
Abstract
Purpose
PUFFIN (NCT02896855), a Chinese bridging study in patients with previously untreated HER2-positive locally recurrent or metastatic breast cancer, assessed consistency of efficacy and safety of pertuzumab plus trastuzumab and docetaxel versus placebo, trastuzumab, and docetaxel, with CLEOPATRA (NCT00567190).
Methods
Eligible patients, n = 243, were randomized 1:1, stratified by visceral disease and hormone receptor status, to pertuzumab, trastuzumab, and docetaxel or placebo, trastuzumab, and docetaxel. Primary endpoint: investigator-assessed progression-free survival (PFS). Secondary endpoints: safety and overall survival (OS). After primary analysis, patients could cross over to the pertuzumab arm.
Results
Updated median PFS: 16.5 months (pertuzumab arm) and 12.5 months (placebo arm), with a hazard ratio (HR) of 0.60 [95% confidence interval (CI) 0.45, 0.81; p = 0.0008]. Median OS was not reached in either arm; the OS HR was 0.68 (95% CI 0.45, 1.03; p = 0.0658). Safety was similar in both arms with no new safety signals: 73.8% (pertuzumab arm) and 69.2% (placebo arm) experienced grade ≥ 3 adverse events. No heart failure, symptomatic left ventricular systolic dysfunction, or left ventricular ejection fraction decline of < 40% were reported.
Conclusions
The PUFFIN final analysis showed, per the primary analysis, that overall efficacy of pertuzumab plus trastuzumab and docetaxel was consistent with CLEOPATRA. Safety remained consistent with the known pertuzumab profile. Overall, PUFFIN contributes to the totality of data with pertuzumab in previously untreated HER2-positive locally recurrent or metastatic breast cancer and supports the favorable benefit–risk profile of pertuzumab in Chinese patients.
Trial registration: ClinicalTrials.gov, NCT02896855, registered 7 September 2016.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pertuzumab and trastuzumab (PERJETA® and Herceptin®; F. Hoffmann-La Roche Ltd, Basel, Switzerland) are monoclonal antibodies that bind to different domains on human epidermal growth factor receptor 2 (HER2). The difference in binding creates complementary antitumor activity by inhibiting signaling and promoting antibody-dependent cellular cytotoxicity [1, 2].
Pertuzumab plus trastuzumab and docetaxel in patients with previously untreated HER2-positive locally recurrent or metastatic breast cancer was shown to improve progression-free and overall survival (PFS and OS) significantly compared with placebo plus trastuzumab and docetaxel in CLEOPATRA (NCT00567190). At CLEOPATRA’s primary analysis, independently assessed median PFS was 18.5 months in the pertuzumab arm and 12.4 months in the placebo arm. The PFS hazard ratio (HR) was 0.62 [95% confidence interval (CI) 0.51, 0.75; p < 0.001] and OS data were immature at the first analysis [3]. At the second OS analysis, median OS was immature in the pertuzumab arm and 37.6 months in the placebo arm, with a HR of 0.66 (95% CI 0.52, 0.84; p = 0.0008) [4]. In the final OS analysis, median OS was 56.5 months and 40.8 months in the respective arms, with a HR of 0.68 (95% CI 0.56, 0.84; p < 0.001) [5]. At the 8-year end-of-study analysis, median OS was 57.1 months and 40.8 months, respectively, with a HR of 0.69 (95% CI 0.58, 0.82; p < 0.0001) [6].
Exploratory analyses of safety showed a higher proportion of patients from Asia [China (including Hong Kong) plus Japan, Korea, the Philippines, Singapore, and Thailand] experienced adverse events (AEs) than patients from Europe, North America, and South America combined. The most common AE was febrile neutropenia, which the authors postulated may have been related to increased diarrhea and mucosal inflammation. Despite these differences, comparable survival benefits across regions were observed [7].
PUFFIN (NCT02896855) was a Chinese bridging study to evaluate the efficacy and safety of pertuzumab and to determine consistency with CLEOPATRA. The primary analysis showed that efficacy data were consistent with CLEOPATRA. Median investigator-assessed PFS was 14.5 months and 12.4 months in the pertuzumab and placebo arms, respectively; OS was relatively immature. The PFS HR was 0.69 (95% CI 0.49, 0.99), which was similar to the investigator-assessed PFS HR in CLEOPATRA; 0.62 (95% CI 0.51, 0.75) [3]. Safety was consistent with the known pertuzumab profile [8]. We present the final analysis of PUFFIN, including updated PFS, OS, and safety data, with an additional 20 + months of follow-up of efficacy and AE reporting.
Methods
PUFFIN was a phase III, randomized, double-blind, placebo-controlled study conducted across 15 centers in China. The protocol was approved by the institutional review board at each participating site. Informed consent was provided from all participants. The study design, eligibility criteria, procedures, assessments, and statistical methods have been reported previously in the primary analysis [8].
Eligible patients were aged ≥ 18 years with HER2-positive (centrally confirmed immunohistochemistry 3 + or in situ hybridization-positive), locally recurrent or metastatic breast cancer. Patients were eligible if they had not received prior treatment for metastatic disease (except for one hormonal regimen before randomization), no prior treatment with HER2-targeting therapies (except trastuzumab in the neoadjuvant or adjuvant settings) or tyrosine kinase inhibitors, and were disease-free for ≥ 12 months. Hormone receptor status was centrally confirmed. Further inclusion criteria included a left ventricular ejection fraction (LVEF) of ≥ 55% at baseline and an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0 or 1. Patients were eligible if they had measurable or non-measurable disease.
Patients were excluded if they had prior exposure to doxorubicin of ≥ 360 mg/m2, had a history of LVEF decline to < 50% during or after trastuzumab in the neoadjuvant or adjuvant settings, or if they had any other conditions that were not controlled and could affect the patient’s ability to comply with the study.
Procedures
Randomization was 1:1, using visceral disease and hormone receptor status to stratify patients to pertuzumab (840 mg loading dose, followed by 420 mg every 3 weeks), trastuzumab (8 mg/kg loading dose, then 6 mg/kg every 3 weeks), and docetaxel (75 mg/m2 every 3 weeks) or placebo, trastuzumab, and docetaxel arms; all drugs were given intravenously. The HER2-targeted therapies were given until disease progression or unacceptable toxicity. The docetaxel dose could be reduced to 55 mg/m2 if febrile neutropenia or severe or cumulative cutaneous reactions occurred. Discontinuation of docetaxel was at the discretion of the patient and treating physician after completion of cycle 6.
After the primary analysis, patients could cross over from the placebo arm to the pertuzumab arm due to pertuzumab showing a clinically significant improvement over placebo.
Statistical methods
PFS and OS were estimated using the Kaplan–Meier approach. HR and 95% CIs were estimated by a Cox proportional hazard model using the stratification factors. To compare the two arms, a two-sided stratified log-rank test was used. Statistical testing was considered exploratory. Safety analyses were descriptive.
Results
Study population
From 13 September 2016 to 28 September 2017, 243 eligible patients were randomized: 122 and 121 to the pertuzumab and placebo arms, respectively [8], as shown in Fig. 1. One patient randomized to the placebo arm discontinued prior to treatment. The safety population comprised 122 and 120 patients in the respective arms. After the primary analysis, 12 patients crossed over from the placebo arm to the pertuzumab arm. Clinical cut-off for this final analysis was 23 October 2020. Median follow-up was 39.3 months in the pertuzumab arm and 33.4 months in the placebo arm. Baseline demographics and disease characteristics were balanced between arms [8]. Eleven patients received ovarian function suppressors, two of whom received them during the study. Five patients underwent an oophorectomy, only one during the study.

Disposition of patients
Progression-free survival
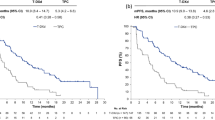
Updated median investigator-assessed PFS was 16.5 months (95% CI 12.7, 20.2) in the pertuzumab arm and 12.5 months (95% CI 10.4, 14.6) in the placebo arm (Fig. 2a). The stratified HR was 0.60 (95% CI 0.45, 0.81; p = 0.008) and the unstratified HR was 0.63 (95% CI 0.47, 0.85; p = 0.0019). The number of patients who had a PFS event was 84/122 (68.9%) in the pertuzumab arm and 99/120 (81.8%) in the placebo arm. Subgroup analyses are shown in Fig. 2b and were consistent with the overall results.

Investigator-assessed PFS in a the intention-to-treat population and b subgroups. CI confidence interval, D docetaxel, ECOG PS Eastern Cooperative Oncology Group Performance Status, ER estrogen receptor, FISH fluorescence in situ hybridization, H trastuzumab, HER2 human epidermal growth factor receptor 2, HR hazard ratio, IHC immunohistochemistry, P pertuzumab, PFS, progression-free survival, PgR progesterone receptor, Pla placebo
Overall survival
Median OS was not reached in either arm. The stratified HR was 0.68 (95% CI 0.45, 1.03; p = 0.0658) and the unstratified HR was 0.70 (95% CI 0.46, 1.06; p = 0.0864) (Fig. 3a). There were 40 OS events (32.8%) in the pertuzumab arm and 50 (41.3%) in the placebo arm. Subgroup analyses are shown in Fig. 3b and were consistent with the overall results.

OS in a the intention-to-treat population and b subgroups. CI confidence interval, D docetaxel, ECOG PS Eastern Cooperative Oncology Group Performance Status, ER estrogen receptor, FISH fluorescence in situ hybridization, H trastuzumab, HER2 human epidermal growth factor receptor 2, HR hazard ratio, IHC immunohistochemistry, OS overall survival, P pertuzumab, PgR progesterone receptor, Pla placebo
Treatment exposure
Prior to crossover, the median number of pertuzumab or placebo cycles administered to patients was 22.0 (range 1–4) and 17.0 (range 1–68), over the respective median duration of 66.1 weeks (range 3–225) and 52.3 weeks (range 3–207). The median number of trastuzumab cycles in the pertuzumab and placebo arms was 22.0 (range 1–74) and 17.0 (range 1–74), respectively. Both arms had a similar median number of docetaxel cycles, with 7.0 (range 1–26) in the pertuzumab arm and 6.5 (range 1–22) in the placebo arm. The median dose of each was 900.0 mg and 826.1 mg. Following crossover, the median number of pertuzumab cycles was 6.0 (range 4–8), with a median duration of 18.1 weeks (range 12–24).
Safety
A summary of the safety profile is shown in Table 1. The number of patients who experienced ≥ 1 AE was 121/122 (99.2%) in the pertuzumab arm and 115/120 (95.8%) in the placebo arm. The most common AE in both arms was leukopenia, reported in 91/122 patients (74.6%) and 87/120 patients (72.5%), respectively. In the pertuzumab arm, AEs with an incidence ≥ 5% higher than with placebo were anemia, alopecia, diarrhea, pyrexia, cough, hypokalemia, hyperglycemia, and stomatitis. There were 111/122 patients (91.0%) in the pertuzumab arm and 104/120 patients (86.7%) in the placebo arm who experienced AEs with a causal relationship to HER2-targeted therapies. Grade ≥ 3 AEs were experienced by 90 patients (73.8%) in the pertuzumab arm and 83 patients (69.2%) in the placebo arm, with the most common being blood and lymphatic system disorders, particularly neutropenia and leukopenia. The number of patients who experienced serious AEs was 30 (24.6%) in the pertuzumab arm and 23 (19.2%) in the placebo arm; the most common were neutropenia and febrile neutropenia. The number of patients who withdrew from treatment due to AEs relating to either pertuzumab or placebo was seven (5.7%) and two (1.7%), respectively. AEs were associated with the deaths of two patients (1.7%) in the placebo arm and four patients (3.3%) in the pertuzumab arm. AEs of interest to pertuzumab are reported in Table 2. No heart failure, symptomatic left ventricle systolic dysfunction, or LVEF decline of < 40% were reported; two patients in the pertuzumab arm experienced grade 2 ejection fraction decreases, which were reported as AEs of special interest, and led to the discontinuation of pertuzumab in both patients. A safety summary for the crossover population is shown in Table 3. There were very few AEs in the crossover group, with five (41.7%) patients experiencing ≥ 1 AE, with only one grade ≥ 3 AE (large intestine polyp).
Discussion
PUFFIN previously met its primary objectives, demonstrating consistency of efficacy with CLEOPATRA [8]. The stratified PFS HR had decreased since the primary analysis [0.60 (95% CI 0.45, 0.81) and 0.69 (95% CI 0.49, 0.99), respectively], with an increased reliability of a narrower CI, and remained consistent with the CLEOPATRA intention-to-treat (ITT) population [HR = 0.62 (95% CI 0.51, 0.75)] [3, 8]. The updated median PFS was closer to the CLEOPATRA ITT population, further demonstrating consistency. As discussed in the primary manuscript, it is important to note that PFS was investigator-assessed per Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 in PUFFIN, whereas in CLEOPATRA, PFS was independent review facility-assessed per RECIST v1.0. The OS HR values were also similar between PUFFIN and CLEOPATRA’s second OS analysis [4]. However, CLEOPATRA had a longer follow-up period; therefore, PUFFIN would require a longer follow-up period to show greater differences between arms and to evaluate median OS. The OS analyses were based on the ITT population, the 12 crossover patients were included in the placebo group for the analysis as randomly assigned. Given the small number of crossover patients, these analyses were not adjusted for crossover to the pertuzumab group. However, they are likely to be conservative in estimating the overall treatment effect.
Subgroup analyses showed consistency with the overall PFS and OS results. Compared with the primary PFS subgroup analysis, the final analysis showed an overall shift toward the pertuzumab group having improved PFS and OS, with little difference between disease type, hormone receptor status, and low or high HER2 status. Patients aged ≥ 65 years showed no difference in PFS between arms. Subgroup analyses showed that patients in the < 65 years age group [PFS HR = 0.58 (95% CI 0.43, 0.80) and OS HR = 0.60 (95% CI 0.39, 0.94)] had better outcomes with pertuzumab. Patients who were ≥ 2 years treatment-free had improved PFS over patients who were < 2 years treatment-free [HR = 0.43 (95% CI 0.25, 0.75) vs. HR = 0.67 (95% CI 0.36, 1.25)]. However, the OS subgroup analysis showed patients who were < 2 years treatment-free had improved OS over patients who were ≥ 2 years treatment-free [HR = 0.30 (95% CI 0.10, 0.92) vs. HR = 0.68 (95% CI 0.32, 1.46)]. Patients with de novo disease had poorer OS with pertuzumab compared with placebo [HR = 1.22 (95% CI 0.58, 2.59)]; this conflicts with CLEOPATRA [HR = 0.65 (95% CI 0.51, 0.82)], which showed that patients with no previous treatment had better prognosis with pertuzumab than with placebo [6]. Additionally, both CLEOPATRA and a retrospective study of patients with de novo, HER2-positive, metastatic breast cancer concluded that trastuzumab as a first-line treatment can improve OS in these patients [6, 9]. Despite PUFFIN showing poorer OS in a subset of patients, new anti-HER2 therapies and regimens may provide improved clinical outcomes following disease progression [10,11,12,13]. Furthermore, the development of screening techniques (such as liquid biopsy) can identify targetable genomic alterations that can be utilized to determine an optimal treatment regimen in patients [14].
The overall safety profile was consistent with the primary analysis, with few AEs reported that required additional follow-up in either arm, and no new safety signals. Leukopenia remained the most common AE in the study, followed by neutropenia. Grade ≥ 3 AEs remained balanced between arms, with neutropenia, leukopenia, febrile neutropenia, anemia, and diarrhea being the most reported (with the addition of hypertension and pneumonia since the primary analysis). No new cardiac AEs were reported since the primary analysis. Comparing the safety profile to the Asian population in CLEOPATRA’s exploratory analysis, the rates of AEs of special interest to pertuzumab were lower, specifically diarrhea (45.9% vs. 74.4% vs.) and febrile neutropenia (4.1% vs. 25.6%) [7]. This suggests that AE management has improved over time, with rates similar to the overall population in CLEOPATRA (diarrhea 66.8% and febrile neutropenia 13.8%) [3]. Therefore, the safety profile remained consistent with that observed in the primary analysis and CLEOPATRA ITT population, and with the established pertuzumab safety profile.
Conclusions
This final analysis of PUFFIN showed, as with the primary analysis, that overall efficacy was consistent with that of the ITT population in CLEOPATRA. Safety of pertuzumab remained consistent with the known pertuzumab profile. Overall, PUFFIN contributes to the totality of data with pertuzumab in previously untreated HER2-positive locally recurrent or metastatic breast cancer and supports the favorable benefit–risk profile of pertuzumab in Chinese patients.
Data availability
For eligible studies, qualified researchers may request access to individual patient-level clinical data through a data request platform. At the time of writing, this request platform is Vivli. https://vivli.org/ourmember/roche/. For up-to-date details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here: https://go.roche.com/data_sharing. Anonymized records for individual patients across more than one data source external to Roche cannot, and should not, be linked due to a potential increase in risk of patient re-identification.
Abbreviations
- AE:
-
Adverse event
- CI:
-
Confidence interval
- D:
-
Docetaxel
- ECOG PS:
-
Eastern Cooperative Oncology Group Performance Status
- ER:
-
Estrogen receptor
- FISH:
-
Fluorescence in situ hybridization
- H:
-
Trastuzumab
- HER2:
-
Human epidermal growth factor receptor 2
- HR:
-
Hazard ratio
- ICH:
-
International Council for Harmonisation
- IEC:
-
Independent ethics committee
- IHC:
-
Immunohistochemistry
- ITT:
-
Intention-to-treat
- LVEF:
-
Left ventricular ejection fraction
- OS:
-
Overall survival
- P:
-
Pertuzumab
- PFS:
-
Progression-free survival
- PgR:
-
Progesterone receptor
- Pla:
-
Placebo
- RECIST:
-
Response Evaluation Criteria in Solid Tumors
References
Nahta R, Hung MC, Esteva FJ (2004) The HER-2-targeting antibodies trastuzumab and pertuzumab synergistically inhibit the survival of breast cancer cells. Cancer Res 64:2343–2346. https://doi.org/10.1158/0008-5472.can-03-3856
Scheuer W, Friess T, Burtscher H, Bossenmaier B, Endl J, Hasmann M (2009) Strongly enhanced antitumor activity of trastuzumab and pertuzumab combination treatment on HER2-positive human xenograft tumor models. Cancer Res 69:9330–9336. https://doi.org/10.1158/0008-5472.Can-08-4597
Baselga J, Cortés J, Kim SB, Im SA, Hegg R, Im YH, Roman L, Pedrini JL, Pienkowski T, Knott A, Clark E, Benyunes MC, Ross G, Swain SM, CLEOPATRA Study Group (2012) Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med 366:109–119. https://doi.org/10.1056/NEJMoa1113216
Swain SM, Kim S-B, Cortés J, Ro J, Semiglazov V, Campone M, Ciruelos E, Ferrero J-M, Schneeweiss A, Knott A, Clark E, Ross G, Benyunes MC, Baselga J (2013) Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA study): overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol 14:461–471. https://doi.org/10.1016/S1470-2045(13)70130-X
Swain SM, Baselga J, Kim SB, Ro J, Semiglazov V, Campone M, Ciruelos E, Ferrero JM, Schneeweiss A, Heeson S, Clark E, Ross G, Benyunes MC, Cortes J, CLEOPATRA Study Group (2015) Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med 372:724–734. https://doi.org/10.1056/NEJMoa1413513
Swain SM, Miles D, Kim SB, Im YH, Im SA, Semiglazov V, Ciruelos E, Schneeweiss A, Loi S, Monturus E, Clark E, Knott A, Restuccia E, Benyunes MC, Cortés J, CLEOPATRA Study Group (2020) Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): end-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol 21:519–530. https://doi.org/10.1016/s1470-2045(19)30863-0
Swain SM, Im YH, Im SA, Chan V, Miles D, Knott A, Clark E, Ross G, Baselga J (2014) Safety profile of pertuzumab with trastuzumab and docetaxel in patients from Asia with human epidermal growth factor receptor 2-positive metastatic breast cancer: results from the phase III trial CLEOPATRA. Oncologist 19:693–701. https://doi.org/10.1634/theoncologist.2014-0033
Xu B, Li W, Zhang Q, Shao Z, Li Q, Wang X, Li H, Sun T, Yin Y, Zheng H, Feng J, Zhang H, Lei G, Restuccia E (2020) Pertuzumab, trastuzumab, and docetaxel for Chinese patients with previously untreated HER2-positive locally recurrent or metastatic breast cancer (PUFFIN): a phase III, randomized, double-blind, placebo-controlled study. Breast Cancer Res Treat 182:689–697. https://doi.org/10.1007/s10549-020-05728-w
Wong Y, Raghavendra AS, Hatzis C, Irizarry JP, Vega T, Horowitz N, Barcenas CH, Chavez-MacGregor M, Valero V, Tripathy D, Pusztai L, Murthy RK (2019) Long-term survival of De Novo stage IV human epidermal growth receptor 2 (HER2) positive breast cancers treated with HER2-targeted therapy. Oncologist 24:313–318. https://doi.org/10.1634/theoncologist.2018-0213
Murthy RK, Loi S, Okines A, Paplomata E, Hamilton E, Hurvitz SA, Lin NU, Borges V, Abramson V, Anders C, Bedard PL, Oliveira M, Jakobsen E, Bachelot T, Shachar SS, Müller V, Braga S, Duhoux FP, Greil R, Cameron D, Carey LA, Curigliano G, Gelmon K, Hortobagyi G, Krop I, Loibl S, Pegram M, Slamon D, Palanca-Wessels MC, Walker L, Feng W, Winer EP (2019) Tucatinib, trastuzumab, and capecitabine for HER2-positive metastatic breast cancer. N Engl J Med 382:597–609. https://doi.org/10.1056/NEJMoa1914609
Modi S, Saura C, Yamashita T, Park YH, Kim S-B, Tamura K, Andre F, Iwata H, Ito Y, Tsurutani J, Sohn J, Denduluri N, Perrin C, Aogi K, Tokunaga E, Im S-A, Lee KS, Hurvitz SA, Cortes J, Lee C, Chen S, Zhang L, Shahidi J, Yver A, Krop I (2019) Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N Engl J Med 382:610–621. https://doi.org/10.1056/NEJMoa1914510
Cortés J, Kim S-B, Chung W-P, Im S-A, Park YH, Hegg R, Kim MH, Tseng L-M, Petry V, Chung C-F, Iwata H, Hamilton E, Curigliano G, Xu B, Huang C-S, Kim JH, Chiu JWY, Pedrini JL, Lee C, Liu Y, Cathcart J, Bako E, Verma S, Hurvitz SA (2022) Trastuzumab deruxtecan versus trastuzumab emtansine for breast cancer. N Engl J Med 386:1143–1154. https://doi.org/10.1056/NEJMoa2115022
Spring LM, Clark SL, Li T, Goel S, Tayob N, Viscosi E, Abraham E, Juric D, Isakoff SJ, Mayer E, Moy B, Supko JG, Tolaney SM, Bardia A (2021) Phase 1b clinical trial of ado-trastuzumab emtansine and ribociclib for HER2-positive metastatic breast cancer. NPJ Breast Cancer 7:103. https://doi.org/10.1038/s41523-021-00311-y
Connors D, Allen J, Alvarez JD, Boyle J, Cristofanilli M, Hiller C, Keating S, Kelloff G, Leiman L, McCormack R, Merino D, Morgan E, Pantel K, Rolfo C, Serrano MJ, Pia Sanzone A, Schlange T, Sigman C, Stewart M (2020) International liquid biopsy standardization alliance white paper. Crit Rev Oncol Hematol 156:103112. https://doi.org/10.1016/j.critrevonc.2020.103112
Acknowledgments
We thank the patients, their families, the nurses, and the investigators who participated in this study. A list of collaborators is provided in the primary analysis [8]. Support for third-party writing assistance for this manuscript, furnished by Eleanor Porteous, MSc of Health Interactions, was provided by F. Hoffmann-La Roche Ltd.
Funding
This work was supported by F. Hoffmann-La Roche Ltd, Basel, Switzerland (no grant number applicable). F. Hoffmann-La Roche Ltd was involved in the study design, data interpretation, and the decision to submit for publication in conjunction with the authors.
Author information
Authors and Affiliations
Contributions
All authors contributed to drafting the work or revising it critically for important intellectual content and read and approved the final the version of the manuscript to be published. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. BX designed the work and provided substantial contributions to the conception. All authors all contributed to the acquisition, analysis, or interpretation of data for the work.
Corresponding author
Ethics declarations
Conflict of interest
BX reports institutional research funding (investigator fees for this study to his institution) from F. Hoffmann-La Roche Ltd; and consultant/advisory roles for Novartis, Roche, and AstraZeneca. WL, QZ, QL, XW, HL, TS, YY, HZhe, and JF all report institutional research funding (investigator fees for this study) from F. Hoffmann-La Roche Ltd. HZhu and HM are employees of F. Hoffmann-La Roche Ltd. AS is an employee of Roche Products Limited. AK is an employee of Roche Products Limited and holds stock in F. Hoffmann-La Roche Ltd. All authors received research funding in the form of third-party writing assistance for this manuscript, provided by F. Hoffmann-La Roche Ltd.
Ethics approval
This study was conducted in full conformance with the International Council for Harmonisation (ICH) E6 guideline for Good Clinical Practice and the principles of the Declaration of Helsinki, or with the laws and regulations of the country in which the research was conducted, whichever afforded the greater protection to the individual. The protocol, the informed consent forms, and any accompanying material provided to patients were submitted to the independent ethics committee (IEC) by the Principal Investigator and reviewed and approved before the study was initiated. A full list of the IECs at participating institutions is provided with the primary analysis [8].
Consent to participate
Written informed consent for participation in the study had to be obtained before performing any study-specific screening tests or evaluations. Informed consent forms for enrolled patients and for patients who were not subsequently enrolled were maintained at the study site.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, B., Li, W., Zhang, Q. et al. Pertuzumab, trastuzumab, and docetaxel for Chinese patients with previously untreated HER2-positive locally recurrent or metastatic breast cancer (PUFFIN): final analysis of a phase III, randomized, double-blind, placebo-controlled study. Breast Cancer Res Treat 197, 503–513 (2023). https://doi.org/10.1007/s10549-022-06775-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-022-06775-1