
Abstract
Purpose
GDC-0810 (ARN-810) is a novel, non-steroidal, orally bioavailable, selective estrogen receptor degrader (SERD) that potentially inhibits ligand-dependent and ligand-independent estrogen receptor (ER)-mediated signaling.
Methods
A phase Ia/Ib/IIa dose escalation, combination treatment with palbociclib or a luteinizing hormone-releasing hormone, and expansion study determined the safety, pharmacokinetics, and recommended phase 2 dose (RP2D) of GDC-0810 in postmenopausal women with ER + (HER2 −) locally advanced or metastatic breast cancer (MBC). Baseline plasma ctDNA samples were analyzed to determine the ESR1 mutation status.
Results
Patients (N = 152) received GDC-0810 100–800 mg once daily (QD) or 300–400 mg twice daily, in dose escalation, expansion, as single agent or combination treatment. Common adverse events regardless of attribution to study drug were diarrhea, nausea, fatigue, vomiting, and constipation. There was one dose-limiting toxicity during dose escalation. The maximum tolerated dose was not reached. GDC-0810 600 mg QD taken with food was the RP2D. Pharmacokinetics were predictable. FES reduction (> 90%) highlighting pharmacodynamic engagement of ER was observed. Outcomes for the overall population and for patients with tumors harboring ESR1 mutations included partial responses (4% overall; 4% ESR1), stable disease (39% overall; 42% ESR1), non-complete response/non-progressive disease (13% overall; 12% ESR1), progressive disease (40% overall; 38% ESR1), and missing/unevaluable (5% overall; 5% ESR1). Clinical benefit (responses or SD, lasting ≥ 24 weeks) was observed in patients in dose escalation (n = 16, 39%) and expansion (n = 24, 22%).
Conclusion
GDC-0810 was safe and tolerable with preliminary anti-tumor activity in heavily pretreated patients with ER + advanced/MBC, with/without ESR1 mutations, highlighting the potential for oral SERDs.
Clinical Trial and registration date April 4, 2013. NCT01823835 .
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer is the leading cause of cancer death in women worldwide, with more than 1,300,000 new cases and nearly 500,000 deaths annually [1]. Hormone receptor-positive (HR +) breast cancer, with tumor expression of the estrogen receptor (ER) and/or progesterone receptor (PR), is the most common form of the disease. Endocrine therapies that either suppress ER signaling or inhibit aromatase in the biosynthesis of estrogen serve as a major treatment strategy. Aromatase inhibitors with or without cyclin-dependent kinase (CDK 4/6) inhibitors are the recommended first-line treatment option for HR + metastatic breast cancers (MBC), although most tumors ultimately develop resistance to the therapeutic regimen. There is mounting evidence that acquisition of mutations in the ESR1 gene is a major contributor toward aromatase inhibitor resistance [2,3,4]. ESR1 mutations in the ligand-binding domain of the ER confer ligand independence to estrogen while retaining dependence on the ER pathway. Consequently, there is interest in selective estrogen receptor degraders (SERDs) that target the ER directly, regardless of the ESR1 mutation status, to potentially inhibit ligand-dependent as well as ligand-independent ER-mediated signaling [5].
GDC-0810 (ARN-810) is a novel, non-steroidal, orally bioavailable SERD that binds to the ER to limit the activity of estrogen and also induces conformational changes leading to receptor degradation, thereby combating ligand-dependent as well as ligand-independent ER signaling in ER + breast cancer. In in vivo MCF-7 breast cancer cell studies, GDC-0810 fully antagonized the response of ER to estrogens and induced proteasomal degradation of ERα [6]. GDC-0810 has also induced tumor regression in tamoxifen-sensitive as well as in tamoxifen-resistant ER + breast cancer xenograft models [6, 7]. Based on preclinical data, we conducted a proof-of-concept phase Ia/Ib/IIa clinical study of GDC-0810 in women who were postmenopausal and with locally advanced or metastatic ER + (HER2 −) breast cancer.
Patients and methods
Study design
This was a multi-institutional phase Ia/Ib/IIa, open-label, dose finding, safety, pharmacokinetics, and proof-of-concept study of GDC-0810 in women with ER + metastatic breast cancer. Phase Ia employed a standard 3 + 3 dose-escalation scheme. Phase Ib was dose escalation as part of combination treatment. Phase IIa explored the recommended phase 2 dose (RP2D) from phase Ia. The protocol was approved by institutional review boards (IRBs) at participating institutions. Written informed consent was obtained from patients prior to performing any procedures; the study was conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice.
Patients
Women who were postmenopausal and over 18 years of age with histologically or cytologically confirmed, locally advanced or metastatic, ER + (HER2 −) breast cancer were eligible for enrollment in phases Ia/Ib/IIa. There were phase-specific inclusion criteria as addressed here, in Supplementary Methods, Supplementary Table S1, and Supplementary Fig. S1. Phase Ia dose escalation required ≥ 2 months since the last use of tamoxifen and ≥ 6 months since the last use of fulvestrant. Phase Ib combination treatments required no prior treatment with CDK4/6 inhibitor in cohort C1. Phase IIa dose expansion at the RP2D did not allow prior fulvestrant in cohorts A1 or B1 but allowed it in cohorts A2 and B2, and required > 2 months since the last use of tamoxifen in cohort A1.
Exclusion criteria for all three study phases are as addressed here, in Supplementary Methods, Supplementary Table S2, and Supplementary Fig. S1. Patients were excluded if they had untreated or symptomatic central nervous system metastases; endometrial disorders (history of endometrial polyps, endometrial cancer, endometrial hyperplasia, and other significant disorders); any significant cardiac dysfunction within 12 months prior to enrollment; active inflammatory bowel disease or chronic diarrhea, short bowel syndrome, or upper gastrointestinal surgery; known human immunodeficiency virus (HIV) infection; known clinically significant history of liver disease; major surgery within 4 weeks prior to enrollment; or radiation therapy within 2 weeks prior to enrollment.
Study treatments
Patients in phase Ia dose-escalation cohorts received oral doses of GDC-0810 during 28-day cycles given once daily (QD) or twice daily (BID), with fasting and without fasting, and with a single dose given on day 7 leading into cycle 1. The starting dose in the first cohort was 100 mg per day based on preclinical studies. Dose escalation to 200 mg and by 200-mg increments in successive cohorts occurred in the absence of dose-limiting toxicities (DLTs) or conditionally in the presence of DLTs.
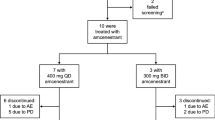
Phase Ib was a dose-escalation study of GDC-0810 starting at 400-mg QD, as combination treatment with 125-mg palbociclib (21 days on/7 days off) or luteinizing hormone-releasing hormone (LHRH) agonist (once every 28 days [Q4W]).
Phase IIa explored the recommended phase 2 dose (RP2D) from phase Ia. All patients received GDC-0810 600 mg under non-fasting conditions.
Patients in all three study phases continued treatment until unacceptable toxicity, disease progression, or consent withdrawal. See Supplementary Methods for other study treatment details.
Study assessments
AEs were assessed using the National Cancer Institute-Common Terminology Criteria for Adverse Events (NCI-CTCAE v4.0). DLTs were assessed during a 35-day DLT window (7 days leading-in + 28 days in cycle 1) and included AEs related to the study drug that were grade ≥ 3 non-hematologic events (excluding alopecia), grade ≥ 3 hematologic events lasting ≥ 7 days, or AEs of any grade that led to study drug interruption for ≥ 7 days. The protocol-defined GDC-0810-specific AEs of special interest (AESI) which were non-serious adverse events that required expedited reporting to the sponsor, included DLTs occurring during the DLT assessment window, grade ≥ 2 vomiting/diarrhea, grade ≥ 3 nausea, grade ≥ 2 thromboembolic events, grade ≥ 2 vaginal or uterine hemorrhage, and grade ≥ 3 elevation of ALT or AST.
Blood samples were collected for pharmacokinetic assessments. Tumor assessments were performed at screening and every 8 weeks from cycle 1, day 1 (C1D1). Radiographic assessment of objective tumor response or disease progression was based on Response Evaluation Criteria in Solid Tumors (RECIST v1.1) [8]. Transvaginal ultrasound scans were performed to monitor endometrial thickness at screening, at every 6 months from C1D1, and at the end of treatment. Imaging with [18F]-fluoroestradiol positron emission tomography (FES-PET) was performed to quantify ER expression in tumors and to assess for pharmacodynamic response. Further assessment details are provided in Supplementary Methods.
Statistical analyses
Analyses of safety, pharmacokinetics, anti-tumor activity, and data from the FES-PET imaging correlative studies were planned. Confirmatory inferential analyses and imputation for missing data were not planned due to the exploratory nature of this study. Descriptive statistics were used to summarize patient data. The number of patients to be enrolled was dependent upon the observed safety and pharmacokinetic profile during dose escalations. The safety- and efficacy-evaluable population included patients who received at least one dose of the study drug. Safety was assessed through summaries of DLTs, AEs, changes in select laboratory test results, vital signs, ECGs, and changes in endometrial thickness. Objective response (with confirmation) and clinical benefit rates (CBR) were derived per RECIST v1.1 and summarized by dose level and cohort.
Results
Study population
Between April 2013 and March 2020, GDC-0810 was administered to 152 female patients who were postmenopausal with ER + (HER2 −) breast cancer, as a single agent (9 dose-escalation cohorts in phase Ia [n = 41] and 4 dose-expansion cohorts in phase IIa [n = 101]) and as combination therapy (2 dose-escalation cohorts in phase Ib [n = 10]). Phases Ia and IIa were conducted in Spain, the Netherlands, South Korea, and the USA; phase Ib was conducted in the USA and South Korea. Patients in phase Ia dose escalation were enrolled in cohorts with or without a fasting regimen, with once or twice daily dosing (Supplementary Fig. S1). The patient population in the 3 phases was predominantly white (n = 127, 84%) with mean age of 60 years (range 31–79) and a high proportion with visceral disease (n = 88, 58%) (Table 1; Supplementary Table S3). Patients were in the metastatic disease setting. Patients in phase Ia dose escalation had received prior MBC therapy (n = 34, 100%) (aromatase inhibitors, tamoxifen, everolimus), chemotherapy (n = 29, 85%), fulvestrant (n = 17, 42%), and cyclin-dependent kinase (CDK 4/6) inhibitor (n = 1, 2%). In phase Ib combination treatments and phase IIa expansion cohorts, patients had received prior MBC therapy (n = 97, 87%) (aromatase inhibitors, tamoxifen, everolimus), chemotherapy (n = 20, 21%), fulvestrant (n = 22, 23%), and CDK 4/6 inhibitor (n = 14, 14%). In phase IIb, 105 of 111 patients had evaluable disease and 6 patients had missing or unavailable measurements.
Pharmacokinetics
The pharmacokinetic profile of GDC-0810 was linear and dose proportional up to 600 mg. GDC-0810 was rapidly absorbed with peak concentrations (median Tmax) achieved at 1–3 h after dosing. The mean terminal half-life was approximately 8 h after 600-mg QD dosing under the non-fasted condition. Minimal drug accumulation was observed following multiple dosing. At the RP2D of 600 mg QD, the average exposure of GDC-0810 at steady state was 1.5-fold (AUC0-24 h) to twofold (Cmax) higher when the drug was administered in non-fasted condition compared to fasted condition. Plasma exposures of glucuronide metabolites were lower than the parent GDC-0810 molecule; the average exposure of GDC-0810-N-glucuronide was approximately 3% and 10% for GDC-0810-acyl-glucuronide at steady state with 600-mg QD dosing under the non-fasted condition. The pharmacokinetic profile and parameters of GDC-0810 and its metabolites, GDC-0810-N-glucuronide and GDC-0810-acyl-glucuronide, following single and multiple doses of GDC-0810, are presented in detail elsewhere [9].
Pharmacodynamics
Pharmacodynamics analyses demonstrated robust target engagement via FES-PET imaging obtained on-treatment versus before GDC-0810 treatment (Fig. 1a), as well as in paired tumor biopsies (Fig. 1b). Overall, complete or near complete (> 90%) suppression of FES uptake was observed in 78% of all patients who had FES-PET scans (24 of 30 phase Ia and 7 of 10 phase IIa dose-expansion patients who had both baseline and C2D3 and C3D3 scans, respectively), including 18 of 26 patients with tumors harboring ESR1 mutations (Fig. 1c). In phase Ia dose escalation, two patients had a single lesion that was considered FES-avid (SUVmax corrected > 1.5) at C2D3; one patient had a subsequent C3 scan which showed no avid lesions. While reduced ER and Ki-67 levels in tumor specimens were noted after treatment with GDC-0810 for one cycle (28 days of treatment) (Fig. 1b), no conclusions could be drawn because baseline and post-dose samples were only available from 3 patients.

Pharmacodynamics of GDC-0810. A Functional imaging with 18F-FES-PET for patient on GDC-0810 600-mg QD (fasting regimen) at baseline and cycle 2 day 3 with best response of stable disease. B ER protein levels and tumor cell proliferation (Ki-67) at baseline and treatment-related reductions after 3 cycles of treatment for patient on GDC-0810 600-mg QD (non-fasting regimen) with best response of stable disease. C Waterfall 18F-FES-PET response for patients in six dose cohorts in phase Ia and cohort A1 in phase IIa
Study drug exposure, RP2D, and MTD
In phase Ia dose escalation, AEs led to dose interruption in 18 (44%) patients, dose reduction in 5 (12%), and study drug withdrawal in 2 (1%). Overall, the average treatment duration was 211 days (range 28–790) (Fig. 2). Twenty (49%) patients remained on treatment for ≥ 24 weeks. Two patients remained on study treatment for 2 years. Although a MTD was not reached in this phase Ia study, the GDC-0810 dose at 800 mg daily was considered intolerable based on the frequency of gastrointestinal AEs (nausea, vomiting, and diarrhea). The GDC-0810 dose of 600 mg QD was declared to be the RP2D when administered under fed conditions, given its overall safety, tolerability, and pharmacokinetic profile. This was the optimal biological dose (600-mg QD, fasting, and non-fasting) where patients demonstrated 80–100% response rates by FES-PET, including patients with tumors harboring ESR1 mutations, discussed in greater detail elsewhere [10].

Patient time on study. Two cohorts in phase Ia, and all cohorts in phase Ib and IIa were dosed under non-fasting conditions
In phase Ib combination treatments, AEs led to dose interruption in 5 (50%) patients, dose reduction in none, and study drug withdrawal in none. The average treatment duration was 413 days (range 56–1144) and 189 days (range 43–504) for cohorts C1 and D1, respectively. Four (40%) patients remained on treatment for ≥ 24 weeks.
In phase IIa expansion cohorts, AEs led to dose interruption in 32 (32%) patients, dose reduction in 4 (4%), and study drug withdrawal in 3 (3%). The average treatment duration was 115 days (range 28–450), 142 days (range 21–791), 216 days (range 8–1586), and 129 days (range 52–504) for cohorts A1, A2, B1, and B2, respectively. Twenty-six (26%) patients remained on treatment for ≥ 24 weeks.
Efficacy
In phase Ia dose escalation, the CBR was 39% as 16 patients had achieved clinical benefit that was defined as RECIST v1.1 responses (complete response [CR], partial response [PR], and/or stable disease [SD]) lasting for ≥ 24 weeks (6 months). For 17 patients in phase Ia who were previously treated with fulvestrant, the CBR was 35% and therefore comparable to that achieved overall (39%). For best confirmed overall outcome, 26 (63%) patients had SD, 12 (29%) had progressive disease (PD), 2 (5%) had confirmed PR, and data for one (2%) patient were missing (Table 2).
In phases Ib combination treatments and IIa expansion cohorts (n = 111), the CBR was 22% (n = 24). The best confirmed overall outcome in phases Ib and IIa included 4 (8%) PR, 33 (30%) SD, 19 (17%) non-complete response or non-progressive disease (non CR/PD), 49 (44%) PD, and 6 (5%) missing or unevaluable (Table 2).
Overall, in the 3 study phases, there were 6 (4%) partial responses, 59 (39%) stable disease, 19 (13%) non-complete response/non-progressive disease, 61 (40%) progressive disease, 5% missing/unevaluable, and the CBR was 26% (n = 40). Patients with clinical benefit did not have significantly different uptake of FES at baseline or a greater reduction in FES uptake on treatment than patients without clinical benefit and a greater reduction in uptake was not associated with a longer time on study.
Efficacy in patients with tumors harboring ESR1 mutations
Among 152 patients with ER + (HER2 −) MBC in the three study phases, 59 (39%) had an outcome of stable disease; data cutoff was April 2016 for phase Ia and March 2020 for phases Ib and IIa. In comparison, among 77 patients with tumors harboring ESR1 mutations, 32 (42%) patients had an outcome of stable disease (Table 2). In patients (n = 77) with tumors harboring ESR1 mutations, there were 3 (4%) partial responses, 32 (42%) stable disease, 9 (12%) non-complete response/non-progressive disease, 29 (38%) progressive disease, and 4 (5%) missing/unevaluable (Table 2; Fig. 3). For patients (n = 77) with tumors harboring ESR1 mutations, the CBR in phase Ia (10 of 23 patients) was 57% and 15% in phase Ib/IIa (8 of 54 patients).

Kaplan–Meier plot of progression-free survival for patients in phase Ia (top) and phases 1b/IIa (bottom)
For the 111 patients in study phases Ib/IIa, the median time to event was 3.5 months (95% confidence interval (CI 1.9, 5.2). In comparison, for the 54 patients in study phases Ib/IIa with tumors harboring ESR1 mutations, the median time to event was the same – 3.5 months (CI 1.9, 5.2) (Fig. 3).
Safety
All patients who received the study drug experienced ≥ 1 AE regardless of causality (Table 3). The most common AEs regardless of attribution across the 3 phases N = 152) in ≥ 25% of patients included diarrhea (n = 95, 63%), nausea (n = 82, 54%), fatigue (n = 77, 51%), vomiting (n = 47, 31%), constipation (n = 42, 28%), and decreased appetite (n = 38, 25%).
During dose escalation, 40 (98%) patients in phase Ia experienced ≥ 1 AE considered related to GDC-0810 by the investigator (Supplementary Table S4). Fifteen (37%) patients in phase Ia experienced AESIs considered related to GDC-0810 by the investigator, including grade ≥ 2 diarrhea (n = 14, 34%), grade ≥ 2 vomiting (n = 7, 17%), grade ≥ 2 thromboembolic event (n = 3, 7%), grade ≥ 3 nausea (n = 1, 2%), and a DLT (diarrhea, 800-mg QD). Thirteen (32%) patients experienced ≥ 1 serious AEs (SAEs) regardless of causality; one of the 20 SAEs was considered related to the study drug by the investigator (Supplementary Table S5) consisting of grade 4 pulmonary embolism.
In phases Ib/IIa, one hundred and one (91%) patients experienced ≥ 1 AE of any grade considered by the investigators to be related to GDC-0810. Diarrhea was the most commonly occurring grade ≥ 3 event (n = 37, 33%). Fifty-five (50%) patients experienced AESIs that included events within the MedDRA SOC of reproductive/breast event (n = 29, 26%), grade ≥ 2 diarrhea (n = 25, 23%), grade ≥ 2 vomiting (n = 9, 8%), grade ≥ 2 thromboembolic event (n = 7, 6%), and grade ≥ 3 elevation of ALT/AST (n = 2, 2%). Three (3%) patients experienced grade 5 AEs including progression of breast cancer (n = 2) and acute renal injury (n = 1); the 3 events were considered unrelated to GDC-0810 by the investigators.
Across all 3 phases, there were 33 patients who reported SAEs regardless of attribution in phase 1a (n = 13), 1b (n = 3), and 2a (n = 17) studies (Supplementary Tables S5 and S6). Six (15%) deaths were reported in phase Ia study, attributed to disease progression (n = 3) and grade 5 SAEs (n = 3), none related to the study treatment. There were no deaths reported in phase Ib and 3 deaths in phase IIa due to progression of the grade 5 AEs described above (breast cancer [n = 2] and acute renal injury [n = 1]).
Among 10 patients in phase Ia dose escalation with a baseline and at least one follow-up scan, 9 (90%) patients showed an increase in the thickness of endometrium compared to baseline, with a median change in thickness of 3.5 mm (range 0 to 11) and mean change of 4.4 mm. There were no treatment discontinuations due to AEs of vagina bleeding or endometrial cancer; a grade 1 vagina hemorrhage in one patient was considered unrelated to GDC-0810 following an endometrial biopsy. In phase Ib combination treatments and IIa dose-expansion studies, 28 of 36 (78%) patients for whom baseline and post-baseline scans were available showed thickening of the endometrium.
Discussion
This proof-of-concept study demonstrated that GDC-0810 was safe and generally well tolerated with predictable pharmacokinetics, evidence of robust target engagement, and encouraging anti-tumor activity in heavily pretreated patients with advanced or metastatic ER + (HER2 −) breast cancer. Most of the AEs were grade 1–2 and manageable with dose interruption, medication, and/or supportive care, with diarrhea being the most common AE with an average onset timeline of 29 days in the phase Ia dose-escalation cohorts. The MTD was not reached; there was one DLT during the DLT window (day 7 to end of cycle 1; 35 days total). Based on safety, pharmacokinetic, and pharmacodynamics data, 600-mg QD was the RP2D for GDC-0810 when administered under non-fasted conditions.
GDC-0810 is a SERD therapy in a landscape where fulvestrant is the only approved first-generation SERD, most frequently used in the second-line setting for ER + advanced or MBC. Combination with CDK4/6 inhibitors has shown a clear benefit over fulvestrant monotherapy in patients who progress on prior endocrine therapy. While fulvestrant has been and continues to be highly impactful in the MBC disease setting, its disadvantages include poor solubility and pharmacokinetic properties requiring intramuscular injections and limited activity in patients with tumors harboring ESR1 mutations [11]. In this study, GDC-0810 demonstrated activity as a single agent including in patients with tumors harboring ESR1 mutations. Furthermore, the clinical benefit rate for GDC-0810 was the same regardless of whether patients received or did not receive prior fulvestrant. In the phase III EMERALD trial investigating elacestrant (a SERD) in patients with MBC who received endocrine + CDK4/6 previously, the PFS was 2.8 months with elacestrant and 1.9 months with standard-of-care (SOC) fulvestrant or an aromatase inhibitor; patients with tumors harboring ESR1 mutations had a median PFS of 3.8 months with elacestrant and 1.9 months with SOC [12, 13]. In the current study evaluating GDC-0810, the median PFS for phase Ib/IIa was 3.5 months (combination treatments in phase Ib; dose expansion at RP2D in phase IIa), and this was the same for patients with or without ESR1 mutations in tumors.
We evaluated combination treatments of GDC-0810 with palbociclib or a LHRH agonist in phase Ib cohorts. Previously, LHRH agonists have been combined with endocrine therapies without significantly affecting their safety profiles in adjuvant and metastatic disease settings for women who were peri- and premenopausal [14, 15]. Guidelines for women in these groups who have advanced breast cancer suggest treatment with ovarian suppression; women are rendered postmenopausal with LHRH agonists and are treated with therapies for the postmenopausal setting. In the current study, patients were postmenopausal without any perceived benefit from LHRH therapy. However, the safety profile of the combination treatment in this study relates to the peri- or premenopausal population who become postmenopausal following LHRH therapy. We found AEs observed for GDC-0810 in combination treatment with palbociclib or LHRH agonist to be similar to those for other endocrine agents; the AEs were amenable to monitoring, manageable, and reversible. Importantly, the benefit from endocrine therapies is expected to be similar for the different patient populations. For example, in the PALOMA-3 study, ~ 80% of the patients were postmenopausal and ~ 20% were peri- or premenopausal; both populations demonstrated similar benefit to combination treatment of a SERD (fulvestrant) and palbociclib, while including LHRH therapy for the latter group [15].
In a patient population with advanced or MBC, FES-PET is a validated method for localizing ER-expressing tumors and has also been reported to predict response to endocrine therapy [16,17,18]. Activity resulting from ER-targeting agents manifests as a decline in FES uptake post-treatment in comparison to baseline and is indicative of ER engagement, although not necessarily of ER degradation. In patients receiving fulvestrant, FES-PET has been used to demonstrate residual ER activity in 38% of patients, thereby signifying inadequate dosing of fulvestrant for targeting the available tumor ERs, which has been associated with early progression [16]. In the current study, ≥ 80% of patients who received GDC-0810 at 600 -g QD (fasting and non-fasting) demonstrated complete or near-complete reduction in FES uptake, including in patients with tumors harboring ESR1 mutations. There was no correlation between FES reduction and clinical benefit in this study, indicating that while high target occupancy may be necessary for ER-targeting agents, achieving clinical benefit and disease control depends on additional factors, such as ER dependency of the tumor.
Breast cancer patients treated with tamoxifen, a selective ER modulator (SERM), have shown clinically significant effects on endometrial thickening. ER is a ligand-inducible transcription factor that contains a central DNA-binding domain; SERMs can have antagonist activity on one domain with agonist activity on another [19]. While highly effective for breast cancer, tamoxifen exhibits ER agonist activity in the uterus and is associated with an increased risk of endometrial hyperplasia and malignancy [20]. Endometrial safety is therefore an important consideration in the development of endocrine agents. In contrast to tamoxifen, fulvestrant is not associated with endometrial thickening. GDC-0810 is a SERD with similarity to fulvestrant, known to suppress ER transcriptional activity by slowing the intra-nuclear mobility of ER [19]. However, endometrial thickening was observed in this study, similar to the class effects of SERM ER agonist activity in the uterus, although no ensuing AEs resulted in treatment discontinuations. The short exposure duration in this study was not expected to result in the development of endometrial cancer. Interestingly, aromatase inhibitors consecutive to or in combination with SERM have been shown to ameliorate the SERM effects of endometrial thickening [21] and could potentially be part of a combination regimen with SERDs. There were no clinically significant ER agonist effects on the lipid profiles of patients treated with GDC-0810.
While the sponsor decision has been to discontinue GDC-0810 development due to an inferior risk/benefit profile relative to other oral SERDs demonstrating early evidence of full ER antagonism and improved tolerability [22, 23], this study has provided important clinical data for continued development of the SERD landscape, including FES-PET utility in the characterization of ER-expressing tumors, activity of this SERD in patients with tumors harboring ESR1 mutations, and the overall clinical safety and pharmacokinetic profile for a SERD. GDC-0810 is the first molecule to be prospectively optimized for ER degradation [6, 7]. This study has contributed to research toward a newer next-generation oral SERDs (GDC-0927 and giredestrant) with improved ER antagonism, no uterine agonism, better toxicity profile, and superior efficacy profile as single agent and combination [22, 24,25,26].
Data availability
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (https://vivli.org/). Other details on Roche’s criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/innovation/process/clinical-trials/data-sharing).
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61(2):69–90. https://doi.org/10.3322/caac.20107[published
Najim O, Seghers S, Sergoynne L et al (1872) 2019 The association between type of endocrine therapy and development of estrogen receptor-1 mutation(s) in patients with hormone-sensitive advanced breast cancer: a systematic review and meta-analysis of randomized and non-randomized trials. Biochim Biophys Acta Rev Cancer 2:188315. https://doi.org/10.1016/j.bbcan.2019.188315[published
Leal MF, Haynes BP, Schuster EF et al (2019) Early enrichment of ESR1 mutations and the impact on gene expression in pre-surgical primary breast cancer treated with aromatase inhibitors. Clin Cancer Res. https://doi.org/10.1158/1078-0432.ccr-19-1129[published
Lopez-Knowles E, Pearson A, Schuster G et al (2019) Molecular characterisation of aromatase inhibitor-resistant advanced breast cancer: the phenotypic effect of ESR1 mutations. Br J Cancer 120(2):247–255. https://doi.org/10.1038/s41416-018-0345-x[published
Sammons S, Shastry M, Dent S, Anders C, Hamilton E (2020) Practical treatment strategies and future directions after progression while receiving CDK4/6 inhibition and endocrine therapy in advanced HR(+)/HER2(-) breast cancer. Clin Breast Cancer 20(1):1–11. https://doi.org/10.1016/j.clbc.2019.06.017[published
Joseph JD, Darimont B, Zhou W et al (2016) The selective estrogen receptor downregulator GDC-0810 is efficacious in diverse models of ER+ breast cancer. Elife. https://doi.org/10.7554/eLife.15828[published
Lai A, Kahraman M, Govek S et al (2015) Identification of GDC-0810 (ARN-810), an orally bioavailable selective estrogen receptor degrader (SERD) that demonstrates robust activity in tamoxifen-resistant breast cancer xenografts. J Med Chem 58(12):4888–4904. https://doi.org/10.1021/acs.jmedchem.5b00054[published
Eisenhauer EA, Therasse P, Bogaerts J et al (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45(2):228–247. https://doi.org/10.1016/j.ejca.2008.10.026[published
Cheung KWK, Yoshida K, Cheeti S et al (2019) GDC-0810 pharmacokinetics and transporter-mediated drug interaction evaluation with an endogenous biomarker in the first-in-human Dose Escalation Study. Drug Metab Dispos 47(9):966–973. https://doi.org/10.1124/dmd.119.087924
Wang Y, Ayres KL, Goldman DA et al (2017) (18)F-fluoroestradiol PET/CT measurement of estrogen receptor suppression during a phase I trial of the novel estrogen receptor-targeted therapeutic GDC-0810: using an imaging biomarker to guide drug dosage in subsequent trials. Clin Cancer Res 23(12):3053–3060. https://doi.org/10.1158/1078-0432.ccr-16-2197[published
Turner NC, Kingston B, Kilburn LS et al (2020) Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): a multicentre, multicohort, phase 2a, platform trial. Lancet Oncol 21(10):1296–1308. https://doi.org/10.1016/s1470-2045(20)30444-7[published
News in brief, no authors listed (2022) Novel SERD Has PFS Edge against Breast Cancer. Cancer Discov 12(2):281. https://doi.org/10.1158/2159-8290.CD-NB2021-406
Bardia A, Neven P, Streich G. Elacestrant. 2021 an oral selective estrogen receptor degrader vs investigator’s choice of endocrine monotherapy for ER+/HER2–advanced/metastatic breast cancer following progression on prior endocrine and CDK4/6 inhibitor therapy: Results of EMERALD phase 3 trial. Proceedings of the 2021 San Antonio Breast Cancer Symposium, San Antonio, TX, USA;8
Pagani O, Regan MM, Walley BA et al (2014) Adjuvant exemestane with ovarian suppression in premenopausal breast cancer. N Engl J Med 371(2):107–118. https://doi.org/10.1056/NEJMoa1404037[published
Turner NC, Ro J, André F et al (2015) Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med 373(3):209–219. https://doi.org/10.1056/NEJMoa1505270[published
van Kruchten M, de Vries EG, Glaudemans AW et al (2015) Measuring residual estrogen receptor availability during fulvestrant therapy in patients with metastatic breast cancer. Cancer Discov 5(1):72–81. https://doi.org/10.1158/2159-8290.cd-14-0697[published
Dehdashti F, Mortimer JE, Trinkaus K et al (2009) PET-based estradiol challenge as a predictive biomarker of response to endocrine therapy in women with estrogen-receptor-positive breast cancer. Breast Cancer Res Treat 113(3):509–517. https://doi.org/10.1007/s10549-008-9953-0
Kurland BF, Peterson LM, Lee JH et al (2017) Estrogen receptor binding (18F-FES PET) and glycolytic activity (18F-FDG PET) Predict progression-free survival on endocrine therapy in patients with ER+ breast cancer. Clin Cancer Res 23(2):407–415. https://doi.org/10.1158/1078-0432.ccr-16-0362
Guan J, Zhou W, Hafner M et al (2019) Therapeutic ligands antagonize estrogen receptor function by impairing its mobility. Cell 178(4):949–63.e18. https://doi.org/10.1016/j.cell.2019.06.026
Pinkerton JV, Goldstein SR (2010) Endometrial safety: a key hurdle for selective estrogen receptor modulators in development. Menopause 17(3):642–653. https://doi.org/10.1097/gme.0b013e3181c4f1d6
Garuti G, Cellani F, Centinaio G, Montanari G, Nalli G, Luerti M (2006) Prospective endometrial assessment of breast cancer patients treated with third generation aromatase inhibitors. Gynecol Oncol 103(2):599–603. https://doi.org/10.1016/j.ygyno.2006.04.004
Kahraman M, Govek SP, Nagasawa JY et al (2019) Maximizing ER-alpha degradation maximizes activity in a tamoxifen-resistant breast cancer model: identification of GDC-0927. ACS Med Chem Lett 10(1):50–55. https://doi.org/10.1021/acsmedchemlett.8b00414
Liang J, Zbieg JR, Blake RA et al (2021) GDC-9545 (Giredestrant): a potent and orally bioavailable selective estrogen receptor antagonist and degrader with an exceptional preclinical profile for ER+ breast cancer. J Med Chem 64(16):11841–11856. https://doi.org/10.1021/acs.jmedchem.1c00847
Kahraman M, Govek SP, Nagasawa JY et al (2018) Abstract 1648: discovery and evolution of orally bioavailable selective estrogen receptor degraders for ER+ breast cancer: from GDC-0810 to GDC-0927. Cancer Res 78(13 Supplement):1648–1748. https://doi.org/10.1158/1538-7445.am2018-1648
Dickler M, Villanueva R, Perez Fidalgo J et al (2018) A first-in-human phase I study to evaluate the oral selective estrogen receptor degrader (SERD) GDC-0927, in postmenopausal women with estrogen receptor positive (ER+), HER2-negative metastatic breast cancer (BC) cancer research. Cancer Res. https://doi.org/10.1158/1538-7445.SABCS17-PD5-10
Jhaveri K, Winer EP, Lim E et al (2020) Abstract PD7–05: a first-in-human phase I study to evaluate the oral selective estrogen receptor degrader (SERD), GDC-9545, in postmenopausal women with estrogen receptor-positive (ER+) HER2-negative (HER2-) metastatic breast cancer. Cancer Res 80(4 Supplement):PD7-05-PD7-05. https://doi.org/10.1158/1538-7445.sabcs19-pd7-05
Acknowledgements
We thank the patients and their families who participated in this clinical trial. Medical writing and editorial support were provided by A. Daisy Goodrich, PhD (Genentech, Inc.) and funded by Genentech, Inc.
Funding
This work was supported by Genentech, Inc., South San Francisco, CA. and K.J. would like to acknowledge a Memorial Sloan Kettering Cancer Center Support Grant (P30 CA008748).
Author information
Authors and Affiliations
Contributions
All authors contributed to manuscript preparation, writing and/or revising the draft, and approving the final manuscript. AB., IM., EW, HML., CXA., BAP., MB., CLA., IC., and KJ. contributed to patient recruitment. SC., C-WC., JF., JMS., HMM., and JG. contributed to data curation. C-WC. contributed to data analysis. LF. contributed to resources and supervision. MG. and IC. contributed to project administration.
Corresponding author
Ethics declarations
Competing interest
A.B. held consulting/advisory board positions at Pfizer, Novartis, Genentech, Merck, Radius Health, Immunomedics/Gilead, Sanofi, Daiichi Pharma/AstraZeneca, Phillips, Eli Lilly, Foundation Medicine, and received contracted research/grant to institution from Genentech, Novartis, Pfizer, Merck, Sanofi, Radius Health, Immunomedics/Gilead, Daiichi Pharma/AstraZeneca, and Eli Lilly. I.M. received consulting fees/ research support from Genentech and is an employee of AstraZeneca. E.W. and H.M.L. received consulting fees and research support from Genentech. C.X.M. held consulting/advisory board positions at Gilead, AstraZeneca, Sanofi-Genzyme, Jacobio, Natera, Novartis, Inivata, Biovica, Athenex, Bayer, Esai, and OncoSignal and received funding from Pfizer and Puma. B.A.P. received contracted research support to the institution from Pfizer, Novartis, Glaxo Smith Kline, Genentech/Roche, and Oncternal Therapeutics Inc., consulting fees from Dare Bioscience, Bioatla Inc. (spouse), EMD Serona (spouse), and Samumed LLC (spouse), and has stock ownership in Merck. M.B. held advisory board positions at Pfizer, Novartis, and Lilly and received speakers bureau fees from Pfizer, Novartis, and Lilly and travel expenses from Pfizer and Roche. C.L.A. held advisory board positions at Novartis, Lilly, Merck, TAIHO Oncology, Immunomedics, Daiichi Sankyo, AstraZeneca, Sanofi, OrigiMed, and Susan G. Komen Foundation, received research grant support from Pfizer, Lilly, and Takeda, and has stock ownership in Provista. S.C., M.G., C.-W.C., J.F., H.M.M., J.G., and I.C. are employees and stockholders of Roche/Genentech. J.M.S. and L.S.F. are former employees and stockholders of Roche/Genentech. E.C.M. is a former employee of Seragon. K.J. received consulting fees from AbbVie, AstraZeneca, Blueprint Medicines, Biotheranostics, BMS, Genentech, Jounce Therapeutics, Lilly Pharmaceuticals, Novartis, Pfizer, Seattle Genetics, SunPharma Pvt Ltd, and Taiho Oncology and contracted research grants to institution from ADC Therapeutics, AstraZeneca, Clovis Oncology, Debio Pharmaceuticals, Genentech, Immunomedics, Novartis, Lilly Pharmaceuticals, Merck/VelosBio, Novartis, Novita Pharmaceuticals, Pfizer, Puma Biotechnology, and Zymeworks.
Ethical approval
The appropriate research ethics committee at each clinic where patients were enrolled approved the study: (1) Memorial Sloan Kettering Cancer Center, 1275 York Ave, New York, NY 10021. (2) Massachusetts General Hospital, 55 Fruit St., Boston, MA 02114. (3) Dana Farber Cancer Institute, 450 Brookline Ave., Boston, MA 02215. (4) Seattle Cancer Care Alliance, 825 Eastlake Ave., Seattle, WA 98109. (5) Washington University, 660 S. Euclid Ave., St. Louis, MO 63110. (6) UCSD Medical Center, 3855 Health Sciences Dr., La Jolla, CA 92093. (7) Vanderbilt University Medical Center, 2220 Pierce Ave., Nashville, TN, 37232. (8) Hospital Universitari Vall d’Hebron, Passeig de la Vall d’Hebron, 119–129, 08035, Barcelona, Spain. (9) Severance Hospital, Yonsei University Health System, 50 Yonsei-ro, Seodaemun-Gu, 120–752, Seoul, South Korea. (10) Centro Integral Oncologico Clara Campal (CIOCC), Calle Oña N°10, 28050, Madrid, Spain. (11) Mount Sinai Medical Center, 1176 Fifth Ave., New York, NY 10029. (12) Vu Medisch Centrum, Dept. Medical Oncology, De Boelelaan 1117, 1081 HV, Amsterdam, the Netherlands. (13) Seoul National University Cancer Hospital, 101 Daehak-ro, Jogno-gu 110–744, Seoul, South Korea. (14) Hospital Clinico Universitario de Valencia, Avda Basco Ibanez 17, 46010, Valencia, Spain.
Consent to participate
Written informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bardia, A., Mayer, I., Winer, E. et al. The oral selective estrogen receptor degrader GDC-0810 (ARN-810) in postmenopausal women with hormone receptor-positive HER2-negative (HR + /HER2 −) advanced/metastatic breast cancer. Breast Cancer Res Treat 197, 319–331 (2023). https://doi.org/10.1007/s10549-022-06797-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-022-06797-9