
Abstract
Purpose
CXCR1, one of the receptors for CXCL8, has been identified as a druggable target on breast cancer cancer stem cells (CSC). Reparixin (R), an investigational oral inhibitor of CXCR1, was safely administered to metastatic breast cancer patients in combination with paclitaxel (P) and appeared to reduce CSC in a window-of-opportunity trial in operable breast cancer. The fRida trial (NCT02370238) evaluated the addition of R to weekly as first-line therapy for metastatic (m) TNBC.
Subjects and Methods
Subjects with untreated mTNBC were randomized 1:1 to R or placebo days 1–21 in combination with weekly P 80 mg/m2 on days 1, 8, 15 of 28-day cycles. The primary endpoint was PFS by central review.
Results
123 subjects were randomized (62 to R + P and 61 to placebo + P). PFS was not different between the 2 groups (median 5.5 and 5.6 months for R + P and placebo + P, respectively; HR 1.13, p = 0.5996). ALDH+ and CD24−/CD44+ CSC centrally evaluated by IHC were found in 16 and 34 of the 54 subjects who provided a metastatic tissue biopsy at study entry. Serious adverse events (21.3 and 20% of subjects) and grade ≥ 3 adverse reactions (ADR) (9.1 and 6.3% of all ADRs) occurred at similar frequency in both groups.
Conclusion
fRida is the first randomized, double-blind clinical trial of a CSC-targeting agent in combination with chemotherapy in breast cancer. The primary endpoint of prolonged PFS was not met.
Clinical Trial Registration/Date of Registration
NCT01861054/February 24, 2015.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cancer stem cells (CSC) have the ability to self-renew and generate the full range of cells that make up a bulk tumor. Experimental models and retrospective clinical observations point to CSC as responsible for tumor initiation, treatment resistance, disease recurrence, and metastasis [1]. An ideal CSC-targeting agent should be a non-toxic molecule that can be safely administered also in combination with chemotherapy to improve disease control over non CSC, bulk tumor cells.
Breast cancer was the first solid tumor where CSC were identified [2]. Two markers are commonly used to identify such cells in clinical specimens, i.e., aldehyde dehydrogenase (ALDH) and CD24/CD44. CXCR1, one of the receptors for CXCL8, has been identified on breast cancer ALDH + CSC [3]. Binding of CXCL8 to CXCR1 on the CSC surface protects CSC from pro-apoptotic signals released in the tumor microenvironment following taxane administration [4]. A CXCL8–CXCR1 axis in breast cancer CSC heightened by taxane administration has been reported by several independent laboratories [5,6,7].
Reparixin, an investigational allosteric inhibitor of CXCR1, reduced CSC in breast cancer (BC) xenografts both as single agent and in combination with taxane chemotherapy [4]. In a phase Ib trial in women with metastatic HER2-negative BC, the combination of escalating doses (400 to 1200 mg three times per day) of reparixin with weekly paclitaxel resulted in a low incidence and severity of adverse reactions, a sizeable response rate and time-to-progression, with some long-term responders [8]. Furthermore, in a window-of-opportunity clinical trial, a 21-day course of reparixin before curative surgery appeared to reduce CSC by flow cytometric analysis in several subjects [9].
Patients with metastatic triple-negative breast cancer (TNBC) receiving single-agent chemotherapy have poor clinical outcomes with median overall survival of around 18 mos. or less [10, 11]. Considering the CSC enrichment/signature of TNBC [12, 13], a phase 2, randomized, double-blind study [fRida (NCT02370238)] evaluating the safety and efficacy of reparixin plus paclitaxel vs. placebo plus paclitaxel in untreated metastatic (m) TNBC was initiated.
Materials and methods
Oversight
The trial was conducted according to GCP and the principles of the Declaration of Helsinki. All the subjects provided written informed consent. Protocol approval was obtained from Independent Review Boards or Ethics Committee at each site. An independent DMC reviewed unblinded safety data every 6 months. All the authors verify that the trial was conducted according to the protocol and vouch for the accuracy and completeness of the data. All the drafts of the manuscript were prepared by the authors. The study agent, reparixin, and placebo were provided by the study sponsor, Dompe, who also worked collaboratively with the study investigators for study design, data collection, analysis, and interpretation.
Subjects
Eligible subjects were female 18 years of age or older and had stage IV, histologically documented TNBC according to ASCO-CAP guidelines [14, 15], as evaluated by local institutions. They were eligible to receive paclitaxel monotherapy and had received no prior systemic therapy for advanced disease. de novo stage IV patients were allowed to be randomized only following protocol amendment nr. 2 in the second half of 2016. Radiation therapy and (neo)adjuvant chemotherapy (including taxanes) were allowed if treatment was completed ≥ 12 and ≥ 6 months for taxane and non taxane regimens, respectively. Measurable disease according to RECIST 1.1 an ECOG performance status ≤ 1 and an adequate hematologic and organ function were also required. Main exclusion criteria were brain metastases (a baseline CT or MRI of the brain was mandatory) and G > 1 peripheral neuropathy. The full eligibility criteria are provided in the protocol, available with the full text of this article at https://www.springer.com/journal/10549.
Trial design and procedures
Subjects were randomly assigned in a 1:1 ratio with an interactive voice–web response system to receive paclitaxel in combination with either reparixin or placebo. Stratification factor was the history or not of (neo)adjuvant chemotherapy. Subjects received paclitaxel 80 mg/m2 on days 1, 8, and 15 and reparixin/placebo oral tablets 1200 mg t.i.d. from day 1 to 21 of 28-day cycles. Subjects received study drugs until disease progression according to RECIST 1.1, withdrawal of consent or unacceptable toxicity, whichever occurred first.
The discontinuation of either reparixin/placebo or paclitaxel and continuation of treatment with either one as single agent was not allowed. Prespecified modifications of the paclitaxel dose were permitted in order to manage the side effects of chemotherapy.
Tumor imaging occurred at baseline and every 8 weeks. Patient management was based upon local radiologist evaluation. Follow-up for survival occurred every 3 months after discontinuation of study treatment.
The primary endpoint was progression-free survival (PFS) as determined by blinded independent radiology review (IRR). Secondary endpoints were overall survival (OS), objective response rate (ORR), and safety of the combination. The primary and secondary efficacy analyses were evaluated primarily for the intention-to-treat (ITT) population, which included all the subjects who had undergone randomization whether or not they received study drugs. The safety population consisted of all subjects who received at least one dose of study treatment.
Exploratory endpoints were time to new metastasis (TTM) and proportion of subjects progressing with new metastatic lesions (both at pre-existing and new sites), the measurement of CSC in metastatic tissue samples, and the incidence and severity of paclitaxel-induced peripheral neuropathy. To this purpose, subjects progressing with no new systemic metastatic lesions were requested to undergo brain imaging to rule out subclinical CNS lesions.
The safety population included all subjects who took at least one dose of the study treatment and was evaluated according to CTCAE version 4.03. Additional details regarding the study design, including key protocol amendments, are available with the protocol.
CSC
A formalin-fixed paraffin-embedded sample of metastatic tissue was obtained, whenever feasible, for prospective centralized measurement/evaluation of CSC. ALDH1 was immunostained using the mouse monoclonal antibody 44/ALDH (Becton, Dickinson & Co, Franklin Lakes, NJ, USA) at a 1:200 dilution, for 30 min at room temperature, followed by incubation with the Bond Polymer Refine Detection Kit (Leica Biosystems, Buccinasco, Italy) on a Bond platform (Leica).
Dual staining for CD44 and CD24 was performed on 3 um thin sections pre-treated with a 10 mM citrate pH 6.0 solution at 98 °C for 30 min followed by a cooling time of 20 min at room temperature.
Sections were then incubated with the primary antibody cocktail (mouse IgG1 anti-Human CD44v6, clone VFF-18, at 1:250 dilution and mouse IgMk anti-Human CD24, clone SN3b, 1:25, Invitrogen, Carlsbad, CA, USA) in a humidity chamber at 4 °C overnight and 1 h at room temperature. Sequential detection steps were performed with Biotin-conjugated Rat anti-Mouse IgG1 (clone M1-14D12, 1:50, eBioscienceTM-Invitrogen) and Streptavidin Protein DyLight 488 (1:200, ThermoFisher Scientific, Waltham, MA, USA) for CD44, and with Alexa Fluor 546-conjugated Goat anti-Mouse IgM (1:250, ThermoFisher Scientific) for 30 min at room temperature. Finally, sections were mounted with Vectashield Antifade Mounting Medium with DAPI (ready to use, Vector Lab, Burlingame, CA, USA) and a coverslip sealed with nail polish. The slides were stored at − 20 °C until screening with a Zeiss AxioImager M2 Microscope, equipped with fluorescence filters DAPI (350/50-460/50), Green (490/40-537/29), and Orange (546/12-590/33), using the Metafer Slide Scanning System software (MetaSystems GmbH, Altlussheim, Germany).
Statistical analysis
The trial was initially designed to randomly assign 156 subjects for the evaluation of a primary endpoint of PFS. 142 PFS events were required to provide 80% power to detect a difference in median PFS from 5 to 8 months corresponding to a hazard ratio of 0.625, when using a logrank statistic having (one-sided) 0.025 false positive error rate.
Due to extreme enrollment difficulties during the first 6 months of 2018, accrual of subjects to the study was terminated early (July 30, 2018) and the final sample size is 123 randomized subjects. No formal recalculation of sample size/required PFS events could be made under these circumstances and the above assumptions remained.
PFS and OS were compared between treatment arms using stratified log-rank test. Hazard ratios for disease progression and death were estimated with the use of a Cox proportional model stratified by randomized sub-group. Kaplan–Meier curves were produced to estimate median PFS, median OS, and median TTM outcomes. Similar methods were applied to the duration of response.
The comparisons of the response rate and the proportion of subjects progressing with new metastatic lesions were made with the use of the Cochran–Mantel–Haenszel (CMH) test, stratified by the patient population (newly diagnosed vs. relapsed).
The CSC markers (CD24−CD44+ and ALDH+ assessed by IHC) within the epithelial cell population were summarized by descriptive statistics (n, mean, standard deviation, median, minimum, and maximum).
The statistical analyses were carried out with SAS version 9.4 (or later versions) from the SAS Institute.
Results
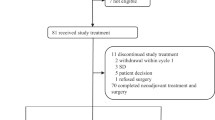
From July 2015 to May 2018, 194 subjects were assessed for eligibility and 123 (ITT population) were randomized to either reparixin (n = 62) or placebo (n = 61) at 47 clinical sites in Europe and USA. A total of 87 subjects were randomized in Europe and 36 in USA. One patient in each group did not receive study treatment and was excluded from the safety population. The disposition of subjects is depicted in Fig. 1. Overall, the characteristics of the subjects at baseline were well balanced between the two treatment groups (Table 1).

Patient disposition: Randomization, trial populations, and follow-up are shown, and so are the numbers of subjects who were receiving study treatment on the data cutoff date (February 20, 2019)
At the clinical cutoff date (February 20, 2019), for subjects in the reparixin–paclitaxel group, the median duration of reparixin and paclitaxel treatment was 16.6 and 16.1 weeks, respectively. For subjects in the placebo–paclitaxel group, the median duration of placebo and paclitaxel treatment was 15.5 and 14.7 weeks, respectively. The mean (± SD) cumulative dose of paclitaxel was 1211.7 ± 944.58 mg/m2 in the reparixin–paclitaxel group and 1344.2 ± 1090.35 mg/m2 in the placebo–paclitaxel group.
Palliative radiation therapy was administered in 1 patient in the reparixin group and in 2 subjects in the placebo group.
Efficacy
At the time of data cutoff, the median follow-up was 14.3 months in the ITT population (14.3 months in the reparixin–paclitaxel arm and 12.9 months in the placebo–paclitaxel arm) and 92 PFS events had been reported by investigators. Progression-free survival by IRR after 74 events (40 in reparixin and 34 in placebo group) was not significantly different between the two treatment groups (median, 5.5 vs. 5.6 months in reparixin–placebo and placebo–paclitaxel arm, respectively; stratified hazard ratio 1.13; 95% confidence interval [CI] 0.71–1.80; p = 0.5996) (Fig. 2). Also, PFS assessed by investigators did not show differences between treatment groups (median 5.5 and 5.8 months in reparixin–paclitaxel and placebo–paclitaxel group, respectively; HR 1.12; 95% CI 0.74–1.70; p = 0.578). Subgroup analyses (i.e., visceral disease, prior taxane, age < 40 years) did not differ from the main analysis (data not shown).

Progression-free survival in the ITT population: Kaplan–Meier estimates of PFS, according to the Response Evaluation Criteria in Solid Tumors, version 1.1, as assessed by the independent radiology review, among subjects in the ITT population. Stratified hazard ratios for disease progression or death are reported along with p values. Tick marks indicate censored data
Subsequent anticancer therapy was administered to 45 patients (73.8%) in the reparixin–paclitaxel group and to 40 (66.7%) in the placebo–paclitaxel arm and was generally balanced between the two groups (Table S1). At the time of data cutoff, 38 of 62 subjects (61.3%) in the reparixin–paclitaxel arm and 34 of 61 (55.7%) in the placebo–paclitaxel arm had died. The median overall survival was 16.0 and 17.4 months in the reparixin–paclitaxel and the placebo–paclitaxel arm, respectively (stratified hazard ratio for death 1.09; 95% CI 0.68–1.75; p = 0.7059] (Fig. 3).

Overall survival in the ITT population: Kaplan–Meier estimates of OS among subjects in the ITT population. Stratified hazard ratios for death are reported along with p values. Tick marks indicate censored data. 6 subjects (4 in reparixin + paclitaxel and 2 in placebo + paclitaxel) did not re-sign ICF to protocol amendment 2 or later and were censored at 1 year after the off treatment visit
In the ITT population, the rate of confirmed objective responses, as assessed by blinded IRR on the ITT population, was 25.8% (95% CI 0.17–0.42) and 22.9% (95% CI 0.15–0.40) in the reparixin–paclitaxel and in the placebo–paclitaxel group, respectively. Only 1 subject, in the reparixin + paclitaxel group, experienced a complete response. The median duration of response (DOR) in the ITT as assessed by IRR was 9.8 (95% CI 3.8–16.8) months in the reparixin–paclitaxel arm and 5.7 (95% CI 3.7–14.8) months in the placebo–paclitaxel arm (p = 0.767).
Time to new metastasis was not different between treatment arms (data not shown). In the ITT population, the number of subjects who at progression displayed new metastatic lesions at existing or new sites was 17/40 (42.5%) in the reparixin–paclitaxel arm and 23/34 (67.6%) in the placebo–paclitaxel arm (p = 0.0305, Chi-square test). A similar proportion of subjects developed brain metastases (data not shown).
Safety
Among subjects in the safety population, treatment-emergent adverse events (TEAE) occurred in 60 (98.4%) and 57 (95%) subjects in the reparixin and placebo arm, respectively. The most common TEAE in the reparixin + paclitaxel group were nausea (37.7% of subjects), alopecia (34.4%), anemia (29.5%), diarrhea (26.2%), and asthenia (26.2%), while in the placebo + paclitaxel group were fatigue (43.3%), nausea (36.7%), alopecia (35%), diarrhea (25.0%), and asthenia (21.7%). Serious TEAEs occurred in 13 (21.3%) and 12 (20%) subjects in the reparixin and placebo group, respectively.
Adverse events that led to withdrawal of study treatment occurred in 7 (11.5%) and 13 (21.7%) subjects in reparixin and placebo arm, respectively.
Fatal TEAE occurred in 3 (4.9%) and 4 (6.7%) subjects in reparixin and placebo group, respectively. One death in the reparixin arm (from peritonitis and intestinal perforation) was considered by the investigators to be possibly related to the study treatment.
The frequency and severity of peripheral neuropathy was similar between the 2 groups (29.5% and 30.0% in reparixin and placebo arm, respectively). However, events grade 2 or greater were more common in placebo group (6.5% with reparixin vs. 16.6%, p = 0.0822 chi-squared test). Two subjects in reparixin + paclitaxel group and 3 subjects in placebo + paclitaxel group discontinued study treatment due to TEAE of peripheral neuropathy.
Interestingly, a statistically significant difference was observed for the TEAE fatigue, which was recorded in 11 (18%) and 26 (43.3%) subjects in the reparixin and placebo group, respectively (p = 0.003, Chi-square test). Also, G3 fatigue was recorded only in 2 subjects in the placebo group. 0 and 3 subjects in the reparixin and placebo group, respectively, discontinued study treatment due to TEAEs including asthenia.
A higher number of ADR (as assessed by the investigator) were recorded in subjects receiving R + P (263) compared to subjects receiving placebo + P (200). The percentage of grade ≥ 3 ADR was similar between the two groups (9.1 and 6.0% recorded in 14 and 6 subjects in reparixin and placebo arm, respectively). Two and 0 subjects experienced G > 3 ADRs in reparixin and placebo arm, respectively. The most common adverse reactions are presented in Table 2.
Cancer stem cells
CSC analysis was performed on metastatic tumor tissue only. An evaluable biopsy of metastatic tissue was obtained at baseline from 54 randomized patients (Table 3). ALDH-1+ and CD24−/CD44+ cells were detected within the epithelial tumor cell populations in 16 and 34 patients, respectively. Only in 9 biopsies, CSC of both phenotype could be detected. Since the presence of CSC was not a stratification factor, subjects with positive biopsies for CSC markers were unevenly distributed in the two treatment groups, i.e., 12 vs. 4 and 22 vs. 12 for ALDH+ and CD24−/CD44+ in reparixin and placebo group, respectively. When PFS was analyzed by the presence or absence of CSC markers (i.e., ALDH+ or CD24−/CD44+ cells), no difference between groups was observed (data not shown). In a post hoc analysis on subjects with ALDH+ CSC, PFS and OS appeared to be longer in reparixin than in placebo group (Figure S1) and so did OS in subjects with CD24−/CD44+ CSC (Figure S2); however, the overlapping 95% CI and the small number of patients in each group do not allow to draw any conclusion.
Discussion
Herein, we report on the first randomized, double-blind, placebo-controlled clinical trial of a CSC-targeting agent in breast cancer. Administered as first-line treatment, the combination of reparixin and paclitaxel did not improve PFS of mTNBC patients over paclitaxel alone. The median PFS in both groups was within the range reported in other contemporary trials in first-line mTNBC treated with single-agent taxanes [16, 17], with the exception of the PAKT trial [18]. Although immature, OS data also did not show a difference between treatment groups.
From a safety standpoint, the combination appeared to be well tolerated and although patients receiving R + P experienced a higher number of adverse reactions, the proportion of severe ADR was similar in the two groups. Following up on preclinical evidence [19] and clinical suggestion [8] of a protective effect of reparixin on paclitaxel-induced peripheral neuropathy, a trend toward a preventive effect of reparixin on peripheral neuropathy grade 2 or greater was observed. However, the trial was not designed to specifically address this question and it is underpowered for the limited number of events recorded. Interestingly, analysis of TEAE revealed that administration of reparixin may lead to a reduction in incidence and severity of cancer-related fatigue. A possible role for IL-8 in CRF has been reported in several studies [20, 21].
Cancer stem cells were the target of reparixin activity, through inhibition of CXCR1 [4]. At study entry, only 54/123 patients provided an evaluable biopsy of metastatic tissue, thus limiting the possibility to fully explore the role of CSC as a therapeutic target in this setting.
Furthermore, only a proportion of subjects’ tumors stained positive for either CSC population and positive biopsies were unevenly distributed in the treatment groups, limiting the power of the study to determine the predictive value of these markers.
Considering the challenges posed by either obtaining serial biopsies of metastatic tissue during treatment [22] or enumerating CSC in peripheral blood [8], anti-CSC activity was also evaluated by means of time to new metastasis and proportion of subjects progressing with new metastatic lesions [23, 24]. The rationale behind this exploratory analysis is that disease progression by enlargement of pre-existing lesions is mainly reflective of therapeutic effects on bulk tumor cell populations, while detection of new lesions may reflect treatment effects on CSC’s. In ITT population, no difference was observed between reparixin and placebo in terms of time to new metastasis. Although a numerically lower proportion of subjects progressed with new lesions in the reparixin–paclitaxel as compared to the placebo–paclitaxel group, this did not reach statistical significance.
The negative results reported for several clinical trials of CSC-targeting agents have called into question on the clinical utility of targeting CSC [25, 26]. Indeed, in this trial, the traditional clinical endpoint of PFS failed to demonstrate a benefit of adding reparixin to paclitaxel in advanced TNBC patients. However, this does not mean that CSC-targeting agents may not have a future role in treatment, as there are possible explanations for this negative result. First, two groups may have been unbalanced for presence of CSC in the metastatic tumor, with reparixin arm having more CSC which convey a worse prognosis [27, 28]. Second, targeting a single CSC survival pathway may not be sufficient. Lastly, only a proportion of ALDH + CSC express CXCR1 at any time point [4] and the clinical schedule of reparixin (21 days followed by 7 days off each 28-day cycle) different than the preclinical administration for 28 consecutive days [4] may have allowed CSC survival. Future CSC studies would benefit of readily available tumor tissue (e.g., breast cancer patients undergoing neoadjuvant treatment) to perform correlative studies with adequate number of sufficient samples from all treatment groups.
Data availability
All data presented can be found in the Clinical Study Report available at Dompé farmaceutici s.p.a., Milano, Italy.
References
Clarke MF (2019) Clinical and therapeutic implications of cancer stem cells. N Engl J Med 380(23):2237–2245. https://doi.org/10.1056/NEJMra1804280
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF (2003) Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 100(7):3983–3988. https://doi.org/10.1073/pnas.0530291100
Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, Hur MH, Diebel ME, Monville F, Dutcher J, Brown M, Viens P, Xerri L, Bertucci F, Stassi G, Dontu G, Birnbaum D, Wicha MS (2009) Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Can Res 69(4):1302–1313. https://doi.org/10.1158/0008-5472.can-08-2741
Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, Wicinski J, Cabaud O, Charafe-Jauffret E, Birnbaum D, Guan JL, Dontu G, Wicha MS (2010) CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Investig 120(2):485–497. https://doi.org/10.1172/jci39397
Bhola NE, Balko JM, Dugger TC, Kuba MG, Sanchez V, Sanders M, Stanford J, Cook RS, Arteaga CL (2013) TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Investig 123(3):1348–1358. https://doi.org/10.1172/jci65416
Samanta D, Gilkes DM, Chaturvedi P, Xiang L, Semenza GL (2014) Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci USA 111(50):E5429-5438. https://doi.org/10.1073/pnas.1421438111
Singh JK, Farnie G, Bundred NJ, Simoes BM, Shergill A, Landberg G, Howell SJ, Clarke RB (2013) Targeting CXCR1/2 significantly reduces breast cancer stem cell activity and increases the efficacy of inhibiting HER2 via HER2-dependent and -independent mechanisms. Clin Cancer Res Off J Am Assoc Cancer Res 19(3):643–656. https://doi.org/10.1158/1078-0432.ccr-12-1063
Schott AF, Goldstein LJ, Cristofanilli M, Ruffini PA, McCanna S, Reuben JM, Perez RP, Kato G, Wicha M (2017) Phase Ib pilot study to evaluate reparixin in combination with weekly paclitaxel in patients with HER-2-negative metastatic breast cancer. Clin Cancer Res Off J Am Assoc Cancer Res 23(18):5358–5365. https://doi.org/10.1158/1078-0432.ccr-16-2748
Goldstein LJ, Perez RP, Yardley D, Han LK, Reuben JM, Gao H, McCanna S, Butler B, Ruffini PA, Liu Y, Rosato RR, Chang JC (2020) A window-of-opportunity trial of the CXCR1/2 inhibitor reparixin in operable HER-2-negative breast cancer. Breast Cancer Res 22(1):4. https://doi.org/10.1186/s13058-019-1243-8
Yardley DA, Coleman R, Conte P, Cortes J, Brufsky A, Shtivelband M, Young R, Bengala C, Ali H, Eakel J, Schneeweiss A, de la Cruz-Merino L, Wilks S, O’Shaughnessy J, Glück S, Li H, Miller J, Barton D, Harbeck N (2018) nab-Paclitaxel plus carboplatin or gemcitabine versus gemcitabine plus carboplatin as first-line treatment of patients with triple-negative metastatic breast cancer: results from the tnAcity trial. Annals Oncol Off J Eur Soc Med Oncol 29(8):1763–1770. https://doi.org/10.1093/annonc/mdy201
Gobbini E, Ezzalfani M, Dieras V, Bachelot T, Brain E, Debled M, Jacot W, Mouret-Reynier MA, Goncalves A, Dalenc F, Patsouris A, Ferrero JM, Levy C, Lorgis V, Vanlemmens L, Lefeuvre-Plesse C, Mathoulin-Pelissier S, Petit T, Uwer L, Jouannaud C, Leheurteur M, Lacroix-Triki M, Cleaud AL, Robain M, Courtinard C, Cailliot C, Perol D, Delaloge S (2018) Time trends of overall survival among metastatic breast cancer patients in the real-life ESME cohort. Eur J Cancer 96:17–24. https://doi.org/10.1016/j.ejca.2018.03.015
Idowu MO, Kmieciak M, Dumur C, Burton RS, Grimes MM, Powers CN, Manjili MH (2012) CD44(+)/CD24(-/low) cancer stem/progenitor cells are more abundant in triple-negative invasive breast carcinoma phenotype and are associated with poor outcome. Hum Pathol 43(3):364–373. https://doi.org/10.1016/j.humpath.2011.05.005
Li H, Ma F, Wang H, Lin C, Fan Y, Zhang X, Qian H, Xu B (2013) Stem cell marker aldehyde dehydrogenase 1 (ALDH1)-expressing cells are enriched in triple-negative breast cancer. Int J Biol Markers 28(4):e357-364. https://doi.org/10.5301/jbm.5000048
Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, Fitzgibbons PL, Francis G, Goldstein NS, Hayes M, Hicks DG, Lester S, Love R, Mangu PB, McShane L, Miller K, Osborne CK, Paik S, Perlmutter J, Rhodes A, Sasano H, Schwartz JN, Sweep FC, Taube S, Torlakovic EE, Valenstein P, Viale G, Visscher D, Wheeler T, Williams RB, Wittliff JL, Wolff AC (2010) American society of clinical oncology/college of american pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol Off J Am Soc Clin Oncol 28(16):2784–2795. https://doi.org/10.1200/jco.2009.25.6529
Wolff AC, Lazar AA, Bondarenko I, Garin AM, Brincat S, Chow L, Sun Y, Neskovic-Konstantinovic Z, Guimaraes RC, Fumoleau P, Chan A, Hachemi S, Strahs A, Cincotta M, Berkenblit A, Krygowski M, Kang LL, Moore L, Hayes DF (2013) Randomized phase III placebo-controlled trial of letrozole plus oral temsirolimus as first-line endocrine therapy in postmenopausal women with locally advanced or metastatic breast cancer. J Clin Oncol Off J Am Soc Clin Oncol 31(2):195–202. https://doi.org/10.1200/jco.2011.38.3331
Kim SB, Dent R, Im SA, Espié M, Blau S, Tan AR, Isakoff SJ, Oliveira M, Saura C, Wongchenko MJ, Kapp AV, Chan WY, Singel SM, Maslyar DJ, Baselga J (2017) Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol 18(10):1360–1372. https://doi.org/10.1016/s1470-2045(17)30450-3
Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Diéras V, Hegg R, Im SA, Shaw Wright G, Henschel V, Molinero L, Chui SY, Funke R, Husain A, Winer EP, Loi S, Emens LA (2018) Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med 379(22):2108–2121. https://doi.org/10.1056/NEJMoa1809615
Schmid P, Abraham J, Chan S, Wheatley D, Brunt AM, Nemsadze G, Baird RD, Park YH, Hall PS, Perren T, Stein RC, Mangel L, Ferrero JM, Phillips M, Conibear J, Cortes J, Foxley A, de Bruin EC, McEwen R, Stetson D, Dougherty B, Sarker SJ, Prendergast A, McLaughlin-Callan M, Burgess M, Lawrence C, Cartwright H, Mousa K, Turner NC (2020) Capivasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer: the PAKT trial. J Clin Oncol Off J Am Soc Clin Oncol 38(5):423–433. https://doi.org/10.1200/jco.19.00368
Brandolini L, Benedetti E, Ruffini PA, Russo R, Cristiano L, Antonosante A, d’Angelo M, Castelli V, Giordano A, Allegretti M, Cimini A (2017) CXCR1/2 pathways in paclitaxel-induced neuropathic pain. Oncotarget 8(14):23188–23201. https://doi.org/10.18632/oncotarget.15533
Li L, Chan SL, Mo F, Hui EP, Koh J, Chan AKC, Tang NLS, Lee KF, Lai PBS, Yu SCH, Yeo W (2019) Correlations of health-related quality of life with serum inflammatory indicators IL-8 and mIBI in patients with hepatocellular carcinoma. Cancer Manage Res 11:2719–2727. https://doi.org/10.2147/cmar.s178482
Reyes-Gibby CC, Wang J, Spitz M, Wu X, Yennurajalingam S, Shete S (2013) Genetic variations in interleukin-8 and interleukin-10 are associated with pain, depressed mood, and fatigue in lung cancer patients. J Pain Symptom Manage 46(2):161–172. https://doi.org/10.1016/j.jpainsymman.2012.07.019
Schott AF, Landis MD, Dontu G, Griffith KA, Layman RM, Krop I, Paskett LA, Wong H, Dobrolecki LE, Lewis MT, Froehlich AM, Paranilam J, Hayes DF, Wicha MS, Chang JC (2013) Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin Cancer Res 19(6):1512–1524. https://doi.org/10.1158/1078-0432.ccr-11-3326
Oxnard GR, Morris MJ, Hodi FS, Baker LH, Kris MG, Venook AP, Schwartz LH (2012) When progressive disease does not mean treatment failure: reconsidering the criteria for progression. J Natl Cancer Inst 104(20):1534–1541. https://doi.org/10.1093/jnci/djs353
Twelves C, Cortes J, Kaufman PA, Yelle L, Awada A, Binder TA, Olivo M, Song J, O’Shaughnessy JA, Jove M, Perez EA (2015) New metastases are associated with a poorer prognosis than growth of pre-existing metastases in patients with metastatic breast cancer treated with chemotherapy. Breast Cancer Res 17(1):150. https://doi.org/10.1186/s13058-015-0657-1
Ramos EK, Hoffmann AD, Gerson SL, Liu H (2017) New opportunities and challenges to defeat cancer stem cells. Trends Cancer 3(11):780–796. https://doi.org/10.1016/j.trecan.2017.08.007
Garber K (2018) Cancer stem cell pipeline flounders. Nat Rev Drug Discov 17(11):771–773. https://doi.org/10.1038/nrd.2018.157
Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G (2007) ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1(5):555–567. https://doi.org/10.1016/j.stem.2007.08.014
Charafe-Jauffret E, Ginestier C, Iovino F, Tarpin C, Diebel M, Esterni B, Houvenaeghel G, Extra JM, Bertucci F, Jacquemier J, Xerri L, Dontu G, Stassi G, Xiao Y, Barsky SH, Birnbaum D, Viens P, Wicha MS (2010) Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin Cancer Res Off J Am Assoc Cancer Res 16(1):45–55. https://doi.org/10.1158/1078-0432.ccr-09-1630
Acknowledgements
We thank the subjects who participated in this trial and the clinical site investigators. We are grateful to the CRO who conducted the study (Pharm Research Associates, Raleigh, NC, US), the Independent Radiology Review team (Bioclinica, Princeton, NJ, US), the DMC members (Kevin J. Carroll, Ph.D., University of Sheffield, UK; Joyce O’Shaughnessy, M.D., Baylor-Sammons Cancer Center, Dallas, TX, US; and Filippo de Braud, M.D., Istituto Nazionale Tumori, Milano, Italy), Guido Fedele for statistical analysis, Marianna Ferrara, MD, PhD and Sonia Amicarella (Dompé farmaceutici s.p.a.) for critically reviewing the manuscript and editorial assistance, respectively. Ricardo H. Alvarez (Southeastern Regional Medical Center, Newnan, GA, US); Anne F. Schott (Rogel Cancer Center, University of Michigan, Ann Arbor, MI, US); Maysa Abu-Khalaf (Thomas Jefferson University, Philadelphia, PA, US); Lori J. Goldstein (Fox Chase Cancer Center, Philadelphia, PA, US); Nuhad Ibrahim, M.D., (Anderson Cancer Center, Houston, TX, US); Jenny C. Chang (The Methodist Hospital Research Institute, Houston, TX, US); Brooke Daniel (Tennesse Oncology, PLLC, Chattanooga, TN); Michael Meshad (Southern Cancer Center, PC, Mobile, AL, US); David Kanamori (Comprehensive Blood and Cancer Center, Bakersfield, CA, US); Amelia Zelnak (Northside Hospital, Inc., Atlanta, GA, US); Mark Graham (Waverly Hematology Oncology, Cary, NC, US); Jason Comer (Overlake Medical Center, Bellevue, WA, US); Manon Huizing (Universitair Ziekenhuis Antwerpen, Edegem, Belgium); Stephanie Henry (CHU UCL NAMUR-Sainte Elisabeth, Namur, Belgium); Francois Duhoux (Cliniques Universitaires Saint LUC UCL, Brussels, Belgium); Vincent Richard (CHU Ambroise Paré, Mons, Belgium); Didier Verhoeven (Algemeen Ziekenhuis Klina, Brasschaat, Belgium); Martin Smakal (Nemocnice Horovice a.s., Horovice, Czech Republic); Jana Prausová (Fakultni nemocnice v Motole, Onkologická klinika 2, LF UK a FN Motol, Prague, Czech Republic); Marta Krasenska (Masarykuv Memorial Cancer Institute, Brno, Czech Republic); Milan Kohoutek (Krajská nemocnice T. Bati, a. s., Zlin, Czech Republic); Martina Zimovjanova (Onkologická klinika VFN a 1. LF UK, Praha, Czech Republic); Eugen Kubala (Fakultni nemocnice, Hrádec Kralové, Czech Republic); Mario Campone (Medicale Centre René Gauducheau, Saint Herblain, France); Jean-Marc Ferrero (Centre Antoine Lacassagne, Nice, France); Anthony Goncalves (Institut Paoli Calmettes, Marseille, France); Laurence Venat-Bouvet (Centre Hospitalier Universitaire (CHU) De Limoges-Hopital Dupuytren, Limoges, France); Christelle Levy (Caen Centre Francois Baclesse, Caen, France); Jacques Medioni (Hôpital Européen Georges Pompidou, Paris, France); Laura Biganzoli (Ospedale Santo Stefano – Azienda USL Toscana Centro, Prato, Italy); Hector Soto Parra (Azienda Ospedaliero-Universitaria Policlinico-Vittorio Emanuele, Catania, Italy); Paolo Pedrazzoli (Fondazione IRCCS Policlinico S. Matteo, Pavia, Italy); Marco Colleoni (Istituto Europeo di Oncologia, Milano, Italy); Mauro Mansutti (Azienda Ospedaliero Universitaria Santa Maria della Misericordia, Udine, Italy); Mauro Moroni (Azienda Socio-Sanitaria Territoriale Santi Paolo e Carlo, Milano, Italy); Dino Amadori (Istituto Scientifico Romagnolo per lo Studia e la Cura dei Tumori, Meldola, Italy); Paolo Morandi (Ospedale dell’Angelo, Venezia, Italy); Saverio Cinieri (Ospedale Di Summa-Perrino, Brindisi, Italy); Piotr Tomczak (Uniwersytetu Medycznego im. Karola Marcinkowskiego o W Poznaniu, Poznan, Poland); Tomasz Sarosiek (Magodent Sp. z o.o, Warszawa, Poland); Marek Wojtukiewicz (Bialostockie Centrum Onkologii im. Marii Sklodowskiej-Curie, Bialystok, Poland); Andrzej Mruk (Mrukmed. Lekarz Beata Madej-Mruk i. Partner Spólka Partnerska Oddzial nr 1 w Rzeszowie, Rzeszow, Poland); Bożena Kukielka-Budny (Centrum Onkologii Ziemi Lubelskiej św. Jana z Dukli, Lublin, Poland); Silvia Antolin Novoa (Complejo Hospitalario Universitario, La Coruňa, Spain); Estela Vega Alonso (Hospital Madrid Centro Integral Oncológico Clara Campal, Madrid, Spain); Miguel Martin Jimenez (Hospital General Universitario Gregorio Maraňon, Madrid, Spain); Isaura Fernandez Perez (Hospital Alvaro Cunqueiro, Vigo, Spain).
Funding
This clinical trial was sponsored by Dompé farmaceutici s.p.a., Milano, Italy.
Author information
Authors and Affiliations
Consortia
Contributions
LJG, AFS, MSW, JCC, and PAR were responsible for the conception and design. GV was responsible for CSC analysis in tumor samples. LJG, MM, CL, JCC, SH, IF-P, JP, ES, MSW, and AFS obtained the data. LJG, MSW, AFS, JCC, PAR, SMC, and BB were responsible for the analysis and interpretation of the data. LJG and MSW drafted the manuscript. All authors critically revised and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
PAR, SMC, and BB are full-time employees of Dompé farmaceutici s.p.a. All other authors declare that they have no competing interests.
Ethical approval
The study was approved by the IRB of each participating site. The study was performed in accordance with the Declaration of Helsinki.
Consent to participate
All subjects signed an IRB approved informed consent form prior to enrollment to the study.
Consent for publication
As part of the consenting process and written within the IRB approved Informed Consent form, participants were informed that the results of this study could be published in an article, but would not include any information that would identify them.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Goldstein, L.J., Mansutti, M., Levy, C. et al. A randomized, placebo-controlled phase 2 study of paclitaxel in combination with reparixin compared to paclitaxel alone as front-line therapy for metastatic triple-negative breast cancer (fRida). Breast Cancer Res Treat 190, 265–275 (2021). https://doi.org/10.1007/s10549-021-06367-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-021-06367-5