lead to more effective treatment strategies for cancer patients.
3 Methodologies in CAF research
Research on CAFs presents challenges due to their high heterogeneity and context-dependent nature. The expanding field of TME research has led to the gradual establishment of a comprehensive methodology for studying CAFs. In in vitro investigations, primary CAFs derived from patients are the preferred choice due to their accessibility and similarity to TME in vivo. Protocols for establishing CAFs in vitro from surgically resected tissues involve steps such as tissue extraction, digestion, CAF dissociation, and incubation [77]. However, limitation persists, including the loss of heterogeneity in isolated CAFs and the limited number of passages for primary CAFs (usually up to 20–25 passages) [78]. To address senescence during cell expansion, some researchers use lentivirus containing human telomerase reverse transcriptase (hTERT) to immortalize patient-derived CAFs [79]. In addition to in vitro studies, CAF-targeted animal models have been developed. For instance, transgenic α-SMA-tk mice are created by ligating a fragment with α-SMA (or other CAF-specific genes) promoter and a truncated version of the herpes simplex 1 virus thymidine kinase (HSV1-tk). Subsequent injection of ganciclovir induces selective depletion of α-SMA myofibroblasts in vivo [80]. Beyond genetic engineering, direct tumor injection of pharmaceuticals and orthotopic xenograft implantation with control/treated CAFs are also effective in studying CAF biology [81].
Detecting the heterogeneity of CAFs is crucial and single-cell analysis has been instrumental in understanding distinct CAF subpopulations and their markers. With the rapid development of single-cell analysis techniques, including multiomic analysis, spatial transcriptomics, and proteomics, more sophisticated methods are applied in CAF detection [82, 83]. Flow cytometry, immunohistochemistry, and immunostaining are also vital for CAF taxonomy. Commonly used probes like α-SMA, vimentin, FAP, FSP1, and PDGFR serve as reliable CAF markers when used in combination [84].
The highlight of CAF research lies in understanding the crosstalk between CAFs and other cell types, particularly cancer cells. Coculture analysis is the most efficient way to explore the interactions. However, traditional 2-dimensional cell line coculture system and patient-derived tumor xenografts model have limitations in mimicking human stromal compartments, immune microenvironment, and organ-specific functions for detailed research [85, 86]. The invention of 3-dimensional organoid technology allows in vitro tumor research to closely replicate the structural and functional aspects of counterpart organs, facilitating personalized precision oncology [87]. Despite the challenges, advanced models like organoid-on-a-chip have demonstrated comparable responses to therapies with greater precision in predicting outcomes in CAF research [88]. As tumor research technologies evolve, it is expected that more accurate and efficient techniques for CAF research will emerge.
4 CAF in cancer progression and metastasis
4.1 CAFs contribute to cancer stemness
Cancer stemness refers to the self-renewal and propagation abilities of cancer stem cells (CSCs), which play a critical role in tumor aggressiveness, drug resistance, and metastasis [89]. CSCs can be identified by several markers, including CD44, CD24, CD133, LGR5, SOX2, AQP5, ESA, PAF1, and CXCR4, although none of these markers is highly specific [90,91,92,93,94]. As a central component of the TME, CAFs are believed to interact with CSCs and maintain a favorable tumor niche, mainly through paracrine signaling [95]. A group of CAF-derived molecules has been found to promote cancer stemness. For instance, Su et al. identified a CD10+GPR77+ subset of CAFs in breast and lung cancer, driven by NF-kB activation, which induces CSC enrichment by secreting IL-6 and IL-8 [60]. Ma et al. found that interferon secreted from bladder cancer cells can induce SLC14A1+ CAFs, which in turn promote stemness of bladder cancer cells via WNT5a/β-catenin pathway [96]. In hepatocellular carcinoma, CAF-derived hepatocyte growth factor (HGF) enhances cancer cell stemness through the extracellular signal-regulated kinase (ERK)1/2-FRA1-HEY1 signaling pathway [95]. Additionally, CAF-secreted exosomes have been shown to sustain cell stemness in various cancers [97, 98]. Furthermore, CAFs indirectly recruit myeloid-derived suppressor cells (MDSCs) and enhance the stemness of CD33+ MDSCs in a FAP-dependent paracrine manner [99]. Thus, targeting these paracrine pathways could be a potentially effective approach to combat tumor stemness. The WNT signaling pathway represents a promising target for specific therapy. Canonical WNT signaling supports the rapidly cycling CSCs, while noncanonical WNT signaling supports the quiescent CSCs [100]. Moreover, WNT signaling broadly mediates the communication between CAFs and CSCs, further emphasizing its importance in tumor stemness regulation.
4.2 CAFs promote angiogenesis
Angiogenesis is a critical process whereby tumors develop new blood vessels to obtain an increased supply of oxygen and nutrients [101]. Hypoxia has been identified as a key driver for tumor angiogenesis. Under hypoxic conditions, cancer cells secrete vascular endothelial growth factor A (VEGFA) which binds to VEGF receptor 2 (VEGFR2) on nearby endothelial cells (ECs) of blood vessels or circulating bone marrow-derived endothelial progenitor cells, and triggering angiogenesis [102]. This process involves the breakdown of the basal lamina and ECM, proliferation of ECs, growth of new vascular sprouts, and vessel maturation. Other signaling molecules, such as delta ligand-like 4 (DLL4) and angiopoietin 2 (ANGPT2), also play crucial roles in angiogenesis [103]. CAFs, originating from the chronic wound-healing response within the tumor, secret pro-angiogenic growth factors that promote angiogenesis. These factors include VEGFA, CXC-chemokine ligand 12 (CXCL12), fibroblast growth factor 2 (FGF2), and platelet-derived growth factor (PDGF) [104]. CXCL12, also known as stromal cell-derived factor 1 (SDF-1), has been shown to enhance tumor growth and angiogenesis through the CXCL12/CXCR4 pathway [105]. This binding initiates divergent signaling pathways, including G-protein/PI3K/AKT/NF-κB axis and Ras-MEK1/2-Erk1/2 axis, resulting in various angiogenic responses [106]. Similarly, FGF2, a member of the heparin-binding growth factor family, binds to FGF receptors (FGFRs) and triggers multiple pro-angiogenic activity, while also participating in crosstalk with VEGF [107]. Additionally, the PDGF/PDGF receptor (PDGFR) signaling plays a significant role in connective tissue development and wound healing [108]. Studies have demonstrated that CAFs with upregulated PDGF-C induce angiogenesis even when VEGF is inhibited, suggesting that the PDGF/PDGFR pathway might compensate for the inhibition of VEGF-mediated angiogenesis [109].
In addition to the direct activation of paracrine ways, CAFs have been reported to promote angiogenesis through various indirect mechanisms. One crucial biomechanical characteristic of the TME, driven by stromal cells, particularly CAFs, is matrix stiffness [110]. CAFs secrete lysyl oxidase (LOX), an enzyme that catalyzes the covalent cross-linking of collagens and elastin, contributing to the determination of matrix stiffness [111]. Additionally, CAFs secrete lysyl hydroxylase 2 (LH2), which induces hydroxylysine aldehyde-derived collagen cross-links in the ECM, further increasing matrix stiffness [112]. Numerous studies have demonstrated a link between matrix stiffness and the production of VEGF. For instance, Sack et al. found that on harder ECM surfaces, endothelial cells exhibit an increased capability of binding VEGF and reduced VEGF internalization, regulated by integrins β1 [113]. Li et al. unraveled a matrix stiffness/integrins β1/Piezo1 activation/Ca2+ influx/HIF-1α ubiquitination/VEGF pathway in hepatocellular carcinoma angiogenesis, with involvement of CXCL16 and IGFBP2 pathways [114]. However, contradictory results were reported by Bao et al., who discovered a YAP/RUNX2/SRSF1 axis in neuroblastoma angiogenesis, wherein VEGF165 secretion is repressed with increasing matrix stiffness [115]. Notably, the disparity in results may be attributed to variations in the stiffness levels of the gels used in the research of Bao et al. (1 kPa, 8 kPa, and 30 kPa, respectively), which might as well be scaled up to a minished range so as to simulate an actual intratumoral microenvironment. Hence, more precise experiments are needed to clarify the functional impact of matrix stiffness on angiogenesis.
As mentioned earlier, hypoxia is a key driver of tumor angiogenesis. Hypoxia-inducible factor (HIF) transcription factors are pivotal in hypoxia signaling in cancer and stromal cells. They translocate to the nucleus in response to the absence of oxygen and activate the expression of hypoxia-related genes, including VEGF [116]. CAFs activated by hypoxic TME can induce abnormalities in the blood vessel by secreting various proangiogenic factors [117]. Furthermore, CAFs produce soluble factors like CCL5, triggering the HIF-1α pathways to promote angiogenesis [118].
4.3 CAFs mediate immunosuppression
Chronic inflammation, immune cell infiltration, and evasion of cancer cells from the immune response are considered some of the hallmarks in cancer progression [35]. Previous studies have established the paradoxical role of the immune system in both promoting and restraining cancer, referred to as “cancer immunoediting.” This dynamic process consists of three sequential phases: elimination, equilibrium, and escape [119]. During the elimination phase, the innate and adaptive immune systems cooperate to recognize and eradicate dysplastic cells before they can develop into clinically detectable tumors. However, if a few variant cancer cells acquire poorly immunogenic or immunoevasive properties that enable them to survive the immune attack, they may enter the equilibrium phase. In this phase, neoplastic cells are still restricted, and their cellular immunogenicity is shaped by the adaptive immune system, primarily involving T cells and related cytokines. As the edited cancer cells face constant immune selection pressure during the equilibrium phase, they may develop immunosuppressive and/or immunoevasive phenotypes, ultimately leading to immune escape. Once in the immune escape phase, the cancer cells are no longer restricted by the immune system, leading to uncontrolled growth, clinically apparent tumors, and even metastasis [120]. Despite its importance, the intricate mechanism of cancer immunoediting is still not fully understood, which presents a challenge for effective immunotherapy targeting cancer.
CAFs, as the major components in the TME, have been reported to mainly exert an immunosuppressive function in facilitating cancer immune evasion. TGF-β, which can be secreted by CAFs, is a significant mediator in the regulation of the immune microenvironment. TGF-β signaling is known to impact T cell differentiation and proliferation by dampening the stimulation of specific transcription factors triggered by Ca2+ influx [121]. In a T cell excluded cohort of ovarian tumor, the upregulation of TGF-β and the activation of stroma are identified as important mechanisms of T cell exclusion. TGF-β can reduce MHC-I expression in ovarian cancer cells in vitro and also activate fibroblasts to induce extracellular matrix production, constructing a physical barrier to hinder T cell infiltration [122]. Additionally, TGF-β has been demonstrated to suppress dendritic cells, inhibit the development of cytolytic natural killer cells (NK cells), and reduce their secretion of IFN-γ. Moreover, it polarizes macrophages towards the M2 phenotype with anti-inflammatory, immune-suppressive, and pro-angiogenic functions [123, 124]. Apart from TGF-β secretion, CAF-derived CXCL12 is a powerful chemokine involved in immunosuppressive regulation. It reduces CD8+ T cells migration, sequestering them from the panstromal compartment, and inhibits NK cell proliferation, maintaining them in a quiescence state [125, 126]. Another essential molecule secreted by CAFs in immune microenvironment is IL-6, which is abundantly expressed in iCAF subtype [127]. IL-6 is associated with the accumulation of tumor-infiltrating lymphocytes and plays a role in regulating the survival, activation, and function of neutrophils through the IL-6/STAT3/PD-L1 signaling pathway [128, 129]. Moreover, CAFs also secret inhibitory immune checkpoints (iICPs) to create an immunosuppressive milieu in the TME, including PD-1 and LAG3 [130]. These pieces of evidence strongly indicate that CAFs play a crucial role in assisting the tumor’s immune escape process.
4.4 CAFs dedicate in metabolic changes in cancer
Despite living in a nutrition-limited TME, cancer cells are highly skilled in perpetual proliferation, which is supported by the metabolic change that occur in the TME. About a century ago, Warburg et al. observed that even in the presence of abundant oxygen, cancer cells exhibited an enhanced and accelerated conversion of glucose to lactate for ATP formation, a phenomenon known as the “Warburg Effect” [131]. Warburg attributed this phenomenon to the dysfunction of mitochondria in tumor cells. As further research in cancer metabolism progressed, it became evident that, in contrast to the Warburg Effect, some tumor cells retain the ability to utilize mitochondria and undergo oxidative phosphorylation (OXPHOS), indicating the dynamic nature of the Warburg Effect in different TME. In some cases, CAFs are reported to adapt their metabolism in response to factors secreted by cancer cells. In this scenario, CAFs switch to aerobic glycolysis and produce high levels of energy‑rich intermediate metabolites, which are then transferred to cancer cells to fuel the mitochondrial tricarboxylic acid cycle and OXPHOS, leading to the production of ATP for cancer cell proliferation [132]. This phenomenon is referred to as the “Reverse Warburg Effect,” acting as a supplementary mechanism to the classic Warburg Effect.
The Reverse Warburg Effect is strongly driven by cancer cell-promoted oxidative stress. Cancer cells release reactive oxygen species (ROS), which reciprocally elevate oxidative stress in the stromal components, enabling autophagosomes to fuse with lysosomes and leading to the destruction of mitochondria in CAFs. This process also results in the degradation of caveolin-1 (Cav-1) through the HIF-1α/ NF-κB pathway [56, 133]. The downregulation of Cav-1 in CAFs, in turn, elevates ROS levels in cancer cells, creating a positive feedback loop that further enhances oxidative stress and impedes NF-kB pathway [134]. TGF‑β, which has a firm and universal association with cancer metabolism, can also regulate the expression of α‑SMA and NOX4 in fibroblasts, thereby influencing ROS levels and stimulating oxidative stress [135]. Through the Reverse Warburg Effect, oxidative cancer cells can receive lactate from hypoxic cancer cells. In addition, CAFs experience oxidative stress due to the cancer cells-secreted ROS, which triggers aerobic glycolysis. As a result, CAFs produce lactate and pyruvate, which can be utilized for further metabolic process in adjacent oxidative cancer cells. Although the transmission of ROS has been substantiated in the Reverse Warburg Effect, there have been few studies elucidating the mechanism by which cancer cells and CAFs perform initiation and adaption to such metabolic changes. Still, targeting the Reverse Warburg Effect, either in cancer cells or through CAFs, is theoretically feasible and could potentially decrease cancer cell metabolism. By disrupting this metabolic interplay between cancer cells and the stromal microenvironment, new therapeutic strategies may be developed to target cancer metabolism and inhibit tumor growth effectively.
4.5 CAFs facilitate cancer metastasis
Cancer metastasis is a complex process involving multiple stages. It begins with tumor cells migrating and invading nearby tissues, followed by intravasation, circulation, and extravasation, and ultimately colonization at the target site [136]. CAFs play a significant role in promoting metastasis through both paracrine signaling ways and physical interactions (Fig. 2).

The role of CAFs in cancer progression and metastasis. ① Several specific subsets of CAFs secrete exosomes, ILs, and Wnt5a to promote cancer stemness. ② CAFs directly produce soluble factors, including VEGFA, CXCL12, FGF2, and PDGF, to trigger angiogenesis by binding to the receptors on endothelial cells. On the other hand, CAFs secrete of LOX and LH2 to enhance ECM stiffness, which facilitates VEGF/VEGFR interaction via integrins. ③ CAF-derived TGF-β can impact T cell differentiation and proliferation, dampen MHC-mediated immune identification, and reduce T cell infiltration through fortifying matrix stiffness. Moreover, TGF-β can also suppress dendritic cells and inhibit the development of cytolytic NK cells. CXCL12 secreted from CAFs can reduce T cells migration and inhibits NK cells proliferation. ④ Various factors produced from CAFs can enhance cancer cell migration and invasion by inducing EMT. Additionally, CAF-promoted matrix stiffness contributes to EMT. ⑤ CAFs secret TGF-β, VEGF, and SOX2 to regulate the intravasation of blood vessels. ⑥ A cCAF subtype is involved in the metastasis through the blood vessels, marked by CD44. ⑦ Metastasis-associated cytokines and exosomes derived from CAFs in primary tumor facilitate the formation of the distant PMN. CAF, cancer-associated fibroblast; HAPLN-1, hyaluronan and proteoglycan link protein 1; PAI-1, plasminogen activator inhibitor-1; IL-6, interleukin 6; IL-8, interleukin 8; CSC, cancer stem cell; VEGFA, vascular endothelial growth factor A; CXCL12, CXC-chemokine ligand 12; NK cell, natural killer cell; LOX, lysyl oxidase; LH2, lysyl hydroxylase 2; EMT, epithelial-to-mesenchymal transition; CTC, circulating tumor cell; cCAF, circulating CAF
The motility of cancer cells is closely related to their ability to migrate and invade, which is often facilitated by the process of EMT. EMT is characterized by the loss of polarity, adhesion, and tight junctions, leading to cancer cells adopting a mesenchymal phenotype that promotes migration and invasion. However, it remains controversial whether EMT is essential for every metastatic event [137, 138]. CAFs have been shown to enhance cancer cell migration and invasion by secreting various factors, including chemokines and exosomes. For example, in gastric cancer, CAFs activated by TGF-β1/Smad2/3 signaling can highly express hyaluronan and proteoglycan link protein 1 (HAPLN1), promoting tumor migration and invasion [139]. In esophageal squamous cell carcinoma, plasminogen activator inhibitor-1 (PAI-1) derived from CAF-like cells enhances migration and invasion abilities through the Akt-Erk1/2 signaling pathways via the PAI-1/low-density lipoprotein receptor-related protein 1 (LRP1) axis [140]. CAF-secreted exosomes containing miR-18b and miR-382-5p have also been reported to promote cancer cell migration and invasion through EMT [141, 142]. Additionally, CAFs can promote EMT induction by increasing matrix stiffness signaling mediators such as TWIST1/G3BP2 pathway and EPHA2/LYN/TWIST1 pathway [143, 144]. Apart from inducing EMT, CAFs can directly drive cancer cell migration through physical forces. Labernadie et al. identified a mechanism in which CAFs exert physical force on cancer cells through the heterophilic adhesion involving N-cadherin on the CAF membrane and E-cadherin on the cancer cell membrane, mediated by β-catenin recruitment and α-catenin/vinculin interaction [145]. Erdogan et al. demonstrated that CAFs produce and align a fibronectin (Fn)-rich matrix via the nonmuscle myosin II/PDGFRα/α5β1-integrin/Fn pathway to mediate CAF-cancer cell association and directional migration [146]. Additionally, CAFs express membrane-anchored metalloproteinases (MT1-MMPs) that have collagenolytic effect, facilitating tumor cell penetration of connective tissue barriers and trafficking within the three-dimensional ECM [147].
Intravasation is a crucial process that occurs before tumor cells can enter the circulation and spread to distant sites. During angiogenesis, the formation of new blood vessels, the vessels are often considered immature, lacking proper junctional contacts between endothelial cells, and are leaky and vulnerable due to abnormal pericyte coverage. These features enable cancer cells to easily intravasate through the blood barrier [148]. Several factors, such as TGF-β, VEGF, and SOX2, have been shown to play roles in regulating both the intravasation and extravasation processes during metastasis [149, 150]. CAFs not only promote hematogenous metastasis (metastasis through the blood vessels) but also play a role in facilitating lymphatic metastasis (metastasis through the lymphatic vessels). This promotion of lymphatic metastasis has been reported to involve various signaling pathways, such as periostin/integrin/FAK/Src/VE-cadherin pathway, VEGFC/VEGFR3 pathway, and IL-6/IL-6R pathway [151,152,153].
Despite their role in motivating tumor cells, CAFs themselves are not quiescent. In 2015, Ao et al. examined a functional subpopulation of CAFs in the peripheral blood of patients with metastatic breast cancer, referred to as circulating CAF (cCAF), the presence of which was associated with clinical metastasis [154]. Sharma et al. also detected this heterotypic cluster of cells in patient blood and preclinical mouse models of breast cancer, and they found that CD44, an adhesion and stemness marker, might be an important mediator in this context [155]. Furthermore, Hurtado et al. utilized a metastasis model in zebrafish and observed that CAFs exert a pro-survival and pro-proliferative effect on circulating tumor cells (CTCs) when they remain joined as cell clusters. This clustering led to production of soluble factors associated with breast cancer cell survival and proliferation [156]. Identifying and targeting cCAFs at an early stage of tumor development could be a potent therapeutic approach to reduce cancer metastasis and relapse, especially since the detection of cCAFs in patients with localized breast cancer has also been reported [154].
CTCs that extravasate at the target site face a challenging microenvironment that is often hostile for their survival. Interestingly, even before the metastasis process begins, the host metastasis site microenvironment at the future metastatic site has already been selectively modified by the remote primary tumor. This modified microenvironment is referred to as pre-metastatic niches (PMN) [157]. The formation of PMN is largely attributed to cytokines and exosomes released by the tumor and the TME. CAFs play a dual role in activating the PMN. On the one hand, metastasis-associated factors derived from CAFs in primary tumor facilitate the formation of pre-metastatic niche. For example, a long non-coding RNA called LncSNHG5 expressed in breast CAFs is found to mediate angiogenesis and vascular permeability in the PMN of the lung through the lncSNHG5-ZNF281-CCL2/CCL5 signaling axis [158]. Similarly, extracellular vesicles (EVs) derived from CAFs in salivary adenoid cystic carcinoma induce remarkable changes in lung fibroblasts, enhancing their tumor-permissive abilities. The uptake of CAF EVs by lung fibroblasts is mediated through integrin α2β1 [159]. On the other hand, fibroblast activation into CAF is recognized as the initial phase during PMN formation. Research by Pein et al. has shown that breast cancer cells secrete IL-1α and IL-1β, which induce lung fibroblasts to produce CXCL9 and CXCL10 via NF-κB signaling, leading to inflammatory phenotypic changes in lung fibroblasts [160]. Besides cytokines, Ji et al. discovered that primary colorectal tumors release integrin beta-like 1 (ITGBL1)-enriched EVs, which stimulate the TNFAIP3-mediated NF-κB signaling pathway to activate remote fibroblasts and transform them into CAFs. These CAFs subsequently induce the formation of the PMN by secreting proinflammatory cytokines such as IL-6 and IL-8 [161]. After activation, CAFs play critical roles in ECM remodeling, metabolic changes, immunosuppression, and angiogenesis, all of which contribute to the formation of the PMN, as discussed earlier [162].
4.6 CAFs reinforce therapeutic resistance
Therapeutic resistance in cancer often leads to a poor prognosis in patients, and the underlying mechanisms behind it remain complex and dynamic. Konieczkowski et al. proposed a convergence-based framework for understanding cancer drug resistance, with pathway reactivation, pathway bypass, and pathway indifference being major causes of resistance [163]. Besides genomic changes in tumor cells, the involvement of CAFs has been extensively demonstrated in cancer therapeutic resistance, with their role being multifaceted.
CAF’s influence on the mechanical TME can promote matrix stiffness, thereby reducing the infiltration of chemical drugs. For instance, gastric CAFs expressing calponin 1 activate ROCK1/MLC pathway, leading to increased matrix stiffness and contributing to 5-fluorouracil (5-Fu) resistance in cancer cells by activating YAP [164]. CAF-derived exosomes also play a significant role in mediating cancer therapy resistance in the TME [165]. Annexin A6 in CAF-derived EVs can activate the integrin β1-focal adhesion kinase (FAK)-YAP signaling pathway, leading to the formation of a tubular network in the ECM, reinforcing chemotherapeutic resistance [166]. In breast cancer, CAF-derived circulating EVs containing the full mitochondrial genome promote estrogen receptor (ER)-independent OXPHOS, inducing therapy-induced dormant cancer stem-like cells and leading to endocrine therapy resistance [167]. Targeting the YAP signaling pathway may hold promise in overcoming the mechanical resistance encountered in targeted therapy. In the context of immunotherapy, CAFs induced by the IL-17/Act1/HIF1α pathway can initiate collagen deposition to enhance PD-L1 resistance, leading to a decrease in cytotoxic T cell infiltration [168]. Another CAF subtype, ecm-myCAF, has been found to upregulate PD-1 and CTLA4 protein levels in regulatory T lymphocytes (Tregs), increasing TGFβ-myCAF cellular content and mediating primary resistance to immunotherapy. Therefore, combining tumor-targeted therapy with CAF-targeted therapy has been considered a potential approach to address resistance. Examples of this approach, such as FAP5-DM1, an anti-FAP monoclonal antibody conjugated to maytansinoid, have shown long-lasting inhibition of tumor growth and complete regressions in xenograft models of multiple cancers [169]. Additionally, CAFs have been found to promote resistance to radiotherapy. Upon irradiation, CAFs are polarized towards the iCAF subtype via IL-1a with oxidative DNA damage, leading to p53-mediated therapy-induced senescence in iCAFs, which in turn results in chemoradiotherapy resistance and disease progression [170].
4.7 tumor-restraining CAF
The tumor-promoting role of CAFs has been extensively studied; however, recent research suggests the existence of specific subtypes of CAFs with tumor-restraining characteristics termed cancer-restraining CAF (rCAF) [171]. The presence of rCAFs may pose a challenge to CAF-depleting therapies, as achieving a balance between tumor-promoting CAFs (pCAFs) and rCAFs is crucial. Despite this, only a few markers for identifying rCAFs have been identified. In 2019, Mizutani et al. discovered Meflin, a glycosylphosphatidylinositol-anchored protein and a marker of mesenchymal stromal/stem cells that maintain their undifferentiated state, to be expressed by PSCs, which are one of the sources of CAFs in PDAC. Meflin+ CAFs were found to be correlated with a favorable patient outcome, and Meflin deficiency promoted the alignment of stromal collagen fibers, which is considered an aggressive tumor signature [172]. Similarly, Bhattacharjee et al. showed that myCAF-expressed type I collagen suppresses tumor growth by mechanically restraining tumor spread, overriding signaling mechanisms induced by matrix stiffness [173]. And deletion of type I collagen accelerates the PDAC emergence via SOX9/Cxcl5 [174]. These findings suggest that collagen fibers produced by CAFs may contribute to their tumor-suppressing properties. In a transgenic mouse model, the depletion of α-SMA+ myofibroblasts in pancreatic cancer mechanistically resulted in a tumor with more progressive and invasive tumor feature. Paradoxically, this was accompanied by a decrease in overall immune infiltration and an increase in the frequency of FoxP3+ Treg cells [175]. These seemingly contradictory findings could be explained by the restraining influence of stromal components, particularly collagen deposition in models characterized by abundant collagen, such as PDAC [176]. Furthermore, the diverse subpopulations of CAFs, originating from different progenitors and influenced by distinct factors, exert varying effects on tumorigenesis depending on the specific context [172, 177]. Therefore, exploring the regulatory relationship between different contexts and CAF subtypes using multiple animal models may offer a promising avenue for breakthrough in therapies targeting CAFs.
5 CAF-targeted cancer treatment
Cancer-targeted therapy has emerged as a viable and remarkable option for cancer patients since the early 2000s, encompassing a range of approaches such as drugs, viruses, and gene therapy [8, 9]. However, due to the genomic instability of cancer cells, the development of therapeutic resistance is inevitable. This has shifted the focus towards targeting non-tumor cells in the TME due to their relatively stable genetical nature, presenting a promising avenue for therapy [178]. Among these non-tumor cells, CAF have been recognized for their significant role in tumor progression and are now an emerging target for precise targeted therapy within the TME. Despite the potential of CAF-targeted therapy, there are ongoing challenges and obstacles. One major challenge it the lack of a specific CAF marker, which hinders the direct depletion of CAFs and makes it challenging to specifically target them. Additionally, there is a concern about potential adverse effect if normal tissue cells are unintentionally damaged during the therapy. Fortunately, as our understanding of CAF biology in cancer continues to advance, several preclinical studies and clinical trials have been reported, demonstrating promising results in this area. In the realm of CAF-targeted cancer therapy, NOX4 inhibition has emerged as a promising strategy, given its demonstrated efficacy in reversing the myCAF phenotype and facilitating intratumoral CD8+ T-cell infiltration in mouse models [179, 180]. Setanaxib (GKT137831), a pharmacologic NOX4 inhibitor, has successfully completed its phase I trial (NCT04327089) and is currently being investigated in combination with pembrolizumab in patients with recurrent or metastatic head and neck squamous cell carcinoma (NCT05323656). Moreover, LRRC15 has been identified as a particularly noteworthy myCAF biomarker due to its significant role in mediating CD8+ T cells infiltration and influencing immunotherapy response [63, 181]. A newly developed antibody–drug conjugate targeting LRRC15, known as ABBV-085, has demonstrated safety, tolerability, and promising anti-solid tumor activity in its phase I study (NCT02565758). These developments highlight the potential for more precise CAF-targeted therapy to be developed [182]. Further details on the evolving CAF-targeted therapies are shown in Table 2.
6 Conclusion
The pivotal role of the TME in cancer progression has long been emphasized, with CAFs being the most well-described components. However, the heterogeneity of CAFs poses challenges for the application of clinical CAF-targeted therapy. This heterogeneity can be attributed to two main factors: (1) diverse origin of CAFs: CAFs can arise from different cell types, including quiescent fibroblasts, MSCs, adipocytes, and pericytes, through various activating pathways; (2) heterogeneous TME: The TME in various tumors is heterogeneous and can induce CAF activation via different signaling pathways, such as TGF-β, interleukin, PDGF, and CXCL12, among others. Phenotypically, this heterogeneity is reflected in the diverse expressing levels of multiple markers in distinct CAF clusters, as well as multifarious functions in ECM remodeling, inflammation, immunoregulation, and antigen presenting. The roles of CAFs in cancer progression, metastasis, and immunosuppression through both physical interactions and paracrine signaling have been extensively studied. Several therapeutic treatments targeting CAFs have been explored and put into clinical trials. However, none of these treatments have shown significant effectiveness or safety in clinical settings. Despite the challenges, the advanced discovery of specific markers and signaling pathways, such as LRRC15, offers hope that CAF-targeted therapy will progress from bench to bedside in the near future. As researchers continue to unravel the complexities of CAF heterogeneity and their precise roles in tumor biology, novel therapeutic strategies may emerge to effectively target CAFs and improve cancer treatment outcomes.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed in the study.
References
Bussard, K. M., Mutkus, L., Stumpf, K., Gomez-Manzano, C., & Marini, F. C. (2016). Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Research: BCR, 18(1), 84. https://doi.org/10.1186/s13058-016-0740-2
Rahmanian, M., Seyfoori, A., Ghasemi, M., Shamsi, M., Kolahchi, A. R., Modarres, H. P. ,…, Majidzadeh-A, K. (2021). In-vitro tumor microenvironment models containing physical and biological barriers for modelling multidrug resistance mechanisms and multidrug delivery strategies. Journal of Controlled Release: Official Journal of the Controlled Release Society, 334, 164–177. https://doi.org/10.1016/j.jconrel.2021.04.024
Marozzi, M., Parnigoni, A., Negri, A., Viola, M., Vigetti, D., Passi, A. ,…, Rizzi, F. (2021). Inflammation, extracellular matrix remodeling, and proteostasis in tumor microenvironment. International Journal of Molecular Sciences, 22(15), 8102. https://doi.org/10.3390/ijms22158102
Hu, D., Li, Z., Zheng, B., Lin, X., Pan, Y., Gong, P. ,…, Wang, L. (2022). Cancer-associated fibroblasts in breast cancer: Challenges and opportunities. Cancer Communications (London, England), 42(5), 401–434. https://doi.org/10.1002/cac2.12291
Bartoschek, M., Oskolkov, N., Bocci, M., Lövrot, J., Larsson, C., Sommarin, M. ,…, Pietras, K. (2018). Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nature Communications, 9(1), 5150. https://doi.org/10.1038/s41467-018-07582-3
Bryce, A. S., Dreyer, S. B., Froeling, F. E. M., & Chang, D. K. (2022). Exploring the biology of cancer-associated fibroblasts in pancreatic cancer. Cancers, 14(21), 5302. https://doi.org/10.3390/cancers14215302
Montori, M., Scorzoni, C., Argenziano, M. E., Balducci, D., De Blasio, F., Martini, F. ,…, Maroni, L. (2022). Cancer-associated fibroblasts in cholangiocarcinoma: Current knowledge and possible implications for therapy. Journal of Clinical Medicine, 11(21), 6498. https://doi.org/10.3390/jcm11216498
Wan, P.K.-T., Ryan, A. J., & Seymour, L. W. (2021). Beyond cancer cells: Targeting the tumor microenvironment with gene therapy and armed oncolytic virus. Molecular Therapy: The Journal of the American Society of Gene Therapy, 29(5), 1668–1682. https://doi.org/10.1016/j.ymthe.2021.04.015
Prasad, V., Fojo, T., & Brada, M. (2016). Precision oncology: Origins, optimism, and potential. The Lancet. Oncology, 17(2), e81–e86. https://doi.org/10.1016/S1470-2045(15)00620-8
Kalluri, R., & Zeisberg, M. (2006). Fibroblasts in cancer. Nature Reviews Cancer, 6(5), 392–401. https://doi.org/10.1038/nrc1877
Kalluri, R. (2016). The biology and function of fibroblasts in cancer. Nature Reviews. Cancer, 16(9), 582–598. https://doi.org/10.1038/nrc.2016.73
Kennel, K. B., Bozlar, M., De Valk, A. F., & Greten, F. R. (2022). Cancer-associated fibroblasts in inflammation and anti-tumor immunity. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, CCR-22–1031. https://doi.org/10.1158/1078-0432.CCR-22-1031
Shi, X., Young, C. D., Zhou, H., & Wang, X. (2020). Transforming growth factor-β signaling in fibrotic diseases and cancer-associated fibroblasts. Biomolecules, 10(12), 1666. https://doi.org/10.3390/biom10121666
Kuzet, S.-E., & Gaggioli, C. (2016). Fibroblast activation in cancer: When seed fertilizes soil. Cell and Tissue Research, 365(3), 607–619. https://doi.org/10.1007/s00441-016-2467-x
Li, Z., Low, V., Luga, V., Sun, J., Earlie, E., Parang, B. ,…, Blenis, J. (2022). Tumor-produced and aging-associated oncometabolite methylmalonic acid promotes cancer-associated fibroblast activation to drive metastatic progression. Nature Communications, 13(1), 6239. https://doi.org/10.1038/s41467-022-33862-0
Fang, T., Lv, H., Lv, G., Li, T., Wang, C., Han, Q. ,…, Wang, H. (2018). Tumor-derived exosomal miR-1247–3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nature Communications, 9(1), 191. https://doi.org/10.1038/s41467-017-02583-0
Ammirante, M., Shalapour, S., Kang, Y., Jamieson, C. A. M., & Karin, M. (2014). Tissue injury and hypoxia promote malignant progression of prostate cancer by inducing CXCL13 expression in tumor myofibroblasts. Proceedings of the National Academy of Sciences of the United States of America, 111(41), 14776–14781. https://doi.org/10.1073/pnas.1416498111
Kalluri, R., & Weinberg, R. A. (2009). The basics of epithelial-mesenchymal transition. The Journal of Clinical Investigation, 119(6), 1420–1428. https://doi.org/10.1172/JCI39104
Marconi, G. D., Fonticoli, L., Rajan, T. S., Pierdomenico, S. D., Trubiani, O., Pizzicannella, J., & Diomede, F. (2021). Epithelial-mesenchymal transition (EMT): The type-2 EMT in wound healing, tissue regeneration and organ fibrosis. Cells, 10(7), 1587. https://doi.org/10.3390/cells10071587
Brabletz, T., Kalluri, R., Nieto, M. A., & Weinberg, R. A. (2018). EMT in cancer. Nature Reviews. Cancer, 18(2), 128–134. https://doi.org/10.1038/nrc.2017.118
Ang, H. L., Mohan, C. D., Shanmugam, M. K., Leong, H. C., Makvandi, P., Rangappa, K. S. ,…, Sethi, G. (2023). Mechanism of epithelial-mesenchymal transition in cancer and its regulation by natural compounds. Medicinal Research Reviews, 43(4), 1141–1200. https://doi.org/10.1002/med.21948
Toledo, B., Picon-Ruiz, M., Marchal, J. A., & Perán, M. (2022). Dual role of fibroblasts educated by tumour in cancer behavior and therapeutic perspectives. International Journal of Molecular Sciences, 23(24), 15576. https://doi.org/10.3390/ijms232415576
Adjuto-Saccone, M., Soubeyran, P., Garcia, J., Audebert, S., Camoin, L., Rubis, M. ,…, Tournaire, R. (2021). TNF-α induces endothelial-mesenchymal transition promoting stromal development of pancreatic adenocarcinoma. Cell Death & Disease, 12(7), 649. https://doi.org/10.1038/s41419-021-03920-4
Potenta, S., Zeisberg, E., & Kalluri, R. (2008). The role of endothelial-to-mesenchymal transition in cancer progression. British Journal of Cancer, 99(9), 1375–1379. https://doi.org/10.1038/sj.bjc.6604662
Pérez, L., Muñoz-Durango, N., Riedel, C. A., Echeverría, C., Kalergis, A. M., Cabello-Verrugio, C., & Simon, F. (2017). Endothelial-to-mesenchymal transition: Cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions. Cytokine & Growth Factor Reviews, 33, 41–54. https://doi.org/10.1016/j.cytogfr.2016.09.002
Choi, J., Cha, Y. J., & Koo, J. S. (2018). Adipocyte biology in breast cancer: From silent bystander to active facilitator. Progress in Lipid Research, 69, 11–20. https://doi.org/10.1016/j.plipres.2017.11.002
Bochet, L., Lehuédé, C., Dauvillier, S., Wang, Y. Y., Dirat, B., Laurent, V. ,…, Muller, C. (2013). Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Research, 73(18), 5657–5668. https://doi.org/10.1158/0008-5472.CAN-13-0530
Iyoshi, S., Yoshihara, M., Nakamura, K., Sugiyama, M., Koya, Y., Kitami, K. ,…, Kajiyama, H. (2021). Pro-tumoral behavior of omental adipocyte-derived fibroblasts in tumor microenvironment at the metastatic site of ovarian cancer. International Journal of Cancer, 149(11), 1961–1972. https://doi.org/10.1002/ijc.33770
Teichert, M., Milde, L., Holm, A., Stanicek, L., Gengenbacher, N., Savant, S. ,…, Augustin, H. G. (2017). Pericyte-expressed Tie2 controls angiogenesis and vessel maturation. Nature Communications, 8, 16106. https://doi.org/10.1038/ncomms16106
Yao, F., Luo, Y., Liu, Y.-C., Chen, Y.-H., Li, Y.-T., Hu, X.-Y. ,…, Jing, J.-H. (2022). Imatinib inhibits pericyte-fibroblast transition and inflammation and promotes axon regeneration by blocking the PDGF-BB/PDGFRβ pathway in spinal cord injury. Inflammation and Regeneration, 42(1), 44. https://doi.org/10.1186/s41232-022-00223-9
Hosaka, K., Yang, Y., Seki, T., Fischer, C., Dubey, O., Fredlund, E. ,…, Cao, Y. (2016). Pericyte-fibroblast transition promotes tumor growth and metastasis. Proceedings of the National Academy of Sciences of the United States of America, 113(38), E5618–5627. https://doi.org/10.1073/pnas.1608384113
Tang, P. C.-T., Chung, J. Y.-F., Xue, V. W.-W., Xiao, J., Meng, X.-M., Huang, X.-R. ,…, Lan, H.-Y. (2022). Smad3 promotes cancer-associated fibroblasts generation via macrophage-myofibroblast transition. Advanced Science (Weinheim, Baden-Wurttemberg, Germany), 9(1), e2101235. https://doi.org/10.1002/advs.202101235
Huang, H., Wang, Z., Zhang, Y., Pradhan, R. N., Ganguly, D., Chandra, R. ,…, Brekken, R. A. (2022). Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell, 40(6), 656–673.e7. https://doi.org/10.1016/j.ccell.2022.04.011
Biffi, G., & Tuveson, D. A. (2021). Diversity and biology of cancer-associated fibroblasts. Physiological Reviews, 101(1), 147–176. https://doi.org/10.1152/physrev.00048.2019
Rimal, R., Desai, P., Daware, R., Hosseinnejad, A., Prakash, J., Lammers, T., & Singh, S. (2022). Cancer-associated fibroblasts: Origin, function, imaging, and therapeutic targeting. Advanced Drug Delivery Reviews, 189, 114504. https://doi.org/10.1016/j.addr.2022.114504
Kim, D., Kim, J. S., Cheon, I., Kim, S. R., Chun, S. H., Kim, J. J. ,…, Ko, Y. H. (2022). Identification and characterization of cancer-associated fibroblast subpopulations in lung adenocarcinoma. Cancers, 14(14), 3486. https://doi.org/10.3390/cancers14143486
Kieffer, Y., Hocine, H. R., Gentric, G., Pelon, F., Bernard, C., Bourachot, B. ,…, Mechta-Grigoriou, F. (2020). Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discovery, 10(9), 1330–1351. https://doi.org/10.1158/2159-8290.CD-19-1384
Mosa, M. H., Michels, B. E., Menche, C., Nicolas, A. M., Darvishi, T., Greten, F. R., & Farin, H. F. (2020). A Wnt-induced phenotypic switch in cancer-associated fibroblasts inhibits EMT in colorectal cancer. Cancer Research, 80(24), 5569–5582. https://doi.org/10.1158/0008-5472.CAN-20-0263
Öhlund, D., Handly-Santana, A., Biffi, G., Elyada, E., Almeida, A. S., Ponz-Sarvise, M. ,…, Tuveson, D. A. (2017). Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. The Journal of Experimental Medicine, 214(3), 579–596. https://doi.org/10.1084/jem.20162024
Chen, Z., Zhou, L., Liu, L., Hou, Y., Xiong, M., Yang, Y. ,…, Chen, K. (2020). Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma. Nature Communications, 11(1), 5077. https://doi.org/10.1038/s41467-020-18916-5
Affo, S., Nair, A., Brundu, F., Ravichandra, A., Bhattacharjee, S., Matsuda, M. ,…, Schwabe, R. F. (2021). Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell, 39(6), 866–882.e11. https://doi.org/10.1016/j.ccell.2021.03.012
Biffi, G., Oni, T. E., Spielman, B., Hao, Y., Elyada, E., Park, Y. ,…, Tuveson, D. A. (2019). IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discovery, 9(2), 282–301. https://doi.org/10.1158/2159-8290.CD-18-0710
Zheng, S., Hu, C., Lin, H., Li, G., Xia, R., Zhang, X., … Chen, R. (2022). circCUL2 induces an inflammatory CAF phenotype in pancreatic ductal adenocarcinoma via the activation of the MyD88-dependent NF-κB signaling pathway. Journal of experimental & clinical cancer research: CR, 41(1), 71. https://doi.org/10.1186/s13046-021-02237-6
Picard, F. S. R., Lutz, V., Brichkina, A., Neuhaus, F., Ruckenbrod, T., Hupfer, A. ,…, Huber, M. (2023). IL-17A-producing CD8+ T cells promote PDAC via induction of inflammatory cancer-associated fibroblasts. Gut, gutjnl-2022–327855. https://doi.org/10.1136/gutjnl-2022-327855
Fuentes, N. R., & Taniguchi, C. M. (2023). Turning down oxygen to turn up inflammation in CAFs. Cancer Research, 83(10), 1560–1562. https://doi.org/10.1158/0008-5472.CAN-23-0523
Schwörer, S., Cimino, F. V., Ros, M., Tsanov, K. M., Ng, C., Lowe, S. W. ,…, Thompson, C. B. (2023). Hypoxia potentiates the inflammatory fibroblast phenotype promoted by pancreatic cancer cell-derived cytokines. Cancer Research, 83(10), 1596–1610. https://doi.org/10.1158/0008-5472.CAN-22-2316
Elyada, E., Bolisetty, M., Laise, P., Flynn, W. F., Courtois, E. T., Burkhart, R. A. ,…, Tuveson, D. A. (2019). Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discovery, 9(8), 1102–1123. https://doi.org/10.1158/2159-8290.CD-19-0094
Wilson, R. B., Archid, R., & Reymond, M. A. (2020). Reprogramming of mesothelial-mesenchymal transition in chronic peritoneal diseases by estrogen receptor modulation and TGF-β1 inhibition. International Journal of Molecular Sciences, 21(11), 4158. https://doi.org/10.3390/ijms21114158
apCAFs are derived from mesothelial cells and induce regulatory T cells. (2022). Cancer Discovery, 12(7), 1609. https://doi.org/10.1158/2159-8290.CD-RW2022-085
Hu, B., Wu, C., Mao, H., Gu, H., Dong, H., Yan, J. ,…, Long, J. (2022). Subpopulations of cancer-associated fibroblasts link the prognosis and metabolic features of pancreatic ductal adenocarcinoma. Annals of Translational Medicine, 10(5), 262. https://doi.org/10.21037/atm-22-407
Lavie, D., Ben-Shmuel, A., Erez, N., & Scherz-Shouval, R. (2022). Cancer-associated fibroblasts in the single-cell era. Nature Cancer, 3(7), 793–807. https://doi.org/10.1038/s43018-022-00411-z
Nurmik, M., Ullmann, P., Rodriguez, F., Haan, S., & Letellier, E. (2020). In search of definitions: Cancer-associated fibroblasts and their markers. International Journal of Cancer, 146(4), 895–905. https://doi.org/10.1002/ijc.32193
Venning, F. A., Zornhagen, K. W., Wullkopf, L., Sjölund, J., Rodriguez-Cupello, C., Kjellman, P. ,…, Madsen, C. D. (2021). Deciphering the temporal heterogeneity of cancer-associated fibroblast subpopulations in breast cancer. Journal of experimental & clinical cancer research: CR, 40(1), 175. https://doi.org/10.1186/s13046-021-01944-4
Weber, C. E., Kothari, A. N., Wai, P. Y., Li, N. Y., Driver, J., Zapf, M. A. C., & Mi, Z. (2015). Osteopontin mediates an MZF1-TGF-β1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene, 34(37), 4821–4833. https://doi.org/10.1038/onc.2014.410
Zhao, L., Chen, J., Pang, Y., Fu, K., Shang, Q., Wu, H. ,…, Chen, H. (2022). Fibroblast activation protein-based theranostics in cancer research: A state-of-the-art review. Theranostics, 12(4), 1557–1569. https://doi.org/10.7150/thno.69475
Sotgia, F., Martinez-Outschoorn, U. E., Howell, A., Pestell, R. G., Pavlides, S., & Lisanti, M. P. (2012). Caveolin-1 and cancer metabolism in the tumor microenvironment: Markers, models, and mechanisms. Annual Review of Pathology, 7, 423–467. https://doi.org/10.1146/annurev-pathol-011811-120856
Li, M., Wang, J., Wang, C., Xia, L., Xu, J., Xie, X., & Lu, W. (2020). Microenvironment remodeled by tumor and stromal cells elevates fibroblast-derived COL1A1 and facilitates ovarian cancer metastasis. Experimental Cell Research, 394(1), 112153. https://doi.org/10.1016/j.yexcr.2020.112153
Lin, T.-Y., Chan, H.-H., Chen, S.-H., Sarvagalla, S., Chen, P.-S., Coumar, M. S. ,…, Cheung, C. H. A. (2020). BIRC5/Survivin is a novel ATG12-ATG5 conjugate interactor and an autophagy-induced DNA damage suppressor in human cancer and mouse embryonic fibroblast cells. Autophagy, 16(7), 1296–1313. https://doi.org/10.1080/15548627.2019.1671643
Lee, K.-W., Yeo, S.-Y., Sung, C. O., & Kim, S.-H. (2015). Twist1 is a key regulator of cancer-associated fibroblasts. Cancer Research, 75(1), 73–85. https://doi.org/10.1158/0008-5472.CAN-14-0350
Su, S., Chen, J., Yao, H., Liu, J., Yu, S., Lao, L. ,…, Song, E. (2018). CD10+GPR77+ cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell, 172(4), 841–856.e16. https://doi.org/10.1016/j.cell.2018.01.009
James, A. W., Hindle, P., Murray, I. R., West, C. C., Tawonsawatruk, T., Shen, J. ,…, Soo, C. (2017). Pericytes for the treatment of orthopedic conditions. Pharmacology & Therapeutics, 171, 93–103. https://doi.org/10.1016/j.pharmthera.2016.08.003
Sauzay, C., Voutetakis, K., Chatziioannou, A., Chevet, E., & Avril, T. (2019). CD90/Thy-1, a cancer-associated cell surface signaling molecule. Frontiers in Cell and Developmental Biology, 7, 66. https://doi.org/10.3389/fcell.2019.00066
Krishnamurty, A. T., Shyer, J. A., Thai, M., Gandham, V., Buechler, M. B., Yang, Y. A. ,…, Turley, S. J. (2022). LRRC15+ myofibroblasts dictate the stromal setpoint to suppress tumour immunity. Nature, 611(7934), 148–154. https://doi.org/10.1038/s41586-022-05272-1
Ni, W.-D., Yang, Z.-T., Cui, C.-A., Cui, Y., Fang, L.-Y., & Xuan, Y.-H. (2017). Tenascin-C is a potential cancer-associated fibroblasts marker and predicts poor prognosis in prostate cancer. Biochemical and Biophysical Research Communications, 486(3), 607–612. https://doi.org/10.1016/j.bbrc.2017.03.021
Werner, S., Lützkendorf, J., Müller, T., Müller, L. P., & Posern, G. (2019). MRTF-A controls myofibroblastic differentiation of human multipotent stromal cells and their tumour-supporting function in xenograft models. Scientific Reports, 9(1), 11725. https://doi.org/10.1038/s41598-019-48142-z
Szot, C., Saha, S., Zhang, X. M., Zhu, Z., Hilton, M. B., Morris, K. ,…, St Croix, B. (2018). Tumor stroma-targeted antibody-drug conjugate triggers localized anticancer drug release. The Journal of Clinical Investigation, 128(7), 2927–2943. https://doi.org/10.1172/JCI120481
Zeltz, C., Alam, J., Liu, H., Erusappan, P. M., Hoschuetzky, H., Molven, A. ,…, Gullberg, D. (2019). α11β1 integrin is induced in a subset of cancer-associated fibroblasts in desmoplastic tumor stroma and mediates in vitro cell migration. Cancers, 11(6), 765. https://doi.org/10.3390/cancers11060765
Hwang, W. L., Jagadeesh, K. A., Guo, J. A., Hoffman, H. I., Yadollahpour, P., Reeves, J. W. ,…, Regev, A. (2022). Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment. Nature Genetics, 54(8), 1178–1191. https://doi.org/10.1038/s41588-022-01134-8
Aghamaliyev, U., Gaitantzi, H., Thomas, M., Simon-Keller, K., Gaiser, T., Marx, A. ,…, Breitkopf-Heinlein, K. (2019). Downregulation of SPARC is associated with epithelial-mesenchymal transition and low differentiation state of biliary tract cancer cells. European Surgical Research. Europaische Chirurgische Forschung. Recherches Chirurgicales Europeennes, 60(1–2), 1–12. https://doi.org/10.1159/000494734
Silini, A., Ghilardi, C., Figini, S., Sangalli, F., Fruscio, R., Dahse, R. ,…, Bani, M. (2012). Regulator of G-protein signaling 5 (RGS5) protein: A novel marker of cancer vasculature elicited and sustained by the tumor’s proangiogenic microenvironment. Cellular and molecular life sciences: CMLS, 69(7), 1167–1178. https://doi.org/10.1007/s00018-011-0862-8
Kan, T., Zhang, S., Zhou, S., Zhang, Y., Zhao, Y., Gao, Y. ,…, Yang, M. (2022). Single-cell RNA-seq recognized the initiator of epithelial ovarian cancer recurrence. Oncogene, 41(6), 895–906. https://doi.org/10.1038/s41388-021-02139-z
Houthuijzen, J. M., de Bruijn, R., van der Burg, E., Drenth, A. P., Wientjens, E., Filipovic, T. ,…, Jonkers, J. (2023). CD26-negative and CD26-positive tissue-resident fibroblasts contribute to functionally distinct CAF subpopulations in breast cancer. Nature Communications, 14(1), 183. https://doi.org/10.1038/s41467-023-35793-w
Daniel, S. K., Seo, Y. D., & Pillarisetty, V. G. (2020). The CXCL12-CXCR4/CXCR7 axis as a mechanism of immune resistance in gastrointestinal malignancies. Seminars in Cancer Biology, 65, 176–188. https://doi.org/10.1016/j.semcancer.2019.12.007
Neuzillet, C., Nicolle, R., Raffenne, J., Tijeras-Raballand, A., Brunel, A., Astorgues-Xerri, L. ,…, Bousquet, C. (2022). Periostin- and podoplanin-positive cancer-associated fibroblast subtypes cooperate to shape the inflamed tumor microenvironment in aggressive pancreatic adenocarcinoma. The Journal of Pathology, 258(4), 408–425. https://doi.org/10.1002/path.6011
Su, H., Na, N., Zhang, X., & Zhao, Y. (2017). The biological function and significance of CD74 in immune diseases. Inflammation Research: Official Journal of the European Histamine Research Society ... [et Al.], 66(3), 209–216. https://doi.org/10.1007/s00011-016-0995-1
Peran, I., Dakshanamurthy, S., McCoy, M. D., Mavropoulos, A., Allo, B., Sebastian, A. ,…, Byers, S. W. (2021). Cadherin 11 promotes immunosuppression and extracellular matrix deposition to support growth of pancreatic tumors and resistance to gemcitabine in mice. Gastroenterology, 160(4), 1359–1372.e13. https://doi.org/10.1053/j.gastro.2020.11.044
Zawieracz, K., & Eckert, M. A. (2022). Isolation of normal and cancer-associated fibroblasts. In P. K. Kreeger (Ed.), Ovarian Cancer (Vol. 2424, pp. 155–165). New York, NY: Springer US. https://doi.org/10.1007/978-1-0716-1956-8_10
Yasuda, T., Koiwa, M., Yonemura, A., Akiyama, T., Baba, H., & Ishimoto, T. (2021). Protocol to establish cancer-associated fibroblasts from surgically resected tissues and generate senescent fibroblasts. STAR protocols, 2(2), 100553. https://doi.org/10.1016/j.xpro.2021.100553
Hu, H., Piotrowska, Z., Hare, P. J., Chen, H., Mulvey, H. E., Mayfield, A. ,…, Engelman, J. A. (2021). Three subtypes of lung cancer fibroblasts define distinct therapeutic paradigms. Cancer Cell, 39(11), 1531–1547.e10. https://doi.org/10.1016/j.ccell.2021.09.003
LeBleu, V. S., Taduri, G., O’Connell, J., Teng, Y., Cooke, V. G., Woda, C. ,…, Kalluri, R. (2013). Origin and function of myofibroblasts in kidney fibrosis. Nature Medicine, 19(8), 1047–1053. https://doi.org/10.1038/nm.3218
Tang, X., Hou, Y., Yang, G., Wang, X., Tang, S., Du, Y.-E. ,…, Liu, M. (2016). Stromal miR-200s contribute to breast cancer cell invasion through CAF activation and ECM remodeling. Cell Death and Differentiation, 23(1), 132–145. https://doi.org/10.1038/cdd.2015.78
Jain, S., Rick, J. W., Joshi, R. S., Beniwal, A., Spatz, J., Gill, S. ,…, Aghi, M. K. (2023). Single-cell RNA sequencing and spatial transcriptomics reveal cancer-associated fibroblasts in glioblastoma with protumoral effects. The Journal of Clinical Investigation, 133(5), e147087. https://doi.org/10.1172/JCI147087
Foster, D. S., Januszyk, M., Delitto, D., Yost, K. E., Griffin, M., Guo, J. ,…, Longaker, M. T. (2022). Multiomic analysis reveals conservation of cancer-associated fibroblast phenotypes across species and tissue of origin. Cancer Cell, 40(11), 1392–1406.e7. https://doi.org/10.1016/j.ccell.2022.09.015
Dvořánková, B, Lacina, L, & Smetana, K (2019). Isolation of normal fibroblasts and their cancer-associated counterparts (CAFs) for biomedical research. Methods in Molecular Biology (Clifton, N.J.), 1879, 393–406 https://doi.org/10.1007/7651_2018_137
Yoshida, G. J. (2020). Applications of patient-derived tumor xenograft models and tumor organoids. Journal of Hematology & Oncology, 13(1), 4. https://doi.org/10.1186/s13045-019-0829-z
Xu, H., Lyu, X., Yi, M., Zhao, W., Song, Y., & Wu, K. (2018). Organoid technology and applications in cancer research. Journal of Hematology & Oncology, 11(1), 116. https://doi.org/10.1186/s13045-018-0662-9
Tuveson, D, & Clevers, H (2019) Cancer modeling meets human organoid technology Science New York, NY, 364 6444, 952–955 https://doi.org/10.1126/science.aaw6985
Zou, Z., Lin, Z., Wu, C., Tan, J., Zhang, J., Peng, Y. ,…, Zhang, Y. (2023). Micro-engineered organoid-on-a-chip based on mesenchymal stromal cells to predict immunotherapy responses of HCC patients. Advanced Science (Weinheim, Baden-Wurttemberg, Germany), 10(27), e2302640. https://doi.org/10.1002/advs.202302640
Nassar, D., & Blanpain, C. (2016). Cancer stem cells: Basic concepts and therapeutic implications. Annual Review of Pathology, 11, 47–76. https://doi.org/10.1146/annurev-pathol-012615-044438
Feng, Y, Abdel Mouti, M, & Pauklin, S (2021) PAF1 Regulates the stemness of pancreatic cancer stem cells. Gastroenterology, 160 6, 2224–2225 https://doi.org/10.1053/j.gastro.2021.01.201
Nallasamy, P., Nimmakayala, R. K., Karmakar, S., Leon, F., Seshacharyulu, P., Lakshmanan, I. ,…, Ponnusamy, M. P. (2021). Pancreatic tumor microenvironment factor promotes cancer stemness via SPP1-CD44 axis. Gastroenterology, 161(6), 1998–2013.e7. https://doi.org/10.1053/j.gastro.2021.08.023
Wang, Y., Yu, H., Yu, M., Liu, H., Zhang, B., Wang, Y. ,…, Xia, Q. (2023). CD24 blockade as a novel strategy for cancer treatment. International Immunopharmacology, 121, 110557. https://doi.org/10.1016/j.intimp.2023.110557
Pandey, S., Lee, M., Lim, J., Park, S., Choung, Y.-H., Kim, J. E. ,…, Chung, J. H. (2023). SMO-CRISPR-mediated apoptosis in CD133-targeted cancer stem cells and tumor growth inhibition. Journal of Controlled Release: Official Journal of the Controlled Release Society, 357, 94–108. https://doi.org/10.1016/j.jconrel.2023.03.023
Zhao, R., He, B., Bie, Q., Cao, J., Lu, H., Zhang, Z. ,…, Zhang, B. (2022). AQP5 complements LGR5 to determine the fates of gastric cancer stem cells through regulating ULK1 ubiquitination. Journal of Experimental & Clinical Cancer Research: CR, 41(1), 322. https://doi.org/10.1186/s13046-022-02532-w
Peng, H., Zhu, E., & Zhang, Y. (2022). Advances of cancer-associated fibroblasts in liver cancer. Biomarker Research, 10(1), 59. https://doi.org/10.1186/s40364-022-00406-z
Ma, Z., Li, X., Mao, Y., Wei, C., Huang, Z., Li, G. ,…, Liu, Z. (2022). Interferon-dependent SLC14A1+ cancer-associated fibroblasts promote cancer stemness via WNT5A in bladder cancer. Cancer Cell, 40(12), 1550–1565.e7. https://doi.org/10.1016/j.ccell.2022.11.005
Zhuang, J., Shen, L., Li, M., Sun, J., Hao, J., Li, J. ,…, Yan, J. (2023). Cancer-associated fibroblast-derived miR-146a-5p generates a niche that promotes bladder cancer stemness and chemoresistance. Cancer Research, 83(10), 1611–1627. https://doi.org/10.1158/0008-5472.CAN-22-2213
Hu, J. L., Wang, W., Lan, X. L., Zeng, Z. C., Liang, Y. S., Yan, Y. R. ,…, Liang, L. (2019). CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Molecular Cancer, 18(1), 91. https://doi.org/10.1186/s12943-019-1019-x
Lin, Y., Cai, Q., Chen, Y., Shi, T., Liu, W., Mao, L. ,…, He, R. (2022). CAFs shape myeloid-derived suppressor cells to promote stemness of intrahepatic cholangiocarcinoma through 5-lipoxygenase. Hepatology (Baltimore, Md.), 75(1), 28–42. https://doi.org/10.1002/hep.32099
Katoh, M., & Katoh, M. (2022). WNT signaling and cancer stemness. Essays in Biochemistry, 66(4), 319–331. https://doi.org/10.1042/EBC20220016
Avraamides, C. J., Garmy-Susini, B., & Varner, J. A. (2008). Integrins in angiogenesis and lymphangiogenesis. Nature Reviews. Cancer, 8(8), 604–617. https://doi.org/10.1038/nrc2353
Chen, L., Endler, A., & Shibasaki, F. (2009). Hypoxia and angiogenesis: Regulation of hypoxia-inducible factors via novel binding factors. Experimental & Molecular Medicine, 41(12), 849–857. https://doi.org/10.3858/emm.2009.41.12.103
Baeriswyl, V., & Christofori, G. (2009). The angiogenic switch in carcinogenesis. Seminars in Cancer Biology, 19(5), 329–337. https://doi.org/10.1016/j.semcancer.2009.05.003
De Palma, M., Biziato, D., & Petrova, T. V. (2017). Microenvironmental regulation of tumour angiogenesis. Nature Reviews. Cancer, 17(8), 457–474. https://doi.org/10.1038/nrc.2017.51
Orimo, A., Gupta, P. B., Sgroi, D. C., Arenzana-Seisdedos, F., Delaunay, T., Naeem, R. ,…, Weinberg, R. A. (2005). Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell, 121(3), 335–348. https://doi.org/10.1016/j.cell.2005.02.034
Teicher, B. A., & Fricker, S. P. (2010). CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 16(11), 2927–2931. https://doi.org/10.1158/1078-0432.CCR-09-2329
Presta, M., Dell’Era, P., Mitola, S., Moroni, E., Ronca, R., & Rusnati, M. (2005). Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine & Growth Factor Reviews, 16(2), 159–178. https://doi.org/10.1016/j.cytogfr.2005.01.004
Papadopoulos, N., & Lennartsson, J. (2018). The PDGF/PDGFR pathway as a drug target. Molecular Aspects of Medicine, 62, 75–88. https://doi.org/10.1016/j.mam.2017.11.007
Crawford, Y., Kasman, I., Yu, L., Zhong, C., Wu, X., Modrusan, Z. ,…, Ferrara, N. (2009). PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell, 15(1), 21–34. https://doi.org/10.1016/j.ccr.2008.12.004
Zhang, J.-Y., Zhu, W.-W., Wang, M.-Y., Zhai, R.-D., Wang, Q., Shen, W.-L., & Liu, L.-K. (2021). Cancer-associated fibroblasts promote oral squamous cell carcinoma progression through LOX-mediated matrix stiffness. Journal of Translational Medicine, 19(1), 513. https://doi.org/10.1186/s12967-021-03181-x
Zhang, W., Zhang, S., Zhang, W., Yue, Y., Qian, W., & Wang, Z. (2021). Matrix stiffness and its influence on pancreatic diseases. Biochimica Et Biophysica Acta Reviews on Cancer, 1876(1), 188583. https://doi.org/10.1016/j.bbcan.2021.188583
Jiang, Y., Zhang, H., Wang, J., Liu, Y., Luo, T., & Hua, H. (2022). Targeting extracellular matrix stiffness and mechanotransducers to improve cancer therapy. Journal of Hematology & Oncology, 15(1), 34. https://doi.org/10.1186/s13045-022-01252-0
Sack, K. D., Teran, M., & Nugent, M. A. (2016). Extracellular matrix stiffness controls vegf signaling and processing in endothelial cells. Journal of Cellular Physiology, 231(9), 2026–2039. https://doi.org/10.1002/jcp.25312
Li, M., Zhang, X., Wang, M., Wang, Y., Qian, J., Xing, X. ,…, Ren, Z. (2022). Activation of Piezo1 contributes to matrix stiffness-induced angiogenesis in hepatocellular carcinoma. Cancer Communications (London, England), 42(11), 1162–1184. https://doi.org/10.1002/cac2.12364
Bao, M., Chen, Y., Liu, J.-T., Bao, H., Wang, W.-B., Qi, Y.-X., & Lv, F. (2022). Extracellular matrix stiffness controls VEGF165 secretion and neuroblastoma angiogenesis via the YAP/RUNX2/SRSF1 axis. Angiogenesis, 25(1), 71–86. https://doi.org/10.1007/s10456-021-09804-7
Kim, I., Choi, S., Yoo, S., Lee, M., & Kim, I.-S. (2022). Cancer-associated fibroblasts in the hypoxic tumor microenvironment. Cancers, 14(14), 3321. https://doi.org/10.3390/cancers14143321
Kugeratski, F. G., Atkinson, S. J., Neilson, L. J., Lilla, S., Knight, J. R. P., Serneels, J. ,…, Zanivan, S. (2019). Hypoxic cancer-associated fibroblasts increase NCBP2-AS2/HIAR to promote endothelial sprouting through enhanced VEGF signaling. Science Signaling, 12(567), eaan8247. https://doi.org/10.1126/scisignal.aan8247
Xu, H., Zhao, J., Li, J., Zhu, Z., Cui, Z., Liu, R. ,…, Xu, Q. (2022). Cancer associated fibroblast-derived CCL5 promotes hepatocellular carcinoma metastasis through activating HIF1α/ZEB1 axis. Cell Death & Disease, 13(5), 478. https://doi.org/10.1038/s41419-022-04935-1
Schreiber, R. D., Old, L. J., & Smyth, M. J. (2011). Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science (New York, NY), 331(6024), 1565–1570 https://doi.org/10.1126/science.1203486
O’Donnell, J. S., Teng, M. W. L., & Smyth, M. J. (2019). Cancer immunoediting and resistance to T cell-based immunotherapy. Nature Reviews. Clinical Oncology, 16(3), 151–167. https://doi.org/10.1038/s41571-018-0142-8
Chen, C.-H., Seguin-Devaux, C., Burke, N. A., Oriss, T. B., Watkins, S. C., Clipstone, N., & Ray, A. (2003). Transforming growth factor beta blocks Tec kinase phosphorylation, Ca2+ influx, and NFATc translocation causing inhibition of T cell differentiation. The Journal of Experimental Medicine, 197(12), 1689–1699. https://doi.org/10.1084/jem.20021170
Desbois, M., Udyavar, A. R., Ryner, L., Kozlowski, C., Guan, Y., Dürrbaum, M. ,…, Wang, Y. (2020). Integrated digital pathology and transcriptome analysis identifies molecular mediators of T-cell exclusion in ovarian cancer. Nature Communications, 11(1), 5583. https://doi.org/10.1038/s41467-020-19408-2
Batlle, E., & Massagué, J. (2019). Transforming growth factor-β signaling in immunity and cancer. Immunity, 50(4), 924–940. https://doi.org/10.1016/j.immuni.2019.03.024
Ghahremanifard, P., Chanda, A., Bonni, S., & Bose, P. (2020). TGF-β mediated immune evasion in cancer-spotlight on cancer-associated fibroblasts. Cancers, 12(12), 3650. https://doi.org/10.3390/cancers12123650
Correia, A. L., Guimaraes, J. C., Auf Der Maur, P., De Silva, D., Trefny, M. P., Okamoto, R. ,…, Bentires-Alj, M. (2021). Hepatic stellate cells suppress NK cell-sustained breast cancer dormancy. Nature, 594(7864), 566–571. https://doi.org/10.1038/s41586-021-03614-z
Ene–Obong, A., Clear, A. J., Watt, J., Wang, J., Fatah, R., Riches, J. C. ,…, Kocher, H. M. (2013). Activated pancreatic stellate cells sequester cd8+ t cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology, 145(5), 1121–1132. https://doi.org/10.1053/j.gastro.2013.07.025
Li, X., Sun, Z., Peng, G., Xiao, Y., Guo, J., Wu, B. ,…, Wang, X. (2022). Single-cell RNA sequencing reveals a pro-invasive cancer-associated fibroblast subgroup associated with poor clinical outcomes in patients with gastric cancer. Theranostics, 12(2), 620–638. https://doi.org/10.7150/thno.60540
Cheng, Y., Li, H., Deng, Y., Tai, Y., Zeng, K., Zhang, Y. ,…, Yang, Y. (2018). Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death & Disease, 9(4), 422. https://doi.org/10.1038/s41419-018-0458-4
Kato, T., Noma, K., Ohara, T., Kashima, H., Katsura, Y., Sato, H. ,…, Fujiwara, T. (2018). Cancer-associated fibroblasts affect intratumoral CD8+ and FoxP3+ T cells via IL6 in the tumor microenvironment. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 24(19), 4820–4833. https://doi.org/10.1158/1078-0432.CCR-18-0205
Eskandari-Malayeri, F., & Rezaei, M. (2022). Immune checkpoint inhibitors as mediators for immunosuppression by cancer-associated fibroblasts: A comprehensive review. Frontiers in Immunology, 13, 996145. https://doi.org/10.3389/fimmu.2022.996145
Warburg, O., Wind, F., & Negelein, E. (1927). The metabolism of tumors in the body. The Journal of General Physiology, 8(6), 519–530. https://doi.org/10.1085/jgp.8.6.519
Liang, L., Li, W., Li, X., Jin, X., Liao, Q., Li, Y., & Zhou, Y. (2022). “Reverse Warburg effect” of cancer-associated fibroblasts (Review). International Journal of Oncology, 60(6), 67. https://doi.org/10.3892/ijo.2022.5357
Jaworska, M., Szczudło, J., Pietrzyk, A., Shah, J., Trojan, S. E., Ostrowska, B., & Kocemba-Pilarczyk, K. A. (2023). The Warburg effect: A score for many instruments in the concert of cancer and cancer niche cells. Pharmacological reports: PR. https://doi.org/10.1007/s43440-023-00504-1
Mao, X., Wong, S. Y. S., Tse, E. Y. T., Ko, F. C. F., Tey, S. K., Yeung, Y. S. ,…, Yam, J. W. P. (2016). Mechanisms through which hypoxia-induced caveolin-1 drives tumorigenesis and metastasis in hepatocellular carcinoma. Cancer Research, 76(24), 7242–7253. https://doi.org/10.1158/0008-5472.CAN-16-1031
Cruz-Bermúdez, A., Laza-Briviesca, R., Vicente-Blanco, R. J., García-Grande, A., Coronado, M. J., Laine-Menéndez, S. ,…, Provencio, M. (2019). Cancer-associated fibroblasts modify lung cancer metabolism involving ROS and TGF-β signaling. Free Radical Biology & Medicine, 130, 163–173. https://doi.org/10.1016/j.freeradbiomed.2018.10.450
Chen, X., & Song, E. (2019). Turning foes to friends: Targeting cancer-associated fibroblasts. Nature Reviews Drug Discovery, 18(2), 99–115. https://doi.org/10.1038/s41573-018-0004-1
Aiello, N. M., Brabletz, T., Kang, Y., Nieto, M. A., Weinberg, R. A., & Stanger, B. Z. (2017). Upholding a role for EMT in pancreatic cancer metastasis. Nature, 547(7661), E7–E8. https://doi.org/10.1038/nature22963
Hao, Y., Baker, D., & Ten Dijke, P. (2019). TGF-β-mediated epithelial-mesenchymal transition and cancer metastasis. International Journal of Molecular Sciences, 20(11), 2767. https://doi.org/10.3390/ijms20112767
Zhang, T., Li, X., He, Y., Wang, Y., Shen, J., Wang, S. ,…, Shen, L. (2022). Cancer-associated fibroblasts-derived HAPLN1 promotes tumour invasion through extracellular matrix remodeling in gastric cancer. Gastric Cancer: Official Journal of the International Gastric Cancer Association and the Japanese Gastric Cancer Association, 25(2), 346–359. https://doi.org/10.1007/s10120-021-01259-5
Sakamoto, H., Koma, Y.-I., Higashino, N., Kodama, T., Tanigawa, K., Shimizu, M. ,…, Yokozaki, H. (2021). PAI-1 derived from cancer-associated fibroblasts in esophageal squamous cell carcinoma promotes the invasion of cancer cells and the migration of macrophages. Laboratory Investigation; a Journal of Technical Methods and Pathology, 101(3), 353–368. https://doi.org/10.1038/s41374-020-00512-2
Sun, L.-P., Xu, K., Cui, J., Yuan, D.-Y., Zou, B., Li, J. ,…, Zhang, B. (2019). Cancer‑associated fibroblast‑derived exosomal miR‑382‑5p promotes the migration and invasion of oral squamous cell carcinoma. Oncology Reports, 42(4), 1319–1328. https://doi.org/10.3892/or.2019.7255
Yan, Z., Sheng, Z., Zheng, Y., Feng, R., Xiao, Q., Shi, L. ,…, Zhang, B. (2021). Cancer-associated fibroblast-derived exosomal miR-18b promotes breast cancer invasion and metastasis by regulating TCEAL7. Cell Death & Disease, 12(12), 1120. https://doi.org/10.1038/s41419-021-04409-w
Fattet, L., Jung, H.-Y., Matsumoto, M. W., Aubol, B. E., Kumar, A., Adams, J. A. ,…, Yang, J. (2020). Matrix rigidity controls epithelial-mesenchymal plasticity and tumor metastasis via a mechanoresponsive EPHA2/LYN complex. Developmental Cell, 54(3), 302–316.e7. https://doi.org/10.1016/j.devcel.2020.05.031
Wei, S. C., Fattet, L., Tsai, J. H., Guo, Y., Pai, V. H., Majeski, H. E. ,…, Yang, J. (2015). Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nature Cell Biology, 17(5), 678–688. https://doi.org/10.1038/ncb3157
Labernadie, A., Kato, T., Brugués, A., Serra-Picamal, X., Derzsi, S., Arwert, E. ,…, Trepat, X. (2017). A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nature Cell Biology, 19(3), 224–237. https://doi.org/10.1038/ncb3478
Erdogan, B., Ao, M., White, L. M., Means, A. L., Brewer, B. M., Yang, L. ,…, Webb, D. J. (2017). Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. The Journal of Cell Biology, 216(11), 3799–3816. https://doi.org/10.1083/jcb.201704053
Sabeh, F., Ota, I., Holmbeck, K., Birkedal-Hansen, H., Soloway, P., Balbin, M. ,…, Weiss, S. J. (2004). Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. The Journal of Cell Biology, 167(4), 769–781. https://doi.org/10.1083/jcb.200408028
Sobierajska, K., Ciszewski, W. M., Sacewicz-Hofman, I., & Niewiarowska, J. (2020). Endothelial cells in the tumor microenvironment. Advances in Experimental Medicine and Biology, 1234, 71–86. https://doi.org/10.1007/978-3-030-37184-5_6
Reymond, N., d’Água, B. B., & Ridley, A. J. (2013). Crossing the endothelial barrier during metastasis. Nature Reviews. Cancer, 13(12), 858–870. https://doi.org/10.1038/nrc3628
Wu, D., Deng, S., Li, L., Liu, T., Zhang, T., Li, J. ,…, Xu, Y. (2021). TGF-β1-mediated exosomal lnc-MMP2–2 increases blood-brain barrier permeability via the miRNA-1207–5p/EPB41L5 axis to promote non-small cell lung cancer brain metastasis. Cell Death & Disease, 12(8), 721. https://doi.org/10.1038/s41419-021-04004-z
Gong, M. M., Lugo-Cintron, K. M., White, B. R., Kerr, S. C., Harari, P. M., & Beebe, D. J. (2019). Human organotypic lymphatic vessel model elucidates microenvironment-dependent signaling and barrier function. Biomaterials, 214, 119225. https://doi.org/10.1016/j.biomaterials.2019.119225
Wei, W.-F., Chen, X.-J., Liang, L.-J., Yu, L., Wu, X.-G., Zhou, C.-F. ,…, Wang, W. (2021). Periostin+ cancer-associated fibroblasts promote lymph node metastasis by impairing the lymphatic endothelial barriers in cervical squamous cell carcinoma. Molecular Oncology, 15(1), 210–227. https://doi.org/10.1002/1878-0261.12837
Tacconi, C., Correale, C., Gandelli, A., Spinelli, A., Dejana, E., D’Alessio, S., & Danese, S. (2015). Vascular endothelial growth factor C disrupts the endothelial lymphatic barrier to promote colorectal cancer invasion. Gastroenterology, 148(7), 1438-1451.e8. https://doi.org/10.1053/j.gastro.2015.03.005
Ao, Z., Shah, S. H., Machlin, L. M., Parajuli, R., Miller, P. C., Rawal, S. ,…, El-Ashry, D. (2015). Identification of cancer-associated fibroblasts in circulating blood from patients with metastatic breast cancer. Cancer Research, 75(22), 4681–4687. https://doi.org/10.1158/0008-5472.CAN-15-1633
Sharma, U., Medina-Saenz, K., Miller, P. C., Troness, B., Spartz, A., Sandoval-Leon, A. ,…, El-Ashry, D. (2021). Heterotypic clustering of circulating tumor cells and circulating cancer-associated fibroblasts facilitates breast cancer metastasis. Breast Cancer Research and Treatment, 189(1), 63–80. https://doi.org/10.1007/s10549-021-06299-0
Hurtado, P., Martínez-Pena, I., Yepes-Rodríguez, S., Bascoy-Otero, M., Abuín, C., Fernández-Santiago, C. ,…, Piñeiro, R. (2023). Modelling metastasis in zebrafish unveils regulatory interactions of cancer-associated fibroblasts with circulating tumour cells. Frontiers in Cell and Developmental Biology, 11, 1076432. https://doi.org/10.3389/fcell.2023.1076432
Peinado, H., Zhang, H., Matei, I. R., Costa-Silva, B., Hoshino, A., Rodrigues, G. ,…, Lyden, D. (2017). Pre-metastatic niches: Organ-specific homes for metastases. Nature Reviews. Cancer, 17(5), 302–317. https://doi.org/10.1038/nrc.2017.6
Zeng, H., Hou, Y., Zhou, X., Lang, L., Luo, H., Sun, Y. ,…, Liu, M. (2022). Cancer-associated fibroblasts facilitate premetastatic niche formation through lncRNA SNHG5-mediated angiogenesis and vascular permeability in breast cancer. Theranostics, 12(17), 7351–7370. https://doi.org/10.7150/thno.74753
Kong, J., Tian, H., Zhang, F., Zhang, Z., Li, J., Liu, X. ,…, Liu, T. (2019). Extracellular vesicles of carcinoma-associated fibroblasts creates a pre-metastatic niche in the lung through activating fibroblasts. Molecular Cancer, 18(1), 175. https://doi.org/10.1186/s12943-019-1101-4
Pein, M., Insua-Rodríguez, J., Hongu, T., Riedel, A., Meier, J., Wiedmann, L. ,…, Oskarsson, T. (2020). Metastasis-initiating cells induce and exploit a fibroblast niche to fuel malignant colonization of the lungs. Nature Communications, 11(1), 1494. https://doi.org/10.1038/s41467-020-15188-x
Ji, Q., Zhou, L., Sui, H., Yang, L., Wu, X., Song, Q. ,…, Li, Q. (2020). Primary tumors release ITGBL1-rich extracellular vesicles to promote distal metastatic tumor growth through fibroblast-niche formation. Nature Communications, 11(1), 1211. https://doi.org/10.1038/s41467-020-14869-x
Dong, G., Chen, P., Xu, Y., Liu, T., & Yin, R. (2023). Cancer-associated fibroblasts: Key criminals of tumor pre-metastatic niche. Cancer Letters, 566, 216234. https://doi.org/10.1016/j.canlet.2023.216234
Konieczkowski, D. J., Johannessen, C. M., & Garraway, L. A. (2018). A convergence-based framework for cancer drug resistance. Cancer Cell, 33(5), 801–815. https://doi.org/10.1016/j.ccell.2018.03.025
Lu, Y., Jin, Z., Hou, J., Wu, X., Yu, Z., Yao, L. ,…, Su, L. (2023). Calponin 1 increases cancer-associated fibroblasts-mediated matrix stiffness to promote chemoresistance in gastric cancer. Matrix Biology: Journal of the International Society for Matrix Biology, 115, 1–15. https://doi.org/10.1016/j.matbio.2022.11.005
Li, I., & Nabet, B. Y. (2019). Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Molecular Cancer, 18(1), 32. https://doi.org/10.1186/s12943-019-0975-5
Uchihara, T., Miyake, K., Yonemura, A., Komohara, Y., Itoyama, R., Koiwa, M. ,…, Ishimoto, T. (2020). Extracellular vesicles from cancer-associated fibroblasts containing annexin A6 induces FAK-YAP activation by stabilizing β1 integrin, enhancing drug resistance. Cancer Research, 80(16), 3222–3235. https://doi.org/10.1158/0008-5472.CAN-19-3803
Sansone, P., Savini, C., Kurelac, I., Chang, Q., Amato, L. B., Strillacci, A. ,…, Bromberg, J. (2017). Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proceedings of the National Academy of Sciences of the United States of America, 114(43), E9066–E9075. https://doi.org/10.1073/pnas.1704862114
Chen, X., Zhao, J., Herjan, T., Hong, L., Liao, Y., Liu, C. ,…, Li, X. (2022). IL-17-induced HIF1α drives resistance to anti-PD-L1 via fibroblast-mediated immune exclusion. The Journal of Experimental Medicine, 219(6), e20210693. https://doi.org/10.1084/jem.20210693
Ostermann, E., Garin-Chesa, P., Heider, K. H., Kalat, M., Lamche, H., Puri, C. ,…, Adolf, G. R. (2008). Effective immunoconjugate therapy in cancer models targeting a serine protease of tumor fibroblasts. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 14(14), 4584–4592. https://doi.org/10.1158/1078-0432.CCR-07-5211
Nicolas, A. M., Pesic, M., Engel, E., Ziegler, P. K., Diefenhardt, M., Kennel, K. B. ,…, Greten, F. R. (2022). Inflammatory fibroblasts mediate resistance to neoadjuvant therapy in rectal cancer. Cancer Cell, 40(2), 168–184.e13. https://doi.org/10.1016/j.ccell.2022.01.004
Miyai, Y., Esaki, N., Takahashi, M., & Enomoto, A. (2020). Cancer-associated fibroblasts that restrain cancer progression: Hypotheses and perspectives. Cancer Science, 111(4), 1047–1057. https://doi.org/10.1111/cas.14346
Mizutani, Y., Kobayashi, H., Iida, T., Asai, N., Masamune, A., Hara, A. ,…, Takahashi, M. (2019). Meflin-positive cancer-associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Research, 79(20), 5367–5381. https://doi.org/10.1158/0008-5472.CAN-19-0454
Bhattacharjee, S., Hamberger, F., Ravichandra, A., Miller, M., Nair, A., Affo, S. ,…, Schwabe, R. F. (2021). Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. The Journal of Clinical Investigation, 131(11), e146987, 146987. https://doi.org/10.1172/JCI146987
Chen, Y., Kim, J., Yang, S., Wang, H., Wu, C.-J., Sugimoto, H. ,…, Kalluri, R. (2021). Type I collagen deletion in αSMA+ myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell, 39(4), 548–565.e6. https://doi.org/10.1016/j.ccell.2021.02.007
Özdemir, B. C., Pentcheva-Hoang, T., Carstens, J. L., Zheng, X., Wu, C.-C., Simpson, T. R. ,…, Kalluri, R. (2014). Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell, 25(6), 719–734. https://doi.org/10.1016/j.ccr.2014.04.005
Rhim, A. D., Oberstein, P. E., Thomas, D. H., Mirek, E. T., Palermo, C. F., Sastra, S. A. ,…, Stanger, B. Z. (2014). Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell, 25(6), 735–747. https://doi.org/10.1016/j.ccr.2014.04.021
Mucciolo, G., Araos Henríquez, J., Jihad, M., Pinto Teles, S., Manansala, J. S., Li, W. ,…, Biffi, G. (2024). EGFR-activated myofibroblasts promote metastasis of pancreatic cancer. Cancer Cell, 42(1), 101–118.e11. https://doi.org/10.1016/j.ccell.2023.12.002
Xiao, Y., & Yu, D. (2021). Tumor microenvironment as a therapeutic target in cancer. Pharmacology & Therapeutics, 221, 107753. https://doi.org/10.1016/j.pharmthera.2020.107753
Ford, K., Hanley, C. J., Mellone, M., Szyndralewiez, C., Heitz, F., Wiesel, P. ,…, Thomas, G. J. (2020). NOX4 inhibition potentiates immunotherapy by overcoming cancer-associated fibroblast-mediated CD8 T-cell Exclusion from tumors. Cancer Research, 80(9), 1846–1860. https://doi.org/10.1158/0008-5472.CAN-19-3158
Hanley, C. J., Mellone, M., Ford, K., Thirdborough, S. M., Mellows, T., Frampton, S. J. ,…, Thomas, G. J. (2018). Targeting the myofibroblastic cancer-associated fibroblast phenotype through inhibition of NOX4. Journal of the National Cancer Institute, 110(1), 109–120. https://doi.org/10.1093/jnci/djx121
Dominguez, C. X., Müller, S., Keerthivasan, S., Koeppen, H., Hung, J., Gierke, S. ,…, Turley, S. J. (2020). Single-cell RNA sequencing reveals stromal evolution into lrrc15+ myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discovery, 10(2), 232–253. https://doi.org/10.1158/2159-8290.CD-19-0644
Demetri, G. D., Luke, J. J., Hollebecque, A., Powderly, J. D., Spira, A. I., Subbiah, V., … Villalobos, V. M. (2021). First-in-human phase I study of abbv-085, an antibody-drug conjugate targeting LRRC15, in sarcomas and other advanced solid tumors. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 27(13), 3556–3566. https://doi.org/10.1158/1078-0432.CCR-20-4513
Herbertz, S., Sawyer, J. S., Stauber, A. J., Gueorguieva, I., Driscoll, K. E., Estrem, S. T. ,…, Lahn, M. M. (2015). Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Design, Development and Therapy, 9, 4479–4499. https://doi.org/10.2147/DDDT.S86621
van den Bulk, J., de Miranda, N. F. C. C., & Ten Dijke, P (2021). Therapeutic targeting of TGF-β in cancer: Hacking a master switch of immune suppression. Clinical Science (London, England: 1979), 135(1), 35–52 https://doi.org/10.1042/CS20201236
Yamazaki, T., Gunderson, A. J., Gilchrist, M., Whiteford, M., Kiely, M. X., Hayman, A. ,…, Young, K. H. (2022). Galunisertib plus neoadjuvant chemoradiotherapy in patients with locally advanced rectal cancer: A single-arm, phase 2 trial. The Lancet. Oncology, 23(9), 1189–1200. https://doi.org/10.1016/S1470-2045(22)00446-6
Wei, Y., Kim, T. J., Peng, D. H., Duan, D., Gibbons, D. L., Yamauchi, M. ,…, Chapman, H. A. (2017). Fibroblast-specific inhibition of TGF-β1 signaling attenuates lung and tumor fibrosis. The Journal of Clinical Investigation, 127(10), 3675–3688. https://doi.org/10.1172/JCI94624
Huang, H., Zhang, Y., Gallegos, V., Sorrelle, N., Zaid, M. M., Toombs, J. ,…, Brekken, R. A. (2019). Targeting TGFβR2-mutant tumors exposes vulnerabilities to stromal TGFβ blockade in pancreatic cancer. EMBO molecular medicine, 11(11), e10515. https://doi.org/10.15252/emmm.201910515
Diwanji, R., O’Brien, N. A., Choi, J. E., Nguyen, B., Laszewski, T., Grauel, A. L. ,…, Jayaraman, P. (2023). Targeting the IL1β pathway for cancer immunotherapy remodels the tumor microenvironment and enhances antitumor immune responses. Cancer Immunology Research, 11(6), 777–791. https://doi.org/10.1158/2326-6066.CIR-22-0290
Díaz-Maroto, N. G., Garcia-Vicién, G., Polcaro, G., Bañuls, M., Albert, N., Villanueva, A., & Molleví, D. G. (2021). The blockade of tumoral IL1β-mediated signaling in normal colonic fibroblasts sensitizes tumor cells to chemotherapy and prevents inflammatory CAF activation. International Journal of Molecular Sciences, 22(9), 4960. https://doi.org/10.3390/ijms22094960
Cazet, A. S., Hui, M. N., Elsworth, B. L., Wu, S. Z., Roden, D., Chan, C.-L. ,…, Swarbrick, A. (2018). Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nature Communications, 9(1), 2897. https://doi.org/10.1038/s41467-018-05220-6
Steele, N. G., Biffi, G., Kemp, S. B., Zhang, Y., Drouillard, D., Syu, L. ,…, Pasca di Magliano, M. (2021). Inhibition of hedgehog signaling alters fibroblast composition in pancreatic cancer. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 27(7), 2023–2037. https://doi.org/10.1158/1078-0432.CCR-20-3715
Zhao, J., Wang, H., Hsiao, C.-H., Chow, D. S.-L., Koay, E. J., Kang, Y., … Li, C. (2018). Simultaneous inhibition of hedgehog signaling and tumor proliferation remodels stroma and enhances pancreatic cancer therapy. Biomaterials, 159, 215–228. https://doi.org/10.1016/j.biomaterials.2018.01.014
Beauchamp, E. M., Ringer, L., Bulut, G., Sajwan, K. P., Hall, M. D., Lee, Y.-C. ,…, Uren, A. (2011). Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. The Journal of Clinical Investigation, 121(1), 148–160. https://doi.org/10.1172/JCI42874
Slusarz, A., Shenouda, N. S., Sakla, M. S., Drenkhahn, S. K., Narula, A. S., MacDonald, R. S. ,…, Lubahn, D. B. (2010). Common botanical compounds inhibit the hedgehog signaling pathway in prostate cancer. Cancer Research, 70(8), 3382–3390. https://doi.org/10.1158/0008-5472.CAN-09-3012
Chen, S., Nishi, M., Morine, Y., Shimada, M., Tokunaga, T., Kashihara, H. ,…, Wada, Y. (2022). Epigallocatechin‑3‑gallate hinders metabolic coupling to suppress colorectal cancer malignancy through targeting aerobic glycolysis in cancer‑associated fibroblasts. International Journal of Oncology, 60(2), 19. https://doi.org/10.3892/ijo.2022.5309
Zhang, Q., Cao, W.-S., Wang, X.-Q., Zhang, M., Lu, X.-M., Chen, J.-Q. ,…, Han, H.-Y. (2019). Genistein inhibits nasopharyngeal cancer stem cells through sonic hedgehog signaling. Phytotherapy research: PTR, 33(10), 2783–2791. https://doi.org/10.1002/ptr.6464
Chaudary, N., Pintilie, M., Hedley, D., Hill, R. P., Milosevic, M., & Mackay, H. (2017). Hedgehog inhibition enhances efficacy of radiation and cisplatin in orthotopic cervical cancer xenografts. British Journal of Cancer, 116(1), 50–57. https://doi.org/10.1038/bjc.2016.383
Hanley, C. J., & Thomas, G. J. (2020). T-cell tumour exclusion and immunotherapy resistance: A role for CAF targeting. British Journal of Cancer, 123(9), 1353–1355. https://doi.org/10.1038/s41416-020-1020-6
Lang, L., & Teng, Y. (2019). Fibroblast Growth factor receptor 4 targeting in cancer: New insights into mechanisms and therapeutic strategies. Cells, 8(1), 31. https://doi.org/10.3390/cells8010031
Zhang, M., Li, X., Wu, W., Gao, J., Han, Q., Sun, Z., & Zhao, R. C. (2022). Regorafenib Induces the apoptosis of gastrointestinal cancer-associated fibroblasts by inhibiting AKT phosphorylation. Stem Cells and Development, 31(13–14), 383–394. https://doi.org/10.1089/scd.2022.0088
Zhang, Z., Yu, W., Zheng, M., Liao, X., Wang, J., Yang, D. ,…, Lu, K. P. (2019). Pin1 inhibition potently suppresses gastric cancer growth and blocks PI3K/AKT and Wnt/β-catenin oncogenic pathways. Molecular Carcinogenesis, 58(8), 1450–1464. https://doi.org/10.1002/mc.23027
Kitami, K., Yoshihara, M., Tamauchi, S., Sugiyama, M., Koya, Y., Yamakita, Y. ,…, Kajiyama, H. (2022). Peritoneal restoration by repurposing vitamin D inhibits ovarian cancer dissemination via blockade of the TGF-β1/thrombospondin-1 axis. Matrix Biology: Journal of the International Society for Matrix Biology, 109, 70–90. https://doi.org/10.1016/j.matbio.2022.03.003
Santos, A. M., Jung, J., Aziz, N., Kissil, J. L., & Puré, E. (2009). Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. The Journal of Clinical Investigation, 119(12), 3613–3625. https://doi.org/10.1172/JCI38988
Li, M., Li, G., Kiyokawa, J., Tirmizi, Z., Richardson, L. G., Ning, J. ,…, Wakimoto, H. (2020). Characterization and oncolytic virus targeting of FAP-expressing tumor-associated pericytes in glioblastoma. Acta Neuropathologica Communications, 8(1), 221. https://doi.org/10.1186/s40478-020-01096-0
Purcell, J. W., Tanlimco, S. G., Hickson, J., Fox, M., Sho, M., Durkin, L. ,…, Chao, D. T. (2018). LRRC15 is a novel mesenchymal protein and stromal target for antibody-drug conjugates. Cancer Research, 78(14), 4059–4072. https://doi.org/10.1158/0008-5472.CAN-18-0327
Huang, T.-X., Tan, X.-Y., Huang, H.-S., Li, Y.-T., Liu, B.-L., Liu, K.-S. ,…, Fu, L. (2022). Targeting cancer-associated fibroblast-secreted WNT2 restores dendritic cell-mediated antitumour immunity. Gut, 71(2), 333–344. https://doi.org/10.1136/gutjnl-2020-322924
Mellone, M., Piotrowska, K., Venturi, G., James, L., Bzura, A., Lopez, M. A. ,…, Thomas, G. J. (2022). ATM regulates differentiation of myofibroblastic cancer-associated fibroblasts and can be targeted to overcome immunotherapy Resistance. Cancer Research, 82(24), 4571–4585. https://doi.org/10.1158/0008-5472.CAN-22-0435
Shen, Y., Wang, X., Lu, J., Salfenmoser, M., Wirsik, N. M., Schleussner, N. ,…, Schmidt, T. (2020). Reduction of liver metastasis stiffness improves response to bevacizumab in metastatic colorectal cancer. Cancer Cell, 37(6), 800–817.e7. https://doi.org/10.1016/j.ccell.2020.05.005
Funding
We would like to acknowledge the support received for this work from the Key Program of Zhejiang Province Natural Science Foundation of China (LZ24H160001), the National Natural Science Foundation of China (82072855) and the Fundamental Research Funds for the Central Universities (2023QZJH54), and 4 + X Clinical Research Project of Women’s Hospital, School of Medicine, Zhejiang University (ZDFY2022-4X202).
Author information
Authors and Affiliations
Contributions
T.G. was a major contributor in writing and editing the original manuscript. J.X. made efforts in project administration and supervision and contributed to reviewing and editing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no known competing financial or personal relationships that could appear to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, T., Xu, J. Cancer-associated fibroblasts: a versatile mediator in tumor progression, metastasis, and targeted therapy. Cancer Metastasis Rev (2024). https://doi.org/10.1007/s10555-024-10186-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10555-024-10186-7