
Abstract
Purpose
This Phase I, multicenter, randomized study (ClinicalTrials.gov NCT01220128) evaluated the safety and immunogenicity of recombinant Wilms’ tumor 1 (WT1) protein combined with the immunostimulant AS15 (WT1-immunotherapeutic) as neoadjuvant therapy administered concurrently with standard treatments in WT1-positive breast cancer patients.
Methods
Patients were treated in 4 cohorts according to neoadjuvant treatment (A: post-menopausal, hormone receptor [HR]-positive patients receiving aromatase inhibitors; B: patients receiving chemotherapy; C: HER2-overexpressing patients on trastuzumab–chemotherapy combination; D: HR-positive/HER2-negative patients on chemotherapy). Patients (cohorts A–C) were randomized (2:1) to receive 6 or 8 doses of WT1-immunotherapeutic or placebo together with standard neoadjuvant treatment in a double-blind manner; cohort D patients received WT1-immunotherapeutic in an open manner. Safety was assessed throughout the study. WT1-specific antibodies were assessed pre- and post-vaccination.
Results
Sixty-two patients were randomized; 60 received ≥ one dose of WT1-immunotherapeutic. Two severe toxicities were reported: diarrhea (cohort C; also reported as a grade 3 serious adverse event) and decreased left ventricular ejection fraction (cohort B; also reported as a grade 2 adverse event). Post-dose 4 of WT1-immunotherapeutic, 10/10 patients from cohort A, 0/8 patients from cohort B, 6/11 patients from cohort C, and 2/3 patients from cohort D were humoral responders. The sponsor elected to close the trial prematurely.
Conclusions
Concurrent administration of WT1-immunotherapeutic and standard neoadjuvant therapy was well tolerated and induced WT1-specific antibodies in patients receiving neoadjuvant aromatase inhibitors. In patients on neoadjuvant chemotherapy or trastuzumab–chemotherapy combination, the humoral response was impaired or blunted, likely due to either co-administration of corticosteroids and/or the chemotherapies themselves.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Immunotherapies are rapidly becoming standard of care for many solid tumors. In the last 5 years, ipilimumab, pembrolizumab, and nivolumab have been approved for many cancer types [1–4]. There is an evolving interest in the immunogenicity of breast tumors and the possible role of immunotherapy in this common cancer [5, 6]. Various immunotherapeutic strategies, including checkpoint inhibitors, vaccines, adoptive T-cell transfer, or cytokine therapy, have been tested for treatment of breast cancer (BC) [6, 7]. Vaccines constitute an attractive immunotherapy approach aiming to stimulate the intrinsic antitumor immune response by presenting tumor antigens recognized by T-cells. Wilms’ tumor 1 (WT1) is a potential target antigen for cancer immunotherapy as it is over-expressed in the majority of solid tumors [8–12]. Owing to its specificity, oncogenicity, immunogenicity, and therapeutic function, WT1 has been classified as one of the most promising targets for cancer immunotherapy [13]. WT1 plays an oncogenic role in BC and is expressed in approximately 33% (range: 3–48.5%) of malignant breast tumors [11, 14–16]. Additionally, high WT1 levels have previously been correlated with poorer outcomes in BC [15, 17].
Combining chemotherapy with immune-based interventions has great potential for optimizing clinical outcomes of BC patients. This study evaluated the safety, immunogenicity, and preliminary clinical activity of the WT1 antigen combined with GSK’s proprietary immunostimulant AS15 (WT1-immunotherapeutic) administered to women with BC during standard neoadjuvant treatment.
Patients and methods
Study design and patients
This study was an international, multicenter, double-blind, randomized, placebo-controlled, Phase I/II clinical trial conducted between 2011 and 2014 in 19 medical centers in Belgium, France, Germany, Italy, the Russian federation, the United Kingdom, and the United States. Phase I initially included three parallel cohorts (A, B, and C), in which patients were randomized in a double-blind manner (2:1) to receive six or eight doses of WT1-immunotherapeutic (WT1 groups) or placebo (placebo groups) at 3-week intervals, together with the standard neoadjuvant treatment (Fig. S1).
The neoadjuvant treatment was chosen according to institutional standards, based on the hormone receptor (HR) and human epidermal growth factor receptor-2 (HER2) status of the tumor. Cohort A received daily aromatase inhibitors (AIs) for 18 or 24 weeks of neoadjuvant treatment; cohorts B and C received WT1-immunotherapeutic/placebo on the same day as chemotherapy (Fig. S2), with patients in cohort C also receiving trastuzumab. Further recruitment beyond Phase I in each cohort depended on the outcome of intermediate assessment of the induced WT1-specific antibody response. Only if a ≥40% response rate (based on post-dose 4 WT1-specific antibody responses in at least six patients in the WT1 group) was achieved, and provided no safety issues were identified, would the cohort proceed to Phase II.
Following the analysis of early immunogenicity results in cohort B (see Results section), a further cohort (D) was opened to investigate an alternative dosing schedule (Fig. S1). Cohort D received WT1-immunotherapeutic on day 14 of each 3-weekly chemotherapy cycle in an open-label manner (Fig. S2).
Patients aged ≥18 years with WT1-positive, histologically confirmed, primary invasive BC were eligible for enrollment. Details of inclusion/exclusion criteria, as well as study treatment and administration, study procedures, data collection, and blood sampling are included in Supplementary materials.
All patients provided written informed consent before any study-related procedures. The study was conducted in accordance with Good Clinical Practice and all applicable regulatory requirements, including the Declaration of Helsinki. The protocol was approved by the national independent ethics committees and institutional review boards of the study centers. The study was registered at www.ClinicalTrials.gov (NCT01220128). A protocol summary is available at http://www.gsk-clinicalstudyregister.com (GSK study ID 113172).
Objectives
Phase I study objectives were the evaluation of safety and immunogenicity of WT1-immunotherapeutic as neoadjuvant therapy administered concurrently with different standard treatments.
Phase II objectives included further assessment of safety and immunogenicity, and a preliminary assessment of the clinical activity of WT1-immunotherapeutic in combination with standard neoadjuvant treatments, i.e., pathological complete response (pCR) rate, disease free survival (DFS), and overall survival (OS); of note, due to early termination of the trial, the analysis of DFS and OS outcomes was not performed.
Safety and immunogenicity assessments
Adverse events (AEs), including severe toxicities (defined in Supplementary materials), and serious adverse events (SAEs) were assessed throughout the study.
WT1-specific antibodies were measured by an enzyme-linked immunosorbent assay (ELISA). WT1-specific humoral response was defined as the appearance of antibodies for baseline seronegative patients, or an at least 2-fold increase in antibody concentrations for baseline seropositive patients. The ELISA assay cut-off was 9 ELISA units (EU)/ml.
Clinical activity assessment
pCR, i.e., complete response (CR) or partial response (PR) in the breast and axillary nodes was assessed at the definitive surgery. pCR in the primary tumor was evaluated according to the Miller/Payne grading system [18], and in lymph nodes, by histopathological examination. The reference pCR rates based on the reported in literature rates under standard treatment for a given patient population were: 5% for cohort A (based on a 3–5% rate), 20% for cohort B (6–30%), and 50% for cohort C (30–65%) (see details in Supplementary materials).
Statistical analyses
Statistical analyses were performed using Statistical Analysis Systems (SAS) Drug and Development with SAS version 9.2.
The total treated cohort (TTC) included all patients who received at least one dose of WT1-immunotherapeutic/placebo. The according-to-protocol (ATP) cohort for immunogenicity included all eligible patients (i.e., those meeting all eligibility criteria for enrollment), who did not report major protocol deviation, who received at least the first four doses of WT1-immunotherapeutic/placebo, and who provided a valid result for immunogenicity measurement within four weeks of post-dose 4 (visit 5). Data collected after major protocol violation were eliminated from ATP immunogenicity analyses.
Descriptive analyses of demographics and baseline characteristics were performed on the TTC. Safety analyses were performed on the TTC, and immunogenicity analyses on the ATP cohort for immunogenicity.
Results
Study patients
Phase I recruitment was completed in March 2013 for cohort A, November 2011 for cohort B, and June 2012 for cohort C. Phase II recruitment for cohort A had been initiated as the protocol criteria were met, but was stopped prematurely in July 2014, following the sponsor’s decision. Enrollment in cohort B did not proceed to the Phase II segment because the protocol-defined immune response success (≥40% of patients showing a humoral response) was not fulfilled. In cohort C, weak immune responses with antibody concentrations close to the assay cut-off values were induced in only a few patients (see Immunogenicity section below) and, although meeting the protocol criteria of success, these immune responses were considered sub-optimal; therefore, Phase II for this cohort was not initiated. Recruitment of cohort D patients was also stopped prematurely at the same time as the Phase II for cohort A.
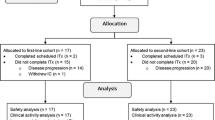
In total, 366 patients were screened for WT1 expression; 127 (34.7%) had WT1-positive tumors. Sixty-two patients were randomized and 60 were treated (cohort A: 22, B: 15, C: 15, D: 8); 47 patients completed the treatment (Fig. 1).

Participant flow N, number of patients; WT1 patients who received WT1-immunotherapeutic; ATP cohort, according-to-protocol cohort for immunogenicity; SAE serious adverse event; pIMD potential immune-mediated disease; PD progressive disease; Cohort A: post-menopausal patients with hormone receptor-positive breast cancer receiving AIs as neoadjuvant therapy; Cohort B: patients receiving neoadjuvant chemotherapy; Cohort C: patients with human epidermal growth factor receptor-2 (HER-2)-overexpressing breast cancer receiving neoadjuvant trastuzumab therapy combined with chemotherapy; Cohort D: patients with hormone receptor-positive/HER2-negative breast cancer receiving neoadjuvant chemotherapy; patients in cohort D received WT1-immunotherapeutic in an open-label manner
The majority of patients (95.0%) were of Caucasian origin; the median age (range) of the patients in WT1 and placebo groups was 72.0 (54–84) and 74.0 (60–80) years in cohort A, 41.0 (37–77) and 62.5 (48–74) years in cohort B, 52 (38–69) and 53.0 (46–61) years in cohort C, respectively, and 47 (42–69) years in cohort D (WT1 group only). The majority of patients enrolled had Stage IIA (38.3%) or IIB (38.3) tumors; 13.3% had Stage IIIA, 8.3%, Stage IIIB, and 1.7%, Stage IIIC tumors.
Safety
Two severe toxicities were reported: diarrhea (cohort C; also reported as a grade 3 SAEs) and decreased left ventricular ejection fraction (cohort B; also reported as a grade 2 AE).
Grade 3 AEs considered by the investigator to be related/possibly related to WT1-immunotherapeutic administration were reported by one patient in cohort A (headache, two separate events) and one patient in cohort C (diarrhea); the latter was also reported as a SAE and as a severe toxicity event (Table 1).
Thirty-seven SAEs were reported by 20 patients (Table 1); two were considered by the investigators to be related/possibly related to WT1-immunotherapeutic administration: grade 2 polymyalgia rheumatica (cohort A; also reported as potential immune-mediated disorder) and diarrhea (mentioned above).
Two patients (WT1 group, cohort B) died during the study. One patient died due to an unknown cause, possibly due to underlying medical conditions of hypertension and thrombosis; this fatal SAE was assessed by the investigators as not causally related to WT1-immunotherapeutic administration. The second patient died due to progressive BC.
The Data Safety Monitoring Committee reviewed safety data every six months during the trial, with the last review in June 2015, and did not identify any potential safety issues.
Immunogenicity
At baseline, all patients were seronegative for WT1-specific antibodies; post-dose 4, all 10 patients from cohort A (100%), 0/8 patients (0.0%) from cohort B, 6/11 (54.5%) patients from cohort C, and 2/3 (66.7%) patients from cohort D were humoral responders.
The highest WT1-specific antibody levels were observed in cohort A, in which patients received AIs as concomitant standard treatment (Fig. 2a). No antibody response was observed in cohort B receiving concomitant chemotherapy (Fig. 2b), while in cohorts C and D, weak WT1-specific antibody responses were only observed in some patients (Fig. 2c–d).

Pre- and post-immunization WT1-specific antibody titers in patients from a cohort A, b cohort B, c cohort C, and d cohort D (ATP cohort for immunogenicity). ATP according-to-protocol; EU/ml, ELISA units per ml (antibody concentration). The cut-off of the ELISA assay was 9 EU/ml. The color lines correspond to individual patients’ antibody titers at indicated timepoints
Of note, different types of antibody responses were observed in cohort C, with some patients presenting no antibody response (similar to cohort B), some having a delayed response, and others, immediate antibody titer development. WT1-specific antibody titers of patients from cohort C who developed an immune response were around 1 log below the results obtained in cohort A. Patients from cohort C who were immediate antibody responders received docetaxel, carboplatin, and trastuzumab (TCH) as concomitant chemotherapy. In cohort B, nearly all patients received sequential chemotherapy, starting with the combination of anthracyclines/cyclophosphamide and finishing with taxane-based therapy (paclitaxel or docetaxel). Patients in cohort C with no or a delayed immune response received the same treatment combination as in cohort B with the addition of trastuzumab.
No conclusions could be drawn for cohort D, as antibody responses were evaluated for only 3/8 patients enrolled in TTC due to the early termination of the study; two of these patients showed positive responses within the same range as those observed in cohort C.
Clinical activity
The clinical activity was evaluated in 51 patients and is shown by treatment group in Table 2. In cohort A, among the 18 evaluable patients, seven patients had PR and 11 had no response. Among 15 patients in cohort B, two had pCR, eight had PR, and five patients had no response. Of the ten evaluable patients in cohort C, nine had pCR, four had PR, and one patient had no response. Among the four patients from cohort D who received WT1-immunotherapeutic in an open manner, one had pCR and three had PR.
Discussion
The role of the host immune response to the tumor in BC has long been debated as, compared to melanoma or renal cell carcinoma, BC has been considered less immunogenic. However, current data suggest that BC, particularly the more aggressive subtypes of HER2-positive and triple-negative BC, can elicit host antitumor immune responses, and that the robustness of the response correlates with prognosis [5, 19–21]. The concept of natural immunogenicity of BC is based on the presence of tumor-infiltrating lymphocytes (TILs) and other immune cells within the tumor microenvironment, on the prognostic value of immune-related gene signatures, and the frequency of genetic instability which leads to higher numbers of somatic mutations and neoantigens [5, 22]. Additionally, the pre-existing immunologic response might enhance the effects of conventional chemotherapy [5, 23].
In cohort A, all patients who received WT1-immunotherapeutic developed WT1-specific antibodies. The antibody titers obtained in this cohort can be considered as reference titers, as only in this cohort patients did not receive chemotherapy or routine corticosteroids. In contrast, none of the patients receiving WT1-immunotherapeutic in cohort B developed antibodies. Analysis of B-cell population dynamics revealed depletion of B-cells in these patients compared to healthy donors, either due to the chemotherapy itself or the corticosteroids which are routinely used as anti-emetics in patients receiving chemotherapy (data not shown). The impact of cancer treatments on all lymphocytic populations, especially B-cells, has been previously described [24–26]. A study in BC patients evaluating the effects of combination chemotherapy regimens with epirubicin (5-fluorouracil, epirubicin, cyclophosphamide) versus doxorubicin (5-fluorouracil, doxorubicin, cyclophosphamide) on immune cells, revealed an increase in cytotoxic T-cell levels and natural killer cell levels, and a dramatic decrease in B-cell levels in the blood following in either regimen [26]. Nevertheless, the lympho-depleting effects induced by chemotherapy are transient and soon after drug discontinuation, a homeostatic rebound overshoot of the lymphocytic pool occurs [24].
In cohort C, a mix of titer profiles was observed, supporting the hypothetic blunting effect of chemotherapy co-administered on day 1, and also suggesting that different chemotherapy agents may have differing immunosuppressive effects. Diverse myelosuppressive effects of specific chemotherapeutic agents have been previously reported [27–29].
Another parameter difficult to discriminate from the chemotherapy effect is the impact of co-administered corticosteroids which were allowed per protocol for the prevention and treatment of chemotherapy-related nausea and hypersensitivity reactions. In cohort C, patients received trastuzumab co-administered with chemotherapy, and in numerous cases, patients receiving chemotherapy also received corticosteroids.
The traditional paradigm that chemotherapeutic agents suppress immune response has been challenged by evidence that chemotherapy induces, and is dependent upon activation of certain immunologic effects and may promote immune-mediated tumor destruction [30–33]. TILs within breast tumors have been shown to correlate with pCR and clinical response to neoadjuvant chemotherapy [34, 35]. The possible immunomodulatory mechanisms involving trastuzumab include inhibition of HER2-mediated signaling and antibody-dependent cell-mediated cytotoxicity [36, 37]. The AI anastrozole was shown to alter the proinflammatory cytokine levels and suppressed differentiation of naive T-cells into regulatory T-cells, which are known to produce immunosuppressive cytokines in the tumor microenvironment [38, 39].
An additional cohort D received WT1-immunotherapeutic on day 14 of each chemotherapy cycle, to evaluate if delaying the immunotherapy administration after the chemotherapy treatment improves the immune response. Day 14 was selected because corticosteroids were not administered on that day and patients were expected to have passed their white cell count nadir. In a study with MAGE-A3 immunotherapeutic in non-small cell lung cancer patients who received concurrent cisplatin/vinorelbine chemotherapy regimen, a robust MAGE-A3-specific antibody response was induced in all patients [40]. However, in this previous study, MAGE-A3 immunotherapeutic was administered on day 8 of each chemotherapy cycle, whereas in cohort B of the current study, chemotherapy was administered on the same day as WT1-immunotherapeutic. This information also reinforced the hypothesis of a differential impact of the chemotherapy types on the immune response. Although our study was stopped before finalization of enrollment in cohort D, from the few data collected, it is apparent that delaying administration of immunotherapy (14 days following the chemotherapy cycle initiation) did not improve the immunogenicity, as antibody titers obtained in cohort D were similar to those obtained in cohort C. In one patient from cohort D, the sequence of chemotherapy was reversed, starting with docetaxel followed by epirubicin/cyclophosphamide combination. In this patient, the WT1-specific antibody level rose immediately while the patient underwent docetaxel chemotherapy, but fell thereafter following epirubicin/cyclophosphamide treatment. Altogether, these data suggest that concomitant corticosteroid administration and/or possibly specific chemotherapies (particularly anthracycline combinations) impacted the WT1-specific antibody generation post-vaccination.
Limitations of our study include the presence of multiple confounding factors and small numbers of patients in each cohort.
In conclusion, concurrent administration of WT1-immunotherapeutic and standard therapy was well tolerated and induced WT1-specific antibody response in BC patients when co-administered with neoadjuvant AIs. In patients on neoadjuvant chemotherapy or a trastuzumab–chemotherapy combination, the humoral response was impaired or blunted, likely due to either co-administration of corticosteroids and/or the chemotherapies themselves.
References
Mahoney KM, Freeman GJ, McDermott DF (2015) The next immune-checkpoint inhibitors: PD-1/PD-L1 Blockade in Melanoma. Clin Ther 37(4):764–782. doi:10.1016/j.clinthera.2015.02.018
Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH Jr, Lao CD, Linette GP, Thomas L, Lorigan P, Grossmann KF, Hassel JC, Maio M, Sznol M, Ascierto PA, Mohr P, Chmielowski B, Bryce A, Svane IM, Grob JJ, Krackhardt AM, Horak C, Lambert A, Yang AS, Larkin J (2015) Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 16(4):375–384. doi:10.1016/S1470-2045(15)70076-8
Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, Agunwamba BU, Zhang X, Lowy I, Inzunza HD, Feely W, Horak CE, Hong Q, Korman AJ, Wigginton JM, Gupta A, Sznol M (2013) Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 369(2):122–133. doi:10.1056/NEJMoa1302369
Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, Castellano D, Choueiri TK, Gurney H, Donskov F, Bono P, Wagstaff J, Gauler TC, Ueda T, Tomita Y, Schutz FA, Kollmannsberger C, Larkin J, Ravaud A, Simon JS, Xu LA, Waxman IM, Sharma P, CheckMate I (2015) Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 373(19):1803–1813. doi:10.1056/NEJMoa1510665
Cimino-Mathews A, Foote JB, Emens LA (2015) Immune targeting in breast cancer. Oncology 29(5):375–385
Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, Pusztai L, Pathiraja K, Aktan G, Cheng JD, Karantza V, Buisseret L (2016) Pembrolizumab in patients with advanced triple-negative breast cancer: phase Ib KEYNOTE-012 Study. J Clin Oncol. doi:10.1200/JCO.2015.64.8931
Emens LA, Asquith JM, Leatherman JM, Kobrin BJ, Petrik S, Laiko M, Levi J, Daphtary MM, Biedrzycki B, Wolff AC, Stearns V, Disis ML, Ye X, Piantadosi S, Fetting JH, Davidson NE, Jaffee EM (2009) Timed sequential treatment with cyclophosphamide, doxorubicin, and an allogeneic granulocyte-macrophage colony-stimulating factor-secreting breast tumor vaccine: a chemotherapy dose-ranging factorial study of safety and immune activation. J Clin Oncol 27(35):5911–5918. doi:10.1200/JCO.2009.23.3494
Sugiyama H (2010) WT1 (Wilms’ tumor gene 1): biology and cancer immunotherapy. Jpn J Clin Oncol 40(5):377–387. doi:10.1093/jjco/hyp194
Nishida S, Koido S, Takeda Y, Homma S, Komita H, Takahara A, Morita S, Ito T, Morimoto S, Hara K, Tsuboi A, Oka Y, Yanagisawa S, Toyama Y, Ikegami M, Kitagawa T, Eguchi H, Wada H, Nagano H, Nakata J, Nakae Y, Hosen N, Oji Y, Tanaka T, Kawase I, Kumanogoh A, Sakamoto J, Doki Y, Mori M, Ohkusa T, Tajiri H, Sugiyama H (2014) Wilms tumor gene (WT1) peptide-based cancer vaccine combined with gemcitabine for patients with advanced pancreatic cancer. J Immunother 37(2):105–114. doi:10.1097/CJI.0000000000000020
Perugorria MJ, Castillo J, Latasa MU, Goni S, Segura V, Sangro B, Prieto J, Avila MA, Berasain C (2009) Wilms’ tumor 1 gene expression in hepatocellular carcinoma promotes cell dedifferentiation and resistance to chemotherapy. Cancer Res 69(4):1358–1367. doi:10.1158/0008-5472.CAN-08-2545
Qi XW, Zhang F, Wu H, Liu JL, Zong BG, Xu C, Jiang J (2015) Wilms’ tumor 1 (WT1) expression and prognosis in solid cancer patients: a systematic review and meta-analysis. Sci Rep 5:8924. doi:10.1038/srep08924
Gillmore R, Xue SA, Holler A, Kaeda J, Hadjiminas D, Healy V, Dina R, Parry SC, Bellantuono I, Ghani Y, Coombes RC, Waxman J, Stauss HJ (2006) Detection of Wilms’ tumor antigen–specific CTL in tumor-draining lymph nodes of patients with early breast cancer. Clin Cancer Res 12(1):34–42. doi:10.1158/1078-0432.CCR-05-1483
Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM, Matrisian LM (2009) The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res 15(17):5323–5337. doi:10.1158/1078-0432.CCR-09-0737
Caldon CE, Lee CS, Sutherland RL, Musgrove EA (2008) Wilms’ tumor protein 1: an early target of progestin regulation in T-47D breast cancer cells that modulates proliferation and differentiation. Oncogene 27(1):126–138. doi:10.1038/sj.onc.1210622
Qi XW, Zhang F, Yang XH, Fan LJ, Zhang Y, Liang Y, Ren L, Zhong L, Chen QQ, Zhang KY, Zang WD, Wang LS, Zhang Y, Jiang J (2012) High Wilms’ tumor 1 mRNA expression correlates with basal-like and ERBB2 molecular subtypes and poor prognosis of breast cancer. Oncol Rep 28(4):1231–1236. doi:10.3892/or.2012.1906
Tuna M, Chavez-Reyes A, Tari AM (2005) HER2/neu increases the expression of Wilms’ Tumor 1 (WT1) protein to stimulate S-phase proliferation and inhibit apoptosis in breast cancer cells. Oncogene 24(9):1648–1652. doi:10.1038/sj.onc.1208345
Miyoshi Y, Ando A, Egawa C, Taguchi T, Tamaki Y, Tamaki H, Sugiyama H, Noguchi S (2002) High expression of Wilms’ tumor suppressor gene predicts poor prognosis in breast cancer patients. Clin Cancer Res 8(5):1167–1171
Ogston KN, Miller ID, Payne S, Hutcheon AW, Sarkar TK, Smith I, Schofield A, Heys SD (2003) A new histological grading system to assess response of breast cancers to primary chemotherapy: prognostic significance and survival. Breast 12(5):320–327
Adams S, Gray RJ, Demaria S, Goldstein L, Perez EA, Shulman LN, Martino S, Wang M, Jones VE, Saphner TJ, Wolff AC, Wood WC, Davidson NE, Sledge GW, Sparano JA, Badve SS (2014) Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol 32(27):2959–2966. doi:10.1200/JCO.2013.55.0491
Ali HR, Provenzano E, Dawson SJ, Blows FM, Liu B, Shah M, Earl HM, Poole CJ, Hiller L, Dunn JA, Bowden SJ, Twelves C, Bartlett JM, Mahmoud SM, Rakha E, Ellis IO, Liu S, Gao D, Nielsen TO, Pharoah PD, Caldas C (2014) Association between CD8+ T-cell infiltration and breast cancer survival in 12,439 patients. Ann Oncol 25(8):1536–1543. doi:10.1093/annonc/mdu191
Loi S, Michiels S, Salgado R, Sirtaine N, Jose V, Fumagalli D, Kellokumpu-Lehtinen PL, Bono P, Kataja V, Desmedt C, Piccart MJ, Loibl S, Denkert C, Smyth MJ, Joensuu H, Sotiriou C (2014) Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: results from the FinHER trial. Ann Oncol 25(8):1544–1550. doi:10.1093/annonc/mdu112
Pardoll D (2003) Does the immune system see tumors as foreign or self? Annu Rev Immunol 21:807–839. doi:10.1146/annurev.immunol.21.120601.141135
Apetoh L, Tesniere A, Ghiringhelli F, Kroemer G, Zitvogel L (2008) Molecular interactions between dying tumor cells and the innate immune system determine the efficacy of conventional anticancer therapies. Cancer Res 68(11):4026–4030. doi:10.1158/0008-5472.CAN-08-0427
Bracci L, Schiavoni G, Sistigu A, Belardelli F (2014) Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ 21(1):15–25. doi:10.1038/cdd.2013.67
Proietti E, Moschella F, Capone I, Belardelli F (2012) Exploitation of the propulsive force of chemotherapy for improving the response to cancer immunotherapy. Mol Oncol 6(1):1–14. doi:10.1016/j.molonc.2011.11.005
Wijayahadi N, Haron MR, Stanslas J, Yusuf Z (2007) Changes in cellular immunity during chemotherapy for primary breast cancer with anthracycline regimens. J Chemother 19(6):716–723. doi:10.1179/joc.2007.19.6.716
del Giglio A, Eniu A, Ganea-Motan D, Topuzov E, Lubenau H (2008) XM02 is superior to placebo and equivalent to Neupogen in reducing the duration of severe neutropenia and the incidence of febrile neutropenia in cycle 1 in breast cancer patients receiving docetaxel/doxorubicin chemotherapy. BMC Cancer 8:332. doi:10.1186/1471-2407-8-332
Kurtin S (2012) Myeloid toxicity of cancer treatment. J Adv Pract Oncol 3(4):209–224
Rivera E, Smith RE Jr (2006) Trends in recommendations of myelosuppressive chemotherapy for the treatment of breast cancer: evolution of the national comprehensive cancer network guidelines and the cooperative group studies. Clin Breast Cancer 7(1):33–41. doi:10.3816/CBC.2006.n.011
Kandalaft LE, Singh N, Liao JB, Facciabene A, Berek JS, Powell DJ Jr, Coukos G (2010) The emergence of immunomodulation: combinatorial immunochemotherapy opportunities for the next decade. Gynecol Oncol 116(2):222–233. doi:10.1016/j.ygyno.2009.11.001
Emens LA (2010) Chemoimmunotherapy. Cancer J 16(4):295–303. doi:10.1097/PPO.0b013e3181eb5066
Galluzzi L, Senovilla L, Zitvogel L, Kroemer G (2012) The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov 11(3):215–233. doi:10.1038/nrd3626
Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G (2013) Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity 39(1):74–88. doi:10.1016/j.immuni.2013.06.014
Demaria S, Volm MD, Shapiro RL, Yee HT, Oratz R, Formenti SC, Muggia F, Symmans WF (2001) Development of tumor-infiltrating lymphocytes in breast cancer after neoadjuvant paclitaxel chemotherapy. Clin Cancer Res 7(10):3025–3030
Hornychova H, Melichar B, Tomsova M, Mergancova J, Urminska H, Ryska A (2008) Tumor-infiltrating lymphocytes predict response to neoadjuvant chemotherapy in patients with breast carcinoma. Cancer Investig 26(10):1024–1031. doi:10.1080/07357900802098165
Jones KL, Buzdar AU (2009) Evolving novel anti-HER2 strategies. Lancet Oncol 10(12):1179–1187. doi:10.1016/S1470-2045(09)70315-8
Minami T, Kijima T, Kohmo S, Arase H, Otani Y, Nagatomo I, Takahashi R, Miyake K, Higashiguchi M, Morimura O, Ihara S, Tsujino K, Hirata H, Inoue K, Takeda Y, Kida H, Tachibana I, Kumanogoh A (2013) Overcoming chemoresistance of small-cell lung cancer through stepwise HER2-targeted antibody-dependent cell-mediated cytotoxicity and VEGF-targeted antiangiogenesis. Sci Rep 3:2669. doi:10.1038/srep02669
Knutson KL, Disis ML (2005) Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother 54(8):721–728. doi:10.1007/s00262-004-0653-2
Wang J, Zhang Q, Jin S, Feng M, Kang X, Zhao S, Liu S, Zhao W (2009) Immoderate inhibition of estrogen by anastrozole enhances the severity of experimental polyarthritis. Exp Gerontol 44(6–7):398–405. doi:10.1016/j.exger.2009.03.003
Pujol JL, Vansteenkiste JF, De Pas TM, Atanackovic D, Reck M, Thomeer M, Douillard JY, Fasola G, Potter V, Taylor P, Bosquee L, Scheubel R, Jarnjak S, Debois M, de Sousa Alves P, Louahed J, Brichard VG, Lehmann FF (2015) Safety and Immunogenicity of MAGE-A3 Cancer Immunotherapeutic with or without Adjuvant Chemotherapy in Patients with Resected Stage IB to III MAGE-A3-Positive Non-Small-Cell Lung Cancer. J Thorac Oncol 10(10):1458–1467. doi:10.1097/JTO.0000000000000653
Acknowledgements
The authors thank the patients who participated in this study and the clinical staff at individual centers without whom the study could not have been performed; the investigators and their clinical teams for their contribution to the study and their support and care to patients. We also thank Urszula Miecielica, PhD for providing medical writing services and Maria Ana de la Grandière, PhD (both XPE Pharma & Science on behalf of GSK Vaccines) for editorial support in preparing this manuscript.
Author contribution
All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. All authors reviewed and commented critical drafts of the manuscript for important intellectual content and gave final approval to submit for publication. MH, GC, VW, MG, PMDSA, FFL, and PG contributed to the study conception and design. MH, GC, SK, GK, MC, TB, PK, VW, MG, PMDSA, LS, SC, FFL, PG, VD, and AF contributed to acquisition of data. MH, GC, SK, MC, TB, MG, PMDSA, FFL, PG, and SC contributed to interpretation of results. MC, PMDSA, and SC provided administrative, technical, and material support. MH, GC, MC, PMDSA, FFL, PG, and SC were involved in study supervision. MH, GC, MC, VW, MG, PMDSA, FFL, PG, and SC performed or supervised the analysis. SK, GK, PAF, MC, TB, PK, LS, VD, AF, and SC provided study materials or subjects. MG and SC provided statistical expertise.
Funding
This work was supported by GlaxoSmithKline Biologicals SA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
MH, SC, LS, GC, GK, MC, AF, PK, SK, and PG have no conflict of interest. TB reports grant, personal fees, and non-financial support from Roche and Novartis outside the submitted work. VD reports personal fees from Lilly for her participation to advisory boards, from Roche Genentech, Novartis, and Pfizer for her participation to advisory boards and symposium, and from GSK for her participation to symposium, outside the submitted work. PAF reports grant from Amgen and Novartis, and personal fees from Amgen, Novartis, Celgene, Pfizer, GSK, and Genomic Health, outside the submitted work. PMDSA and FFL were employees of the GSK group of companies during the conduct of the study. VW and MG are employees of the GSK group of companies. VW and FFL hold shares in the GSK group of companies as part of her/his employee remuneration.
Ethical standards
All procedures performed in this study were in accordance with the ethical standards of the national independent ethics committees and institutional review boards of the study centers. The study was conducted in accordance with Good Clinical Practice and all applicable regulatory requirements, including the Declaration of Helsinki.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Higgins, M., Curigliano, G., Dieras, V. et al. Safety and immunogenicity of neoadjuvant treatment using WT1-immunotherapeutic in combination with standard therapy in patients with WT1-positive Stage II/III breast cancer: a randomized Phase I study. Breast Cancer Res Treat 162, 479–488 (2017). https://doi.org/10.1007/s10549-017-4130-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-017-4130-y