
Abstract
Accumulation of misfolded proteins in the endoplasmic reticulum (ER) induces the unfolded protein response (UPR). The UPR promotes cell survival by adjusting ER protein folding capacity but if homeostasis cannot be re-established, apoptosis is induced. The execution of life/death decisions is regulated by the three UPR branches (IRE1, PERK, ATF6) and their downstream effectors. Events that offset the balance of the UPR branches can have devastating consequences, and UPR misregulation has been correlated with various diseases, including metabolic and neurodegenerative diseases and cancer. In cancer, upregulation of the UPR is thought to provide a growth advantage to tumor cells. In contrast to this prevailing view, we report here an analysis of data obtained by others indicating that all three UPR branches appear selectively down-regulated in mouse models of prostate tumorigenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Activation of the unfolded protein response
In eukaryotic cells, most secreted and plasma membrane proteins enter the lumen of the endoplasmic reticulum (ER), where they must be modified and properly folded. Cytotoxic stresses, such as hypoxia, nutrient deprivation, redox and Ca2+ misregulation, can induce accumulation of misfolded or unfolded proteins. Cells respond rapidly with adaptive programs to restore protein homeostasis, indicating a strong selection for mechanisms that maintain fidelity in ER protein folding and assembly. The unfolded protein response (UPR) [1] re-establishes homeostasis by reducing protein translation thereby suppressing protein loading into the ER, up-regulating ER abundance of its resident chaperones and protein modifying enzymes, and targeting misfolded proteins for degradation via ER-associated degradation (ERAD) or autophagy [1–4]. The UPR has been linked to various physiological processes, including cell differentiation, apoptosis, and inflammation [3], and UPR deregulation is correlated with a variety of diseases.
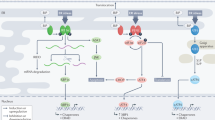
Three ER-resident transmembrane proteins (IRE1, PERK and ATF6) sense unfolded proteins in the ER lumen and activate transcriptional programs in the nucleus [3] (Fig. 1a).

(a) The three branches of the UPR balance between cell survival and cell death. IRE1, PERK, and ATF6 surveys the level of unfolded proteins in the ER lumen and relays this information to transcriptional factors through mRNA splicing, translational regulation, and proteolysis, respectively. Subsequently, the transcription factors (XBP1, ATF4, and ATF6f) coordinate the expression of UPR target genes that in part programs cells for survival or death. (b) Specific down-regulation of UPR regulators during prostate cancer development in Nkx3.1:Pten mutant mice. Right panel: The positive control (+ control) represents a set of 22 known prostate cancer markers [11]. The negative control (- control) includes a panel of 20 randomly generated genes (http://rsat.ulb.ac.be/rsat/random-genes_form.cgi) and 20 random housekeeping genes [19]. All Atg genes represent autophagy regulators or related genes. All the UPR regulators assessed are indicated in the right panel. The relative expression of the genes was obtained from the microarrays performed by Ouyang et al [11], transformed to Log2 scale, and the relative transcript levels of each individual gene set were clustered and averaged as a group (graphs are plotted with s.e.m). For those genes with multiple probes in the microarrays, the probes that exhibited a difference in expression compared to the normal samples are used. The expression of + control and UPR regulator gene sets exhibited a statistical difference (P<0.05, t-test) in expression in the LGPIN, HGPIN, cancer, AI-HGPIN, and AI-Cancer compared to the normal samples. Left panel: The UPR regulators with decreased (P<0.05, t-test) or no difference in expression within the HGPIN compared to the normal prostate samples are highlighted in gray and white, respectively. (c) Expression of target genes induced by the three branches (ATF6, IRE1, PERK) of the UPR is compromised during prostate tumorigenesis in Nkx3.1:Pten mutant mice. The ATF6 UPR targets (25 genes) represent tunicamycin inducible genes in which their expression was up-regulated by ATF6 over-expression or impaired with ATF6 depletion [6, 15]. XBP1 and ATF4 UPR targets are used to represent IRE1 and PERK responsive genes, respectively. XBP1 UPR targets (24 genes) are genes that were up-regulated by tunicamycin, induced by XBP1 over-expression, and down-regulated in Xbp1 −/− cells [16]. ATF4 UPR targets (40 genes) are genes inducible by tunicamycin with decreased expression upon ATF4 ablation [5]. The genes were analyzed and plotted as described in Fig. 1b (d) Expression of UPR inducible targets is reduced in prostate cancers driven by Myc expression in mice. The set of UPR inducible targets examined are the same as those in Fig. 1c. Transcripts with differential expression (P<0.05) [17] in normal and PIN/cancer prostate tissues (isolated from wildtype and Myc transgenic mice, respectively) are included. Genes highlighted in gray are those with reduced expression in the PIN/cancer tissues. The values represent the fold change of the genes in the PIN/cancer tissues from the study of Ellwood-Yen et al. [17]
-
IRE1 is a bi-functional transmembrane kinase/endoribonuclease that upon activation initiates the non-conventional splicing reaction of the mRNA encoding the UPR transcriptional activator XBP1, resulting in translation of a form of XBP1 that is competent for transcriptional regulation [1]. XBP1 up-regulates chaperones to enhance protein folding and genes that mediate ERAD to target degradation of misfolded proteins [3]. IRE1 also activates procaspase–12 and associates with other apoptotic proteins TRAF2, BAK, and BAX. Thus IRE1 has been linked to both cytoprotective functions and apoptosis [4].
-
PERK acts as a conventional transmembrane kinase that phosphorylates eIF2α to inhibit translation-initiation thus promoting global attenuation of protein translation [4]. The post-translational modification of eIF2α permits an immediate adaptation to stress by acutely reducing the influx of proteins into the ER. Some mRNAs, including those encoding the UPR transcriptional activator ATF4, are selectively translated under conditions of limiting eIF2α activity. ATF4 controls expression of genes linked to amino acid import and oxidative stress [5] as well as genes that mediate apoptosis, such as Chop (Gadd153) that suppress the expression of the anti-apoptotic protein BCL2 [1].
-
ATF6 is a transmembrane protein that upon ER stress is transported to the Golgi apparatus where it is cleaved by S1P and S2P proteases. The proteolytically severed cytosolic domain, ATF6f, is a UPR transcriptional activator [4]. A family of ATF6-related CREB proteins, including OASIS (CREB3L1) and CREBH (CREB3L3), are regulated analogously [1]. ATF6f regulates expression of target genes containing ER stress element (ERSE) [3], including ER chaperones, those that target misfolded proteins (DERLINs and EDEMs) for protein degradation in the ERAD pathway [3, 6], and apoptotic genes such as Chop [1].
Thus, all three branches of the UPR facilitate seemingly paradoxical transcriptional responses, both aimed at cell survival by decreasing the misfolded protein load and preparing the cell for apoptosis if ER stress cannot be mitigated in an acceptable way (Fig. 1a).
2 Implications of the UPR in disease pathogenesis
Many lines of evidences point towards a role for the UPR in the genesis or progression of different pathologies. For example, many familial protein folding and aggregation disorders and neurodegenerative diseases display evidence for misregulation of UPR [3], emphasizing the critical importance of this pathway. The UPR has also been associated with other diseases, including metabolic disease. Indeed, Xbp1 +/− mice and those harboring eIF2α with a mutation at the PERK phosphorylation site display insulin resistance typical of type II diabetes when challenged with a high-fat diet, and Perk −/− mice develop early onset diabetes [3]. Finally, many viruses exploit the UPR to allow ER expansion in order to accommodate the vast amounts of membrane protein biogenesis required for their reproduction.
Here we focus on the putative role of the UPR in cancer. Several reports show that specific genes that either control the UPR pathway or are target of the UPR are over-expressed in various types of tumors [2]. For example, increased expression of Bip (GRP78) protein has been observed in breast, colon, and adenocarcinoma cancer cell lines, as well as in ex vivo human primary and animal model tissues [4]. Elevated Bip (GRP78) Xbp1, and Atf6 mRNAs expression were found in hepatocarcinomas [4]. Functional importance of UPR in tumorigenesis is suggested by the observation that increased expression of Bip (Grp78) and Grp94 correlates with larger tumor size and enhanced metastatic capability in mouse models [4]. Moreover, a transgenic mouse model of multiple myeloma has been created using a B-cell specific transgene of active spliced Xbp1 mRNA that closely resembles the human disease [7]. The transgenic mice displayed hypergammaglobulinemia, bone lytic lesions, and spontaneous multiple myeloma in aged animals. Thus, over-expression of UPR effectors can induce cancer. The Xbp1 transgenic B cells exhibited increased proliferation that underscores the importance of the UPR in maintaining cell survival as an inappropriate, constitutive activation of the IRE1 branch in B cells allows them to grow out of control [7]. Similarly, XBP1 protein appears to be required for growth transformed fibroblasts in a hypoxic environment when such cells were xenografted into mice [2]. Collectively, these studies suggest that increased expression of UPR-regulating genes functionally correlates with specific cancers, presumably providing cancer cells with a growth advantage.
Prostate cancer is one the leading cause of deaths in men within Europe and the U.S. Androgen ablation is the most common therapy for treatment of advanced prostate cancer. However, tumor cells often become relatively hormone-independent. No direct studies have examined the connection between the UPR and prostate cancer. Indirectly, a link between the UPR and prostate tumorigenesis is implicated by the observation of altered Bip (Grp78) mRNA expression in human castration-resistant prostatic tumors [8], and enhanced Bip (Grp78) mRNA expression has been associated with decreased survival rate in patients [9]. Also, evidence based on a limited number of human prostate cancer tissues suggests that Xbp1 expression may be decreased [10]. In this study, however, it was not determined whether or not the XBP1 mRNA was spliced; thus it is unknown whether the cancer cells contained elevated concentrations of the active transcription factor. A comprehensive characterization of UPR gene expression and activity at different developmental stages during prostate tumorigenesis has not been described.
3 Down-regulation of the UPR during prostate tumorigenesis in mouse models
To examine the UPR during prostate cancer progression, we review here the expression of genes involved in the UPR pathway at various stages of prostate tumorigenesis. Mice harboring single or multiple disruptions of Nkx3.1 and Pten recapitulate different human prostate cancer stages in an age and hormone-dependent manner [11]. For instance, Nkx3.1 −/− mice develop dysplasia whereas Nkx3.1 +/−:Pten +/− animals acquire low-grade prostatic intraepithelial neoplasia (LGPIN) by 6 months of age and later develop high-grade prostatic intraepithelial neoplasia (HGPIN) followed by invasive adenocarcinoma with metastatic potential (cancer). Using these animals, Gao et al. [11] had elaborately profiled the gene expression patterns of prostate epithelial cells harvested from distinct prostate cancer stages: normal prostate, dysplasia, LGPIN, HGPIN, cancer, androgen-independent HGPIN (AI-HGPIN), androgen-independent cancer (AI-cancer) [11]. Interestingly, compared to normal prostate samples, the average expression level of 19 genes involved in the UPR pathway [1–3, 12–14] was strikingly down-regulated in association with disease progression, from LGPIN, HGPIN to cancer (Fig. 1b, red squares). Decreased expression of these genes was also observed in the androgen independent samples (AI-HGPIN and AI-cancer), indicating that the effect is general and not correlated with hormone dependence. Importantly, the average expression of a set of negative control genes (grey diamonds), autophagy genes (Atg) (grey crosses), and heat-shock protein genes (Hsp) (grey squares) were similar amongst all samples whereas a set of known human prostate cancer markers [11] were up-regulated in the tumorigenic samples (black triangles), demonstrating the specificity of the expression patterns. Notably, a large number (11 of the 19 or 58%) of genes in the UPR pathway were statistically (P<0.05) decreased in the HG-PIN samples compared to normal; the remainders were not statistically different. PIN is a precancerous stage frequently associated with development of prostate cancer. Thus, it is tempting to speculate that the decreased expression of the UPR genes during LGPIN and HGPIN is a marker for the onset of prostate tumorigenesis. The down-regulated genes in the HGPIN samples included Atf6, Ire1, and Atf4 (the transcriptional effectors of the PERK pathway) (Fig. 1b), suggesting that the functions of all three branches of the UPR is altered during prostate cancer progression.
To gain further evidence that all three branches (ATF6, IRE1, PERK) of the UPR are impaired during prostate tumorigenesis, we analyzed the expression of their target genes. Indeed, consistent with the observation of Atf6 down-regulation (Fig. 1b), 18 of the 25 assessed (72%) UPR targets inducible by ATF6 [11, 15] were statistically (P<0.05) lower in HGPIN compared to normal prostate samples, and the decreased average expression level persisted as tumorigenesis progressed to AI-cancer cells (Fig. 1c, black diamonds). Similarly, the overall average expressions of UPR targets inducible by XBP1 [16] and ATF4 [5]—the transcription factors that mediate the IRE1 and PERK pathway, respectively—also exhibited a similar down-regulatory trend in the precancerous and cancerous prostate cell samples (Fig. 1c, red squares and blue triangles). Together, these data show that the ability of all three branches of the UPR to induce their transcription programs is profoundly impaired in pre-malignant and malignant prostate samples, further supporting the notion that the function of the UPR is attenuated during prostate tumorigenesis.
In addition, the expression of UPR genes and their targets were down-regulated in prostate tissues in other mouse prostate cancer models. Transgenic over-expression of Myc in mice results in phenotypes resembling human PIN and eventually progresses to prostate cancer [17]. Ellwood-Yen et al. had previously performed microarray analysis to compare gene expression of normal murine prostate tissues versus those in PIN/cancer transition isolated from Myc transgenic mice [17]. Similar to the observation seen in the Nkx3.1:Pten null mouse models, a panel of genes in the UPR pathway (Atf6, Edem1, Herpud1, Dnajc3) was also down-regulated in the MYC-induced PIN/cancer compared to normal prostate samples [17]. Quantitatively, the expression of 89% (25 of 28) of inducible UPR targets [5, 6, 15, 16] that were different in expression level was also reduced in the PIN/cancer samples (Fig. 1d), consistent with the notion that the function of the UPR is disrupted during prostate tumorigenesis.
4 Summary and concluding remarks
In contrast to other human tumors and animal tumor models that demonstrate activation of the UPR [2], the UPR appears dampened in mouse prostate cancer models. Two independent prostate cancer models (Nkx3.1:Pten mutant and Myc-transgenic) show downregulated transcripts of a panel of UPR genes, correlating with compromised expression of UPR inducible target genes. In the mouse prostate cancer models, all three branches of the UPR appeared disrupted. In some other cancers, the deregulation of the UPR could be specific to a particular branch. Given that the UPR balances between pro-survival and apoptotic decisions, both events that up- or down-regulate the UPR may offset the balance and potentially produce detrimental biological outcome [18]. The up-regulation of UPR in some cancers may be beneficial for the tumor cells by increasing the protein folding capacity and prolonging life; on the contrary, the down-regulation of UPR in other cancer cells (e.g prostate cancer cells) may allow them to escape the apoptotic pathway and favor tumorigenesis. Thus, we surmise that any potential pharmacologic intervention would need to be tailored to specific types of cancers. In particular, it seems possible that depending on the circumstances either upregulation or downregulation of the UPR could be of potential therapeutic value. We anticipate that as UPR pharmacological mediators are discovered, components of the UPR will serve as important biomarkers for selection of targeted therapy.
References
Bernales, S., Papa, F. R., & Walter, P. (2006). Intracellular signaling by the unfolded protein response. Annual Review of Cell and Developmental Biology, 22, 487–508.
Ma, Y., & Hendershot, L. M. (2004). The role of the unfolded protein response in tumour development: friend or foe? Nature Review, Cancer, 4, 966–977.
Malhotra, J. D., & Kaufman, R. J. (2007). The endoplasmic reticulum and the unfolded protein response. Seminars Cell & Development Biology, 18, 716–731.
Scriven, P., Brown, N. J., Pockley, A. G., & Wyld, L. (2007). The unfolded protein response and cancer: a brighter future unfolding? Journal of Molecular Medicine, 85, 331–341.
Harding, H. P., Zhang, Y., Zeng, H., Novoa, I., Lu, P. D., Calfon, M., et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Molecular Cell, 11, 619–633.
Adachi, Y., Yamamoto, K., Okada, T., Yoshida, H., Harada, A., & Mori, K. (2008). ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Structure & Function, 33, 75–89.
Carrasco, D. R., Sukhdeo, K., Protopopova, M., Sinha, R., Enos, M., Carrasco, D. E., et al. (2007). The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell, 11, 349–360.
Pootrakul, L., Datar, R. H., Shi, S. R., Cai, J., Hawes, D., Groshen, S. G., et al. (2006). Expression of stress response protein Grp78 is associated with the development of castration-resistant prostate cancer. Clinical Cancer Research, 12, 5987–5993.
Daneshmand, S., Quek, M. L., Lin, E., Lee, C., Cote, R. J., Hawes, D., et al. (2007). Glucose-regulated protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and survival. Human Pathology, 38, 1547–1552.
Takahashi, S., Suzuki, S., Inaguma, S., Ikeda, Y., Cho, Y. M., Nishiyama, N., et al. (2002). Down-regulation of human X-box binding protein 1 (hXBP-1) expression correlates with tumor progression in human prostate cancers. Prostate, 50, 154–161.
Ouyang, X., Jessen, W. J., Al-Ahmadie, H., Serio, A. M., Lin, Y., Shih, W. J., et al. (2008). Activator protein-1 transcription factors are associated with progression and recurrence of prostate cancer. Cancer Research, 68, 2132–2144.
Liang, J., Yin, C., Doong, H., Fang, S., Peterhoff, C., Nixon, R. A., et al. (2006). Characterization of erasin (UBXD2): a new ER protein that promotes ER-associated protein degradation. Journal of Cell Science, 119, 4011–4024.
van Huizen, R., Martindale, J. L., Gorospe, M., & Holbrook, N. J. (2003). P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2alpha signaling. Journal of Biological Chemistry, 278, 15558–15564.
van Laar, T., van der Eb, A. J., & Terleth, C. (2001). Mif1: a missing link between the unfolded protein response pathway and ER-associated protein degradation? Current Protein and Peptide Science, 2, 169–190.
Okada, T., Yoshida, H., Akazawa, R., Negishi, M., & Mori, K. (2002). Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochemical Journal, 366, 585–594.
Lee, A. H., Iwakoshi, N. N., & Glimcher, L. H. (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Molecular and Cell Biology, 23, 7448–7459.
Ellwood-Yen, K., Graeber, T. G., Wongvipat, J., Iruela-Arispe, M. L., Zhang, J., Matusik, R., et al. (2003). Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell, 4, 223–238.
Lin, J. H., Li, H., Yasumura, D., Cohen, H. R., Zhang, C., Panning, B., et al. (2007). IRE1 signaling affects cell fate during the unfolded protein response. Science, 318, 944–949.
Eisenberg, E., & Levanon, E. Y. (2003). Human housekeeping genes are compact. Trends Genetics, 19, 362–365.
Acknowledgments
We thank Dr. Cory Abate-Shen for assistance with reviewing the microarray data. This review was supported by FONDECYT 1080449 and Conicyt PFB-16.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
So, A.YL., de la Fuente, E., Walter, P. et al. The unfolded protein response during prostate cancer development. Cancer Metastasis Rev 28, 219–223 (2009). https://doi.org/10.1007/s10555-008-9180-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-008-9180-5