
Abstract
Purpose
Despite the therapeutic success of existing HER2-targeted therapies, tumors respond quite differently to them. This study aimed at figuring out genetic mutation profile of Chinese HER2-positive patients and investigating predictive factors of neoadjuvant anti-HER2 responses.
Methods
We employed two cohorts. The first cohort was comprised of 181 HER2-positive patients treated at Guangdong Provincial People’s Hospital from 2012 to 2018. The second cohort included 40 patients from the first cohort who underwent HER2-targeted neoadjuvant chemotherapy. Genetic mutations were characterized using next-generation sequencing. We employed the most commonly used definition of pathological complete response (pCR)-eradication of tumor from both breast and lymph nodes (ypT0/is ypN0).
Results
In Chinese HER2-positive breast cancer patients, TP53 (74.6%), CDK12 (64.6%) and PIK3CA (46.4%) have the highest mutation frequencies. In cohort 2, significant differences were found between pCR and non-pCR groups in terms of the initial Ki67 status, TP53 missense mutations, TP53 LOF mutations, PIK3CA mutations and ROS1 mutations (p = 0.028, 0.019, 0.005, 0.013, 0.049, respectively). Furthermore, TP53 LOF mutations and initial Ki67 status (OR 7.086, 95% CI 1.366–36.749, p = 0.020 and OR 6.007, 95% CI 1.120–32.210, p = 0.036, respectively) were found to be predictive of pCR status.
Conclusion
TP53 LOF mutations and initial Ki67 status in HER2-positive breast cancer are predictive of pCR status after HER2-targeted NACT.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
HER2 positivity accounts for about 15–20% of breast cancers and the development of HER2-targeted therapies has profoundly changed the course of these patients [1]. More and more HER2-targeted drugs, such as trastuzumab, pertuzumab, T-DM1 and neratinib, have become available for treatment of HER2-positive breast cancer. Despite this progress, however, many patients still die of HER2-positive breast cancer, calling for the identification and investigation of genetic profiles of HER2-positive breast cancer and predictors of responses to HER2-targeted therapies.
Neoadjuvant chemotherapy (NACT) is used commonly to downstage locally advanced cancer to allow breast-conserving surgery and to predict responses to systemic therapy based on pathological assessment. Based on the recent progress, NACT has gained momentum as an ideal setting in which to investigate predictive biomarkers of treatment responses. The Collaborative Trials in Neoadjuvant Breast Cancer (CTNeoBC) pooled analysis has confirmed NACT to be a good way to discriminate patients who have different clinical outcomes by their responses to the therapies [2]. Patients who attain complete eradication of tumor after NACT in both breast and lymph nodes have improved survival, which is defined as pathological complete response (pCR) [2, 3]. The NeoSphere and NeoALTTO trials both tried to seek higher pCR rates by different anti-HER2 combinations [4, 5] and new treatments in the past decade have significantly improved the prognosis of HER2-positive breast cancer with a pCR rate as high as 75% [6]. Despite these achievements, however, HER2-positive breast cancer patients still have a high death rate [6]. According to the latest interim analysis of the landmark KATHERINE trial, patients with HER2-positive early breast cancer who had residual invasive disease after NACT have improved survival when they receive adjuvant T-DM1 therapy after surgery [7]. As these great trials have provided effective regimens for patients with different responses, it is important to investigate the response predictors.
Large-scale, next-generation sequencing studies have provided large amounts of genetic information and produced valuable insights into the genomic landscape of primary breast cancers [8,9,10,11,12]. These studies have highlighted that TP53 and PIK3CA were the two most prevalent mutated genes in HER2-positive breast cancers [8] and enriched in residual tissues after HER2-targeted therapies. PIK3CA mutation rate in HER2-positive breast cancers is about 23%, and patients with PIK3CA mutations have a lower pCR rate after HER2-targeted NACT [13].
In this study, we investigated the mutation profiles of HER2-positive breast cancer patients in China and analyzed the mutation differences between primary HER2-positive breast cancers with pCR and non-pCR after HER2-targeted NACT. We hypothesized that there would be mutations predictive of the anti-HER2 therapy responses and could be used as biomarkers for guiding treatment decisions.
Methods
Study cohorts
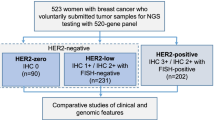
This study was comprised of two cohorts. The first cohort included 181 HER2-positive patients treated at GPPH from 2012 to 2018. The second cohort included 40 patients from the first cohort who underwent HER2-targeted neoadjuvant therapy (NACT). Twenty-three patients received NACT with docetaxel 75 mg/m2, carboplatin (6 mg/min/ml carboplatin AUC area under curve) and trastuzumab (8 mg per kilogram intravenously as a loading dose, followed by 6 mg per kilogram intravenously every 3 weeks), while oral lapatinib was added in other 17 patients. Lapatinib was given daily at a dose of 750 mg (250 mg tablets) for the first week, followed by 1000 mg daily for a year. All 40 patients completed the scheduled 6 NACT cycles.
Detailed information of our study cohorts is listed in Table 1.
Pathological examination of tumor specimens was performed in the Department of Pathology at GPPH. ER, PR, ROS1 and HER2 status were reconfirmed by two experienced pathologists based on IHC and fluorescence in situ hybridization (FISH) [14]. The cutoff for ER-negative and PR-negative IHC status was less than 1% staining in the nuclei. HER2 status was considered negative when an IHC score was 0 or 1 or when HER2 amplification was absent (ratio < 2.2) by FISH analysis. If any disagreements arose during the evaluation of the IHC and FISH results, a third pathologist was consulted.
Next-generation sequencing
NGS library preparation
DNA fragmentation was performed using Covaris M220, followed by end repair, phosphorylation and adaptor ligation. Fragments of size 200–400 bp were selected by bead (Agencourt AMPure XP Kit, Beckman Coulter, California, USA) followed by hybridization with capture probes baits, hybrid selection with magnetic beads and PCR amplification. Subsequently, a high-sensitivity DNA assay was performed to assess the quality and size of the fragments. Indexed samples were sequenced on Nextseq500 sequencer (Illumina, Inc., California, USA) with pair-end reads.
Capture-based targeted DNA sequencing
Genomic profiling was performed using a panel covering 520 cancer-related genes (Burning Rock Biotech Ltd.). Among them, whole exons of 312 genes and critical exons, introns and promoter regions of the remaining 208 genes were captured.
Sequence data analysis
Sequence data were mapped to the human genome (hg19) using BWA aligner 0.7.10. Local alignment optimization, variant calling and annotation were performed using GATK 3.2, MuTect and VarScan. Variants were filtered using the VarScan filter pipeline, with loci with depth less than 100 filtered out. At least 5 supporting reads were needed for INDELs, while 8 supporting reads were needed for SNVs to be called. According to the ExAC, 1000 Genomes, dbSNP and ESP6500SI-V2 database, variants with population frequency over 0.1% were grouped as SNP and excluded from further analysis. Remaining variants were annotated with ANNOVAR and SnpEff v3.6. DNA translocation analysis was performed using both Tophat2 and Factera 1.4.3.
Statistical analyses
Statistical analyses were performed using GraphPad Prism version 7.00 for Mac (GraphPad Software, La Jolla California, USA). Pearson’s Chi-square test and Yate’s continuity-corrected Chi-square test were employed for significance of differences between groups. A two-sided p value less than 0.05 was considered significant unless otherwise stated. To determine which covariates affected pCR, we used pathological and mutational variables by univariate and multivariate regression. We included variables which are < 0.05 for p value and those supposed to affect pCR status.
Results
Clinicopathologic features and genetic mutations of Chinese HER2-positive breast cancer patients
The patients’ clinicopathological parameters are listed in Table 1. In cohort 1, the median age was 48, and 56.9% of patients were pre-menopausal. The population was comprised of stage I, II and III patients, which account for 20.4%, 59.7% and 19.9%, respectively. The cohort 2 was derived from cohort 1, in which the median age was 53, more II, III stage patients were included and there was no difference in hormone receptor expression. No difference of pCR status was found between the two treatment groups. However, the two groups of Ki67 < 40% versus ≥ 40% had different pCR rates of 63.2% and 28.6% (p < 0.028) (Table 1).
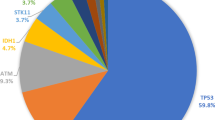
We next analyzed the mutation profiles in Chinese HER2-positive breast cancers (Fig. 1 and Table 2). The most commonly mutated genes in Chinese HER2-positive breast cancer patients were TP53 (74.6%), CDK12 (64.6%) and PIK3CA (46.4%). Most of the TP53 mutations were missense mutations (40.35%) and LOF mutations (21.5%). Interestingly, the ROS1 mutations were only detected in HR + HER2 + patients. Mutation sites of TP53, PIK3CA and ROS1 are listed in Fig. 1. No TP53 mutation hotspots were found in these Chinese HER2 + patients, while p.H1047R was investigated to be hotspot in PIK3CA mutations (Fig. 1 and Table 3). In cohort 2 (Table 2 and Fig. 2), TP53 (90.0%), CDK12 (77.5%) and PIK3CA (55%) were still the most mutated genes. And 5 patients got ROS1 mutations, all of whom belong to HR + HER2 + subgroup. The mutation sites of TP53, PIK3CA and ROS1 in cohort 2 are also listed in Fig. 2.

The mutational landscape of 181 Chinese patients with HER2-positive breast cancer (cohort 1) elucidated using a 520-gene panel (OncoScreen Plus, Burning Rock Biotech). a Oncoprint summarizing the mutational landscape of the cohort. Only somatic alterations with a frequency of 5% or greater in the whole cohort are displayed. The HR status of the patients was indicated at the bottom of the oncoprint, wherein green represents patients with HR-negative (HR−/HER2+) status and red represents patients with HR-positive (HR+/HER2+) status. Each column represents a patient and each row represents a gene. Numbers on the left represent the percentage of patients with mutations in a specific gene. Top plot represents the overall number of mutations a patient carried. Different colors denote different types of mutations. b Lollipop diagrams depicting the type and specific locations of TP53, PIK3CA and ROS1 mutations in cohort 2. Colored boxes depict the different functional domains along the gene. Colored circles denote the type of mutation, while the location of the circle specifies the mutation site. The length of the lollipop represents the number of patients harboring a specific variant. The legend on the right side summarizes the total number of mutation types such as missense, truncating, inframe and other mutations

The mutational landscape of 40 Chinese patients with early-stage HER2-positive breast cancer who received HER2 inhibitors as neoadjuvant therapy (cohort 2) elucidated using a 520-gene panel (OncoScreen Plus, Burning Rock Biotech). a Oncoprint summarizing the mutational landscape of the cohort. Only somatic alterations with a frequency of 5% or greater in the whole cohort are displayed. The pathologic complete response (pCR) and HR status of the patients were indicated at the bottom of the oncoprint, wherein red represents patients achieving pCR (n = 18), pink represents patients with non-pCR (n = 22); cyan represents patients with HR-positive (HR+/HER2+) status (pCR, n = 11; non-pCR, n = 9) and green represents patients with HR-negative (HR−/HER2+) status (pCR, n = 7; non-pCR, n = 13). Each column represents a patient and each row represents a gene. Numbers on the left represent the percentage of patients with mutations in a specific gene. Top plot represents the overall number of mutations a patient carried. Different colors denote different types of mutations. b Lollipop diagrams depicting the type and specific locations of TP53, PIK3CA and ROS1 mutations in cohort 2. Colored boxes depict the different functional domains along the gene. Colored circles denote the type of mutation, while the location of the circle specifies the mutation site. The length of the lollipop represents the number of patients harboring a specific variant. The legend on the right side summarizes the total number of mutation types such as missense, truncating, inframe and other mutations
Genetic mutation difference between HER2-positive breast cancer patients with and without pCR
In the NACT group cohort 2, mutations were frequently identified in 48 genes shown in the oncoprint (Fig. 2) and some are selected in Table 2. The genetic mutation profiles were quite different between HER2-positive breast cancer patients with and without pCR. In pCR group, mutation rates of the most frequently mutated genes, such as TP53, PIK3CA, CDK12, SPOP, FGF3, FGF4 and FGF19, were 88.9%, 33.3%, 72.2%, 11.1%, 11.1%, 11.1% and 11.1%, while in non-pCR group their mutation rates were 81.8%, 72.7%, 72.7%, 22.7%, 18.2%, 18.2% and 18.2%.
Then we detected the differences between the pCR and non-pCR groups according to pathological and mutational variables and found significant differences in terms of the initial Ki67 status, TP53 missense mutations, TP53 LOF mutations, PIK3CA mutations and ROS1 mutations (p = 0.028, 0.019, 0.005, 0.013, 0.049, respectively, Tables 1, 2).
One PIK3CA mutation hotspot was examined to be p.H1047R which belongs to be a missense mutation. Three mutation sites (p.V650M, p.E545G and p.E542K) were detected to be located in the PI3Ka subunit which has been found to be crucial for PIK3CA function.
There was a significant difference of PIK3CA mutation frequency between pCR and non-pCR group (33.3% vs 72.7%; p = 0.013, Table 2 and Fig. 3).

Differences between pCR and non-pCR groups in terms of genetic mutations. a–d Pearson’s Chi-square test and Yate’s continuity-corrected Chi-square test analyzed the associations of TP53 missense and LOF mutations, PIK3CA mutations and ROS1 amplifications with pCR or non-pCR of HER2-positive breast cancers after NACT. *Indicates 0.01 < p < 0.05, **indicates p < 0.01
ROS1 amplification was only investigated in 5 hormone receptor-positive patients who all got pCR. The ROS1 amplification breast cancers were found to have higher pCR rate (p = 0.049) (Table 2 and Fig. 2). However, when we go further to investigate whether ROS1 had amplification in protein level by immunohistochemistry, the result was negative (Figs. 3, 4).

ROS1 expression in protein level. a, b Positive and negative control of ROS1 expression by IHC. c ROS1 expression in samples with ROS1 amplification (negative). d ROS1 expression in samples without ROS1 amplification (negative)
Predictors for pCR or non-pCR
As shown above, some mutations and pathological factors seemed to affect pCR status and, to determine which of those are the predictive factors, we did univariate and multivariate regression. For pCR status, three covariates were significant in the univariate analyses (Tumors size, TP53 missense mutations, TP53 LOF mutations), but only one was retained and a new one appeared after forward selection in the multivariate analysis—TP53 LOF mutations and initial Ki67 status (OR 7.086, 95% CI 1.366–36.749, p = 0.020 and OR 6.007, 95% CI 1.120–32.210, p = 0.036, respectively, in Table 4). Some factors selected in the univariate model seemed to be highly correlated and for this reason were rejected or appeared in the multivariate model.
Discussion
Our findings indicate significant associations (p = 0.020 and p = 0.036) of TP53 LOF mutations and lower initial Ki67 status (< 40%) with a high probability of pCR in HER2-positive breast cancer patients receiving NACT.
TP53 has been reported to have heterogenous types of mutations which include attenuation of function, separation of function or neomorphic function [15,16,17]. There are gain-of-function mutations mostly TP53 missense mutations and LOF mutations which have a common characteristic of losing functions of wild-type TP53 and composed of splice site, frame shift and nonsense mutations [16, 18].
HER2-positive breast cancer has a high frequency of TP53 mutations (up to 72%) [8], and in our previous study, the mutation frequency of TP53 in the triple-positive HR + /HER2 + group is 66.1% and up to 89.3% in the HR-/HER2 + group in Chinese breast cancer patients [19]. Interestingly, the TP53 mutation rate was similar in those of HER2-positive patients who achieved a pCR or not (88.9% v 81.8%), and there was no statistical difference of TP53 mutation rate between two groups (p = 0.859). However, as far as specific types of TP53 mutations were considered, such as missense mutations, LOF mutations and so on, quite significant mutational differences were manifested. TP53 missense mutation rates in pCR and non-pCR groups were quite different (22.2% vs 59.1%, p = 0.019, Table 2 and Fig. 3). Similarly, patients in pCR group were detected to have much higher LOF mutation rate (66.7% vs 22.7%, p = 0.005, Table 2 and Fig. 3). Univariate logistic regression showed TP53 missense mutation and TP53 LOF mutation were significantly associated with pCR status (OR 0.198, 95% CI 0.049–0.801, p = 0.023 and OR 6.800, 95% CI 1.680–27.522, p = 0.007). However, when multivariate logistic regression was applied, only TP53 LOF mutation was retained to be predictive of pCR status.
And why is that? How TP53 LOF mutation mediates a good response to NACT is complicated. TP53 has been reported to be activated in response to mitotic stress caused by agents such as taxanes that disrupt microtubules [20]. Evidences from breast cancer models and tumors have shown p53 directs cells to undergo cell cycle arrest and senescence [21, 22]. One mechanism that contributes to senescent cell survival and persistence in the residual disease after chemotherapy treatment is the engulfing and cannibalizing of neighboring cells [23]. We believe that TP53 LOF mutations lose capacity to activate cell cycle arrest and senescence to escape apoptosis induced by therapy, and as a result mediate a good response to NACT.
Some TP53 missense mutations are associated with enhanced characteristics of invasion and metastasis when they acquire a gain-of-function effect [24]. More than 80% of TP53 alterations are missense mutations that will produce a stable but transcriptionally deficient protein. These mutant-TP53-expressing tumors are aggressive and associated to poor prognosis [25, 26]. Compelling evidences have proved that TP53 missense mutations promote cell migration and metastasis and dramatically influence tumor progression [27,28,29]. In our study, TP53 missense mutations showed significant difference between pCR and non-pCR groups, and also a predictor for non-pCR by univariate regression. We speculated that TP53 missense may affect therapy response depending on other variables.
TP53 mutations were not predictive of neoadjuvant chemotherapy response in the EORTC 10,994/BIG 1-00 trial [30]. However, in that study, patients with HER2 + breast cancers were randomly assigned to different NACT groups without HER2-targeted therapy, and the yeast assay was used to assess TP53 mutations which does not distinguish between pure loss-of-function mutations compared to mutations with simultaneous gain and loss-of-function [31, 32]. Several molecular alterations are thought to contribute to trastuzumab resistance, including TP53 mutation [33, 34] and PIK3CA alteration [13, 35, 36], but results evaluating these biomarkers as response predictors have been inconsistent.
Two retrospective studies [37, 38] reported TP53 mutations were significantly predictive of HER2 + patient treatment response (pCR) to neoadjuvant chemotherapies. Soley et al. report that for patient samples with concordant BluePrint/MammaPrint and PAM50 data, the pCR plus non-pCR rate among patients whose tumors were TP53 mutant was 17/39 (44%), whereas in patients whose tumors were TP53 wild type, it was 5/31 (16%), p = 0.020 [37]. And Stefan et al. report that the response rate among TP53-mutated patients was 30%, significantly higher than TP53 wild-type patients (10%; p = 0.0032) [38]. However, both studies used the AmpliChip TP53 assay (Roche Molecular Systems, Pleasanton, CA), a DNA microarray-based resequencing assay designed to detect single-base substitutions and single-base deletions in all coding regions of the TP53 gene, which needs a reference sequence and is unable to detect all the possible mutations like a NGS assay [39, 40]. Two other studies found no associations of TP53 mutations with HER2 + NACT treatment response [41, 42]. However, all these studies only examined associations of TP53 mutations as a whole with treatment response, despite the fact that TP53 missense mutations and loss-of-function mutations have quite different functions during breast cancer progression [16, 24, 26]. In our study, we assessed the associations of TP53 missense mutations and LOF mutations separately with treatment response and found quite different predictive characteristics.
From a tumor biological point of view, Ki67 should be viewed as a continuous variable, as it reflects the percentage of proliferating cells in the tumor, which can reach any value between 0 and 100%. The fact of defining our cut points should not be seen as a limitation of the marker but point to a strength of Ki67, as studies have shown a wide range of cut points was significant for various endpoints and subgroups [43]. Therefore in our study, Ki67 is still a predictive biomarker for HER2-positive breast cancer subgroup receiving chemotherapy and HER2-targeted therapy.
Tumor size was analyzed to be a predictor of pCR status by univariate regression and not by multivariate regression. It is easy to understand that tumor size is a factor susceptible to other pathological and mutational factors.
In conclusion, our study reports TP53 LOF mutations and initial Ki67 status predict pCR status for HER2-positive breast cancer patients receiving NACT. As this study is an exploratory retrospective study of small size, further prospective clinical research with large sample is still needed.
References
Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, Allred DC, Bartlett JM, Bilous M, Fitzgibbons P et al (2013) Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol 31(31):3997–4013
Cortazar P, Zhang L, Untch M, Mehta K, Costantino JP, Wolmark N, Bonnefoi H, Cameron D, Gianni L, Valagussa P et al (2014) Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet 384(9938):164–172
Untch M, Fasching PA, Konecny GE, Hasmuller S, Lebeau A, Kreienberg R, Camara O, Muller V, du Bois A, Kuhn T et al (2011) Pathologic complete response after neoadjuvant chemotherapy plus trastuzumab predicts favorable survival in human epidermal growth factor receptor 2-overexpressing breast cancer: results from the TECHNO trial of the AGO and GBG study groups. J Clin Oncol 29(25):3351–3357
Gianni L, Pienkowski T, Im Y-H, Roman L, Tseng L-M, Liu M-C, Lluch A, Staroslawska E, de la Haba-Rodriguez J, Im S-A et al (2012) Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol 13(1):25–32
Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, de Azambuja E, Aura C, Gómez H, Dinh P, Fauria K, Van Dooren V et al (2012) Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet 379(9816):633–640
Loibl S, Gianni L (2017) HER2-positive breast cancer. Lancet 389(10087):2415–2429
von Minckwitz G, Huang C-S, Mano MS, Loibl S, Mamounas EP, Untch M, Wolmark N, Rastogi P, Schneeweiss A, Redondo A et al (2019) Trastuzumab emtansine for residual invasive HER2-positive breast cancer. N Engl J Med 380(7):617–628
Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, Fulton LL, Dooling DJ, Ding L, Mardis ER et al (2012) Comprehensive molecular portraits of human breast tumours. Nature 490(7418):61–70
Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR et al (2012) The landscape of cancer genes and mutational processes in breast cancer. Nature 486(7403):400–404
Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y et al (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486(7403):346–352
Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ et al (2016) The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun 7:11479
Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC et al (2016) Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534(7605):47–54
Majewski IJ, Nuciforo P, Mittempergher L, Bosma AJ, Eidtmann H, Holmes E, Sotiriou C, Fumagalli D, Jimenez J, Aura C et al (2015) PIK3CA mutations are associated with decreased benefit to neoadjuvant human epidermal growth factor receptor 2-targeted therapies in breast cancer. J Clin Oncol 33(12):1334–1339
Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, Fitzgibbons PL, Francis G, Goldstein NS, Hayes M et al (2010) American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol 28(16):2784–2795
Kastan MB, Berkovich E (2007) p53: a two-faced cancer gene. Nat Cell Biol 9(5):489–491
Kastenhuber ER, Lowe SW (2017) Putting p53 in context. Cell 170(6):1062–1078
Freed-Pastor WA, Prives C (2012) Mutant p53: one name, many proteins. Genes Dev 26(12):1268–1286
MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, Jostins L, Habegger L, Pickrell JK, Montgomery SB et al (2012) A systematic survey of loss-of-function variants in human protein-coding genes. Science 335(6070):823–828
Zhang G, Wang Y, Chen B, Guo L, Cao L, Ren C, Wen L, Li K, Jia M, Li C et al (2019) Characterization of frequently mutated cancer genes in Chinese breast tumors: a comparison of Chinese and TCGA cohorts. Ann Transl Med 7(8):179
Sablina AA, Chumakov PM, Levine AJ, Kopnin BP (2001) p53 activation in response to microtubule disruption is mediated by integrin-Erk signaling. Oncogene 20(8):899–909
Jackson JG, Pant V, Li Q, Chang LL, Quintas-Cardama A, Garza D, Tavana O, Yang P, Manshouri T, Li Y et al (2012) p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 21(6):793–806
Tonnessen-Murray C, Ungerleider NA, Rao SG, Wasylishen AR, Frey WD, Jackson JG (2018) p53 mediates vast gene expression changes that contribute to poor chemotherapeutic response in a mouse model of breast cancer. Transl Oncol 11(4):930–940
Tonnessen-Murray CA, Frey WD, Rao SG, Shahbandi A, Ungerleider NA, Olayiwola JO, Murray LB, Vinson BT, Chrisey DB, Lord CJ et al (2019) Chemotherapy-induced senescent cancer cells engulf other cells to enhance their survival. J Cell Biol 218(11):3827–3844
Bougeard G, Sesboue R, Baert-Desurmont S, Vasseur S, Martin C, Tinat J, Brugieres L, Chompret A, de Paillerets BB, Stoppa-Lyonnet D et al (2008) Molecular basis of the Li-Fraumeni syndrome: an update from the French LFS families. J Med Genet 45(8):535–538
Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS et al (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 98(19):10869–10874
Soussi T, Beroud C (2001) Assessing TP53 status in human tumours to evaluate clinical outcome. Nat Rev Cancer 1(3):233–240
Girardini JE, Napoli M, Piazza S, Rustighi A, Marotta C, Radaelli E, Capaci V, Jordan L, Quinlan P, Thompson A et al (2011) A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 20(1):79–91
Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo V et al (2009) A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 137(1):87–98
Caulin C, Nguyen T, Lang GA, Goepfert TM, Brinkley BR, Cai WW, Lozano G, Roop DR (2007) An inducible mouse model for skin cancer reveals distinct roles for gain- and loss-of-function p53 mutations. J Clin Invest 117(7):1893–1901
Bonnefoi H, Piccart M, Bogaerts J, Mauriac L, Fumoleau P, Brain E, Petit T, Rouanet P, Jassem J, Blot E et al (2011) TP53 status for prediction of sensitivity to taxane versus non-taxane neoadjuvant chemotherapy in breast cancer (EORTC 10994/BIG 1–00): a randomised phase 3 trial. Lancet Oncol 12(6):527–539
Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M (2007) Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat 28(6):622–629
Oren M, Rotter V (2010) Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol 2(2):a001107
Darb-Esfahani S, Denkert C, Stenzinger A, Salat C, Sinn B, Schem C, Endris V, Klare P, Schmitt W, Blohmer JU et al (2016) Role of TP53 mutations in triple negative and HER2-positive breast cancer treated with neoadjuvant anthracycline/taxane-based chemotherapy. Oncotarget 7(42):67686–67698
Fountzilas G, Giannoulatou E, Alexopoulou Z, Zagouri F, Timotheadou E, Papadopoulou K, Lakis S, Bobos M, Poulios C, Sotiropoulou M et al (2016) TP53 mutations and protein immunopositivity may predict for poor outcome but also for trastuzumab benefit in patients with early breast cancer treated in the adjuvant setting. Oncotarget 7(22):32731–32753
Carey LA, Berry DA, Cirrincione CT, Barry WT, Pitcher BN, Harris LN, Ollila DW, Krop IE, Henry NL, Weckstein DJ et al (2016) Molecular heterogeneity and response to neoadjuvant human epidermal growth factor receptor 2 targeting in CALGB 40601, a randomized phase III trial of paclitaxel plus trastuzumab with or without lapatinib. J Clin Oncol 34(6):542–549
Guarneri V, Dieci MV, Frassoldati A, Maiorana A, Ficarra G, Bettelli S, Tagliafico E, Bicciato S, Generali DG, Cagossi K et al (2015) Prospective biomarker analysis of the randomized CHER-LOB study evaluating the dual anti-HER2 treatment with trastuzumab and lapatinib plus chemotherapy as neoadjuvant therapy for HER2-positive breast cancer. Oncologist 20(9):1001–1010
Bayraktar S, Royce M, Stork-Sloots L, de Snoo F, Gluck S (2014) Molecular subtyping predicts pathologic tumor response in early-stage breast cancer treated with neoadjuvant docetaxel plus capecitabine with or without trastuzumab chemotherapy. Med Oncol 31(10):163
Gluck S, Ross JS, Royce M, McKenna EF Jr, Perou CM, Avisar E, Wu L (2012) TP53 genomics predict higher clinical and pathologic tumor response in operable early-stage breast cancer treated with docetaxel-capecitabine +/- trastuzumab. Breast Cancer Res Treat 132(3):781–791
Grollman AP, Shibutani S, Moriya M, Miller F, Wu L, Moll U, Suzuki N, Fernandes A, Rosenquist T, Medverec Z et al (2007) Aristolochic acid and the etiology of endemic (Balkan) nephropathy. Proc Natl Acad Sci USA 104(29):12129–12134
L'Esperance S, Popa I, Bachvarova M, Plante M, Patten N, Wu L, Tetu B, Bachvarov D (2006) Gene expression profiling of paired ovarian tumors obtained prior to and following adjuvant chemotherapy: molecular signatures of chemoresistant tumors. Int J Oncol 29(1):5–24
Singla H, Kaur RP, Shafi G, Vashistha R, Banipal RPS, Kumar V, Munshi A (2019) Genomic alterations associated with HER2+ breast cancer risk and clinical outcome in response to trastuzumab. Mol Biol Rep 46(1):823–831
Loi S, Michiels S, Lambrechts D, Fumagalli D, Claes B, Kellokumpu-Lehtinen PL, Bono P, Kataja V, Piccart MJ, Joensuu H et al (2013) Somatic mutation profiling and associations with prognosis and trastuzumab benefit in early breast cancer. J Natl Cancer Inst 105(13):960–967
Denkert C, Loibl S, Müller BM, Eidtmann H, Schmitt WD, Eiermann W, Gerber B, Tesch H, Hilfrich J, Huober J et al (2013) Ki67 levels as predictive and prognostic parameters in pretherapeutic breast cancer core biopsies: a translational investigation in the neoadjuvant GeparTrio trial. Ann Oncol 24(11):2786–2793
Funding
Research Funds from Guangzhou Municipal Science and Technology Project (201707010418). The funding body has no roles in the design of the study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Ethical approval
Primary tumor biopsies were obtained using an Institutional Review Board approved protocol, and this study had been approved by the Ethics Committee of Guangdong Provincial People’s Hospital (no. GDREC2014122H). All patients provided written informed consent for translational research.
Informed consent
We thank all the patients and their families for participation. Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, K., Liao, N., Chen, B. et al. Genetic mutation profile of Chinese HER2-positive breast cancers and genetic predictors of responses to Neoadjuvant anti-HER2 therapy. Breast Cancer Res Treat 183, 321–332 (2020). https://doi.org/10.1007/s10549-020-05778-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-020-05778-0