
Abstract
Survivin overexpression, frequently found in breast cancers and others, is associated with poor prognosis. Its dual regulation of cell division and apoptosis makes it an attractive therapeutic target but its exact functions that are required for tumor maintenance are still elusive. Survivin protects cancer cells from genotoxic agents and this ability is generally assigned to a universal anti-apoptotic function. However, a specific role in cancer cell protection from DNA damage has been overlooked so far. We assessed DNA damage occurrence in Survivin-depleted breast cancer cells using γH2AX staining and comete assay. QPCR data and a gene conversion assay indicated that homologous recombination (HR) was impaired upon Survivin depletion. We conducted the analysis of Survivin and HR genes’ expression in breast tumors. We revealed BRCAness phenotype of Survivin-depleted cells using cell death assays combined to PARP targeting. Survivin silencing leads to DNA double-strand breaks in breast cancer cells and functionally reduces HR. Survivin depletion decreases the transcription of a set of genes involved in HR, decreases RAD51 protein expression and impairs the endonuclease complex MUS81/EME1 involved in the resolution of Holliday junctions. Clinically, EME1, RAD51, EXO1, BLM expressions correlate with that of BIRC5 (coding for Survivin) and are of prognostic value. Functionally, Survivin depletion triggers p53 activation and sensitizes cancer cells to of PARP inhibition. We defined Survivin as a constitutive actor of HR in breast cancers, and implies that its inhibition would enhance cell vulnerability upon PARP inhibition.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the last decade, Survivin has attracted considerable attention as a therapeutic target for anticancer strategies because of its dual role in regulating cell division and apoptosis, which are both involved in tumor development [1]. Survivin is a mitotic protein that associates with AuroraB kinase, INCENP and borealin to form chromosomal passenger complex [2] and also play a role in mitotic checkpoint as a sensor of kinetochore-microtubule attachment [3]. Survivin promotes cell survival, especially in cancer cells as part of various molecular networks encompassing major regulators of apoptosis such as caspases, XIAP and its endogenous inhibitor SMAC [4, 5]. Survivin has been consistently identified as a risk-associated gene in various malignancies, carrying unfavorable implications for cancer prognosis, disease recurrence, and abbreviated survival [6]. Of note, Survivin overexpression has been identified in breast cancer cells [7] and together with other gene signatures; this information is now being used in the clinic for the risk assessment of breast cancer patients [8, 9]. Survivin may play a particular role in oncogenic processes involved in, at least, the subset of breast cancers defined as triple negative cancers [10, 11]. Finally, Survivin has been frequently associated with resistance of cancer cells to chemo- or radiotherapy [12, 13].
Dealing with DNA lesions, such as DNA double-strand breaks (DSB), is particularly challenging for cancer cells and their capacity to repair them will finally determine their fate between survival and death upon overwhelming DNA damage. Immediately following the generation of a DSB, a highly conserved DNA damage response (DDR) pathway is activated to ensure cell survival including halt cell cycle progression and repair of the lesion. The molecular DDR begins with the recognition of the DSB and subsequent activation of the PIKK kinases ATM, ATR, or DNA-PK, rapidly followed by the recruitment of early markers such as γH2AX or 53BP1 that will further nucleate the assembly of various effectors. DSB repair relies on two major pathways, homologous recombination (HR) or non-homologous end joining (NHEJ), each with distinct and overlapping roles in maintaining genomic integrity. The phosphorylation of the histone variant H2AX on S139 by activated PIKK kinases is a key initiating step in the DDR, since it creates an epigenetic signal that increases DNA accessibility and leads to the recruitment and accumulation at DNA ends of specific proteins involved in DDR [14]. The choice between the two main DSB repair pathways relies on the initial process of DSB resection which is required for HR but not for NHEJ. DNA resection comprising the DNA end processing by the MRN complex in conjunction with the nuclease EXO1 and auxiliary factors such as 53BP1 or BRCA1, triggers DNA repair by HR pathway. The RAD51 nucleofilaments then mediate homology search and strand invasion in the sister chromatid. After the action of DNA polymerases and ligases, DNA helicase and resolvase enzymes mediate the cleavage and resolution of HR intermediates to yield intact repaired DNA molecules [15].
Previous studies have suggested a link between Survivin and the DDR. Survivin promotes DNA repair after radiation exposure in glioma [12] and decreases radiation-induced DSB in squamous cell carcinoma cancer cells [16]. In the same line, interference with Survivin results in reduced DNA repair [17]. These studies report that Survivin physically interacts with the NHEJ DNA repair complex thus modulating DNA repair after irradiation but whether Survivin participates in DNA repair by HR, and by this way contributes to cell adaptation to genotoxic stress, remains largely unexplored.
We herein investigated a role of Survivin in genome maintenance, by specifically studying DNA repair modulation and its consequences in breast cancer cells depleted in Survivin by RNA interference. Overall, our results indicate that Survivin is required for efficient DNA repair by HR and they imply that it participates in cell adaptation to endogenous genotoxic stress. Importantly, its depletion creates a state of BRCAness (a phenotype that some tumors shared with familial-BRCA cancers [18]) that could be exploited in patients with BRCA-proficient cancer in combining Survivin and PARP1 inhibitors.
Methods
Cell culture
The human breast cancer cell lines MCF7 and MDAMB-231 cell lines were purchased from ATCC (Manassas, VA) and Cal51 cell line from DMSZ (Braunschweig, Germany). They were maintained in DMEM medium (Invitrogen, Cergy Pontoise, France) all supplemented with 5 % FCS, 2 mM l-glutamine.
Reagents, antibodies and treatments
z-VAD-fmk was purchased from Promega (Fitchburg, Wisconsin) and used at 50 μM as 1 h pretreatment prior transfection and maintained during the experiment. Cisplatin and Paclitaxel from Sigma-Aldrich were used at 6 μM and 700 nM, respectively.
Survivin antibody was purchased from R&D Systems (Minneapolis, MN, USA), γH2AX from Upstate (Hampshire, UK), Actin from Chemicon International (Billerica, MA, USA), RAD51 from Genetex (Irvine, CA, USA), MUS81 and EME1 from Abcam (Paris, France), p53 from BD Biosciences (Pont de Claix, France), NOXA from Enzo Life Science (Villeurbanne, France), BAX from Dako (Courtaboeuf, France), p21, Ser15 p53, and PUMA from Cell Signaling (Molsheim, France), CyclinB1 from Santa Cruz (Heidelberg, Germany). A 2 Gray γ-irradiation was performed using the Faxitron CP160 apparatus and cells were harvested 30 min after.
Transfection
RNA interference was performed by transfection using Lipofectamine RNAiMax (Invitrogen, France), according to the manufacturer’s instructions. The following siRNAs were used: control siRNA (D-001810) from Dharmacon (Lafayette, CO, USA), SiRNA Survivin (Ambion), siRNA CDC20 (4392420) from Life Technologies (Carlsbad, CA, USA), siRNA BRCA1 (SMARTpool ON-TARGETplus L-003461), and siRNA AuroraB (SMARTpool ON-TARGETplus L-003326) from Thermo Fisher Scientific (Whaltam, MA, USA).
Plasmids coding for human Survivin or for GFP-fused, c-terminal fragment of 53BP1 containing both TUDOR and BRCT domains’ sequences (pG-AcGFP-53BP1c) were generous gifts from Dr Dario Altieri (Wistar Institute, Philadelphia, PA, USA) and Dr Thomas Von Zglinicki (New Castle University, UK), respectively.
Flow cytometry analysis
For cell death assay, cells were analyzed after immunostaining using the APO2.7 antibody (Beckman Coulter, Villepinte, France), according to the manufacturer’s instructions. For cell cycle analysis, cells were fixed and permeabilized with ethanol 70 % overnight, then stained with propidium iodide (PI). Flow-cytometry analysis was performed on a FACSCalibur using the CellQuestPro software (Becton–Dickinson, San Jose, CA, USA).
Immunoblot analysis
Cells were resuspended in lysis and sonicated as previously described [19]. 50 μg of proteins were loaded for each lane and separated by SDS-PAGE, then electrotransfered to PVDF membranes. Western blot analysis was performed by standard techniques with ECL detection (Bio-Rad, Marne-la coquette, France).
Immunocytochemistry (ICC)
53BP1 expressing Cal51 cells were obtained after transient transfection using the GFP-53BP1c coding plasmid [20]. They were then plated onto glass coverslip and either treated by Cisplatin (6 mM) or transfected with siRNA control or targeting Survivin for 48 h.
For γH2AX staining, cells were seed onto coverslips then transfected with siRNA (control or Survivin) for 48 h. Coverslips were fixed, permeabilized (PBS-1 % SDS 10 min) and blocked. Coverslips were then incubated with the anti-γH2AX antibody and with goat anti-mouse Alexafluor.
Cells were counterstained with DAPI and images were viewed on a Zeiss Axiovert 200 M microscope in the MicroPIcell imaging facility (SFR Bonamy, FED4203/Inserm UMS 016/CNRS 3556, Nantes, France). Images were imported into MetaMorph software and γH2AX scoring was done on the base of a nucleus recognition by DAPI staining and the score was expressed on γH2AX threshold area percentage (mean ± SEM).
Recombination assay
RG37 cell line that contains a single copy of a substrate that specifically monitors gene conversion induced by double-stranded cut targeted by the meganuclease I-SceI was a gift from Bernard Lopez (IGR, Paris, France) [21]. RG37cells were first transfected with a I-Sce-I coding plasmid using jetPEI reagent, then 24 h later, with Survivin, BRCA1, or control siRNA for 48 h. Expression of a functional GFP gene reflecting powerful recombination was then measured by flow cytometry and % of GFP positive cells and data presented herein correspond to six independent experiments.
Single cell electrophoresis comet assay
The comet assay was performed in alkaline or neutral buffer, as previously described [22]. CometSlide™ was visualized using a Leica microscope, and images were analyzed into Comet Assay IV software (Perceptive instruments, Suffolk, UK). The score was expressed on tail moment with arbitrary units.
RT-qPCR
RNA was isolated from cell lines and 500 ng RNA was reverse transcribed as previously described [19]. Quantitative PCR was done using the Maxima SYBR Green/ROX qPCR Master Mix (Life Technologies) and the MX4000 instrument (Stratagene, Basel, Switzerland), according to the manufacturer’s instructions. The endogenous housekeeping genes RPLPO, HPRT1, and ACTB1 were used for normalization. Relative quantification was carried out using the ΔΔCt method.
Gene expression and statistical analysis
Cancer datasets were downloaded from Breast Cancer Gene-Expression Miner v3.1 (http://bcgenex.centregauducheau.fr/BC-GEM/GEM_Accueil.php?js=1) [23, 24].
Statistical analysis
Statistical analysis was performed using paired Student’s t test on GraphPad Prism. Errors bars represent standard errors of mean (SEM). The following symbols are used: *, **, *** that correspond to a p value inferior to 0.05, 0.01, or 0.001, respectively, and ns for non-statistically significant.
Results
Survivin depletion in breast cancer cell lines induces γH2AX activation in response to DSB formation
We first evaluated the impact of Survivin depletion on DNA damage occurrence in the breast cancer cell lines MCF7, MDAMB-231, and Cal51, using the Ser139 phospho-H2AX (γH2AX) marker of DSB either by immunoblot or by immunofluorescence. Survivin depletion clearly increased levels of γH2AX compared to the control condition (siCt) in the three cell lines as did the genotoxic agent cisplatin used as positive control (Fig. 1a). Moreover, γH2AX staining observed upon Survivin depletion, mainly localized in nuclear foci typical of chromatin-associated foci observed in DDR, as observed in γ irradiated cells used as positive control (Fig. 1b). γH2AX activation was also detected in cells transfected with 3 other Survivin siRNA sequences including 2 targeting the BIRC5 3′UTR sequence (Supplementary Fig. 1 and data not shown). Importantly, ectopic Survivin reconstitution performed in rescue experiments using these latter siRNA sequences could prevent Survivin-depleted cells from DNA damage. These results clearly eliminated a potential off-target (Supplementary Fig. 1a). To directly assess DNA breaks, Survivin-depleted cells were further analyzed in a single cell gel electrophoresis comet assay in comparison with siControl cells. As shown in Fig. 1c, Survivin depletion induced comet formation (in either alcali or neutral lysis buffer) and significant increase of the tail moment, in a range comparable to 2 Gray γ-irradiation. Finally, a series of experiments indicate that, the early DNA repair marker 53BP1 localized on nuclear foci in Survivin-depleted cells, as we described above for γH2AX. Indeed, using engineered cells expressing a GFP-fused 53BP1c protein [20], GFP nuclear foci could be evidenced in Survivin-depleted cells compared to control cells, as observed in cisplatin-treated cells (Fig. 1d).

Survivin knockdown induces DNA breaks and DNA damage response in breast cancer cell lines. DNA damage was evaluated in breast cancer cells 48 h after Survivin depletion using siRNA by γH2AX detection by immunoblot (a) and immunocytochemistry (b) and by single cell comet assay (c). a γH2AX and Survivin immunoblot analysis of Cal51 cells (left panel) treated by Cisplatin 6 mM (as positive control, lane 2) or not (untreated, lane 1), and transfected with siRNA control (siControl) (lane 3) or siSurvivin (lane 4). MDAMB-231 cells (middle panel) and MCF7 cells (right panel) depleted in Survivin (siSurv) or not (siCt). γH2AX staining by ICC (b) or single cell gel electrophoresis in alkaline buffer comet assay (c) were performed in 48 h Survivin-depleted or SiCt MDAMB-231 cells. 2 Gy-irradiated cells (b and c) were used as positive control. Representative images are shown (n = 5). Corresponding quantitative comet parameters were calculated using the comet assay software and comet tail moment was shown in c (below panel). d GFP nuclear foci were assessed in GFP-53BP1 expressing Cal51 cells 48 h after Survivin depletion or control. Representative images are shown in the upper panel. % of positive cells based on GFP nuclear foci were counted in each condition. Cisplatin treatment was used as positive control
Eventhough Survivin depletion triggered low if any apoptosis in the experiments, we nevertheless evaluated DNA break occurrence in presence of the pan-caspase inhibitor z-VAD-fmk. This treatment could not significantly prevent DSB formation and arguing against a role of caspases in the generation of DNA breaks (Supplementary Fig. 1b). These data imply that Survivin depletion induces accumulation of DSB impacting on early DDR (since early DNA repair markers are recruited) and independently of an apoptotic process.
Survivin depletion impairs DNA repair by homologous recombination
To gain insight into how Survivin depletion triggers DNA breaks, we examined whether the DNA damage repair is affected in Survivin-depleted cells. We first evaluated the transcription of various genes involved in DNA repair pathways by qPCR after Survivin depletion. We identified six genes over 19, whose transcription was consistently impaired in Survivin-depleted cells, namely EME1, BLM, EXO1, BRCA1, BRCA2, and RAD51 (Fig. 2a). Interestingly, most of them are involved in the homologous recombination (HR) pathway. To directly assess the impact of Survivin depletion on HR, we then used a gene conversion assay based on the RG37 cell line containing a single chromosomally integrated copy of a GFP substrate whose conversion following double-stranded cut targeted by the meganuclease I-Sce-I, monitors the occurrence of HR [21]. We measured the number of GFP positive cells by flow cytometry after transfection of I-Sce-I coding plasmid followed by depletion of Survivin or BRCA1 as positive control, in RG37 cells, and we found that Survivin depletion repressed gene conversion as efficiently as did BRCA1 depletion (Fig. 2b).

Survivin silencing impaired DNA repair by homologous recombination. a qPCR analysis of a set of genes involved in DNA damage repair in Cal51, MDAMB-231, and MCF7 cells depleted or not in Survivin. Data are presented as means (±sem) of ratios normalized to controls from three independent experiments. b HR activity was evaluated by GFP gene conversion assay using the genetically modified RG37 cell line. RG37 cells were first transfected with I-Sce-I coding plasmid, and 24 h later depleted in Survivin or BRCA1 using specific siRNA. After 48 h, % of GFP positive cells was assessed in each condition by flow cytometry and presented as means (±sem) from six independent experiments. c Breast cancer cell lines were depleted in Survivin (SiSurvivin) or not (SiCt) for 48 h and immunoblot analysis were performed to evaluate expression of MUS81/EME1 complex and RAD51 proteins
Because RAD51 and EME1/MUS81 support major activity required in HR, as recombinase and endonuclease, respectively, we focused our study on these proteins. Using immunoblot analysis we pointed out that in both cases, their expression was severely impaired upon Survivin depletion. The RAD51 protein expression decreased upon Survivin depletion consistently with qPCR results (Fig. 2c). In contrast, eventhough qPCR data indicated significant impact of Survivin depletion on EME1 mRNA level, we could not evidence any substantial decrease of EME1 protein level in these cells raising the hypothesis that this protein may be of particular high stability. On the contrary, MUS81 protein (that carries the nuclease activity in the complex) was dramatically reduced upon Survivin depletion in all three cell lines. As qPCR data that did not detect significant modification of MUS81 mRNA in Survivin-depleted cells, this suggests post-translational regulation of MUS81 expression in these cells (Fig. 2c). Collectively, these results strongly argue that Survivin depletion targets both recombinase and nuclease activity required during HR, through at least both modulation of RAD51 and MUS81/EME1 expression.
Prognostic informativity and correlative expression of BIRC5 and HR genes in primary breast tumors
As our data on cell lines suggest that Survivin expression influences the expression of HR genes, we investigated whether the expression of these genes correlate in publically available data using the Breast Cancer Gene-Expression Miner v3.1 microarray database and its correlation module analysis. A gene correlation targeted analysis between all genes identified above, pointed out significative linear dependences between BIRC5, EME1, EXO1, BLM as shown in the correlation map and table corresponding to analysis of all patients, in Fig. 3a and Supplementary Fig. 2a. Altogether, these results from primary breast tumors corroborate those we revealed using breast cancer cell lines, and underscore a positive correlation between the level of expression of BIRC5 and EME1 or RAD51 or EXO1 mRNA.

Gene expression analysis of BIRC5 and HR genes in primary breast tumors. a The correlation map illustrating pairwise correlations among BIRC5, RAD51, EME1, EXO1, BLM, and BRCA1 genes were established using the Breast Cancer Gene-Expression Miner v3.1 web tool. b Kaplan–Meier curves regarding expression of BIRC5, RAD51, EME1, EXO1, BLM, and BRCA1 genes in tumors from patients with breast cancer, were established using the Breast Cancer Gene-Expression Miner v3.1 web tool
Using the same web tool, we also performed a series of gene prognostic meta-analysis including BIRC5, EME1, MUS81, EXO1, RAD51 expression in primary breast tumors applying a Cox univariate method. As suspected, BIRC5 expression has a high prognostic informativity (p < 0.001) as a pejorative gene in a cohort of 2 413 patients with breast cancer. These results, presented as a Kaplan–Meier curves in Fig. 3b, indicate that patients with high BIRC5 expression have a higher risk of metastatic relapse than those with low BIRC5 expression (Hazard Ratio HR 1.64 with 95 %CI 1.40–1.92). Interestingly, RAD51, EME1, EXO1, BLM, and BRCA1 gene expression also significantly separate cohorts of breast tumors in patients with low or high risk of metastatic relapse (Fig. 3b). In contrast, MUS81 gene expression revealed no prognostic informativity (Supplementary Fig. 2b).
AuroraB depletion more faithfully recapitulates the effects of Survivin depletion than that of CDC20
In an attempt to determine whether its function as member of the CPC or its capacity to block cell cycle in G2/M phase, play a role in Survivin effect on HR, we compared the effects of its depletion to those of AuroraB or CDC20 depletion. Cell cycle analysis indicates that Survivin depletion leads to the accumulation of cycling cells in G2/M phase, as observed in AuroraB-depleted cells (Fig. 4a). CDC20 depletion also increases cells in G2/M phase [19]. However, in the case of CDC20 depletion, cells were blocked in mitosis with accumulation of CyclinB1 protein and CDK1 activity assessed by MPM-2 staining (Fig. 4b and [19]). In contrast, Survivin or AuroraB-depleted cells mainly exited mitosis without division, in relation with cytokinesis failure (data not shown). Expression of HR-related genes, including EME1, EXO1, BLM, BRCA1, RAD51, MUS81 was quantified by qPCR analysis in each conditions for the three cell lines and presented as a heatmap in Fig. 4c. These results show that AuroraB depletion more closely mimics the effects of Survivin depletion on gene expression than that of CDC20. However, since AuroraB depletion potently decreases BIRC5 expression and vice versa, dissociating their respective functions is difficult. Collectively, these results indicate that HR defects detected in Survivin-depleted cells better coincides with cytokinesis failure, also observed in AuroraB-depleted cells, than with mitotic block, as induced by CDC20 depletion.

AuroraB depletion partially recapitulates the effects of Survivin depletion on HR genes' expression a Cell cycle progression was assessed by PI staining and flow cytometry analysis, in Cal51 and MDAMB-231 cells upon Survivin (SiSurvivin), AuroraB (SiAuroraB) depletion. Data are presented as % of cells in cell cycle phases, obtained at 1, 2, and 3 days after RNA interference. b CyclinB1 expression was studied by immunoblot in Survivin (SiSurvivin), AuroraB (SiAuroraB), or CDC20 (SiCDC20) depleted cells. c Expression of HR genes, BIRC5, AURKB, and CDC20 was assessed by qPCR after Survivin (SiSurvivin), AuroraB (SiAuroraB), or CDC20 (SiCDC20) depletion. Data are presented as means of ratios obtained using data from control condition (SiCt) in a heatmap. The green scale indicates decreased mRNA, and the red scale increased mRNA. Statistical significativity was indicated as following ***p < 0.001, **p < 0.05, *p < 0.01. (ns not significative)
Survivin depletion initiates a p53 response and sensitizes breast cancer cells to PARP inhibitors
Since DNA damage accumulates upon Survivin depletion, we evaluate whether this coincides with an activation of a typical p53 response. In unmutated TP53 cancer cells (Cal51 and MCF7), Survivin depletion led to both p53 protein accumulation and its S15 phosphorylation, as it could be detected under paclitaxel or cisplatin genotoxic treatment (Fig. 5a). An increased transcription of p53 targets, BAX, BBC3 (PUMA), PMAIP1 (NOXA), and CDKN1A (p21) was detected at mRNA levels as well as protein levels (except for NOXA) (Fig. 5a–b). These results argue for the activation of the p53 pathway in response to DNA damage triggered by Survivin depletion.

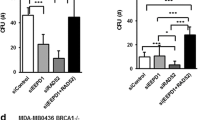
Survivin depletion triggers an active p53 response in p53 proficient cells and modulates response to PARP inhibitors. a and b DNA damage response including BAX (BAX), NOXA (PMAIP1), p21(CDKN1A), PUMA (BBC3) p53 targets, was evaluated in Cal51 cells, 48 h after Survivin depletion (SiSurvivin) or not (SiCt), using either qPCR (a) or immunoblot analysis, this latter including Paclitaxel (700 nM) or Cisplatin (6 mM) treatments as positive controls (b). c Survivin depletion sensibilized cancer cells to the PARP inhibitor Olaparib. MCF7, MDAMB-231 and Cal51 cells were depleted in Survivin (SiSurvivin) or in BRCA1 (SiBRCA1), as previously described, and treated with olaparib at the indicated concentrations, for 4–6 days. Cell death assays were then performed and analyzed by flow cytometry
Since HR defect may lead to PARP dependency for DNA repair and subsequently survival, we further evaluated whether Survivin silencing modifies breast cancer cells sensitivity to PARP inhibitors such as olaparib. Of major importance, combining Survivin depletion and olaparib treatment led to significantly increased cell death in breast cancer cells (Fig. 5c). As expected, this synergic effect was also detectable when olaparib treatment was combined to BRCA1 depletion.
Discussion
Survivin regulates multiple intersecting pathways required for tumor maintenance. Our data show for the first time a role for Survivin in endogenous DNA damage repair by HR in cancer cells. We indeed observed that Survivin participates in the control of HR network, by modulating protein expression of both main HR actors, the recombinase RAD51 and the nuclease MUS81. Our results are consistent with the results of a genome-wide siRNA screen performed in Hela cells that identified Survivin depletion among other targets, as one of the cause of γH2AX activation [25]. Importantly, our results further revealed that Survivin is required for efficient DNA repair by HR since its depletion not only decreased the expression of major actors of DNA repair by HR but also impaired the gene conversion necessary to its onset. Recent studies suggest that Survivin modulates the repair of radiation-induced DSB in supporting the recruitment of repair proteins to the site of DNA damage and interacts with the NHEJ pathway [17]. Collectively, these observations highlight the capability of Survivin to contribute to DNA repair in cancer cells. In this report, we identified the recombinase RAD51 and the endonuclease EME1/MUS81 as major targets controlled by Survivin levels. Interestingly, the endonuclease activity of the complex is usually regulated by phosphorylation of its regulatory subunit EME1 [26]. We provide evidence in this report that the protein MUS81 can also undergo a strong control of its expression upon Survivin depletion. To our knowledge, ubiquitination is the sole modification of MUS81 that has been reported so far, and this in the specific context of HIV infection [27]. Since it also plays a fundamental role in promoting correct chromatid separation and mitotic progression [28] defining which mechanisms govern its expression during cell cycle would be important to better understand its role in DNA repair and mitosis progression.
Importantly, meta-analysis of transcriptomic data from patients with primary breast tumors also reveals not only that BIRC5 expression significantly correlates with expression of EME1, EXO1, BLM, RAD51, or BRCA1 genes (HR gene cluster), but also that high expression of these genes independently associate with pejorative prognosis. In addition, as BIRC5 expression also aggregates within a proliferation gene cluster [8], our results suggest that Survivin stands at the cross-road of proliferation and maintenance of genome integrity. Interestingly, eventhough Survivin does not contain a recognizable DNA binding domain, its ortholog BIR-1 in C. Elegans can regulate gene transcription through its effect on histone phosphorylation [29]. It is in addition puzzling that Survivin can specifically bind to phosphorylated Histone3 during mitosis [30]. Finally Tang and colleagues proposed that Survivin acts as a transcription factor or cofactor since they observed that it binds to CDKN1A promoter where it interacts with p53 [31]. Thus, this is tempting to speculate that Survivin may regulate HR genes’ transcription. Further experiments are needed to validate this hypothesis.
Of note, DSB formation and HR repression upon Survivin depletion occurred in cells independently of their p53 status. Moreover, Survivin depletion triggered a p53 response in proficient cells. This, coinciding with HR repression, excludes a global gene transcription repression upon Survivin silencing.
Finally, we pointed out that Survivin-depleted cells were prone to cell death when treated by the PARP inhibitor olaparib, in the same extent as in BRCA1-depleted cells. We thus provide evidence that Survivin depletion induces an operative BRCAness phenotype in breast cancer cells and we propose that combining Survivin targeting with PARP inhibitors may improve breast cancer therapy and may benefit to patients without inherited BRCA1/2 mutations.
Prognosis and clinical significance of Survivin in breast cancers have been recently evaluated based on 23 articles, revealing overall a higher risk of recurrence and decreased OS rates in patients with higher Survivin expressing tumors [32]. In addition, Survivin expression may be predictive of response to therapy based on various preclinical studies using organotypic human breast tumors for doxorubicin treatment [33] or breast cancer cell lines whose in vitro drug sensitivity was restored by the decrease of Survivin expression induced by miR-218 targeting [34]. Survivin also contributes to radioresistance in various preclinical models of cancers [12, 35, 36]. In this way, clinical use of so-called Survivin suppressors such as YM155, currently in clinical trials in cancer therapy [37] or in development [38], already indicate that YM155 could sensitize cancer cells to ionizing irradiation [39]. Thus, eventhough recent reports (reviewed [37]) and our results [40] suggest that Survivin may not be the only target of this potent cytotoxic agent, it would be interesting to evaluate YM155 activity relies at least in part in induction of HR deficiency.
References
Altieri DC (2008) New wirings in the Survivin networks. Oncogene 27:6276–6284
Ruchaud S, Carmena M, Earnshaw WC (2007) Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol 8:798–812
Lens SM, Wolthuis RM, Klompmaker R, Kauw J, Agami R, Brummelkamp T, Kops G, Medema RH (2003) Survivin is required for a sustained spindle checkpoint arrest in response to lack of tension. EMBO J 22:2934–2947
Dohi T, Xia F, Altieri DC (2007) Compartmentalized phosphorylation of IAP by protein kinase A regulates cytoprotection. Mol Cell 27:17–28
Song Z, Yao X, Wu M (2003) Direct interaction between Survivin and Smac/DIABLO is essential for the anti-apoptotic activity of Survivin during taxol-induced apoptosis. J Biol Chem 278:23130–23140
Altieri DC (2008) Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer 8:61–70
Brennan DJ, Rexhepaj E, O’Brien SL, McSherry E, O’Connor DP, Fagan A, Culhane AC, Higgins DG, Jirstrom K, Millikan RC, Landberg G, Duffy MJ, Hewitt SM, Gallagher WM (2008) Altered cytoplasmic-to-nuclear ratio of Survivin is a prognostic indicator in breast cancer. Clin Cancer Res 14:2681–2689
Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MG, Watson D, Park T, Hiller W, Fisher ER, Wickerham DL, Bryant J, Wolmark N (2004) A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 35:2817–2826
Jézéquel P, Loussouarn D, Guérin-Charbonnel C, Campion L, Vanier A, Gouraud W, Lasla H, Guette C, Valo I, Verrièle V, Campone M (2015) Gene-expression molecular subtyping of triple-negative breast cancer tumours: importance of immune response. Breast Cancer Res 17:43
Lee CW, Raskett CM, Prudovsky I, Altieri DC (2008) Molecular dependence of estrogen receptor-negative breast cancer on a notch-Survivin signaling axis. Cancer Res 68:5273–5281
Wang RH, Zheng Y, Kim HS, Xu X, Cao L, Luhasen T, Lee MH, Xiao C, Vassilopoulos A, Chen W, Gardner K, Man YG, Hung MC, Finkel T, Deng CX (2008) Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol Cell 32:11–20
Chakravarti A, Zhai GG, Zhang M, Malhotra R, Latham DE, Delaney MA, Robe P, Nestler U, Song Q, Loeffler J (2004) Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms. Oncogene 23:7494–7506
Lu J, Tan M, Huang WC, Li P, Guo H, Tseng LM, Su XH, Yang WT, Treekitkarnmongkol W, Andreeff M, Symmans F, Yu D (2009) Mitotic deregulation by Survivin in ErbB2-overexpressing breast cancer cells contributes to Taxol resistance. Clin Cancer Res 15:1326–1334
Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y (2008) gammaH2AX and cancer. Nat Rev Cancer 8:957–967
Krejci L, Altmannova V, Spirek M, Zhao X (2012) Homologous recombination and its regulation. Nucleic Acids Res 40:5795–5818
Jiang G, Ren B, Xu L, Song S, Zhu C, Ye F (2009) Survivin may enhance DNA double-strand break repair capability by up-regulating Ku70 in human KB cells. Anticancer Res 29:223–228
Reichert S, Rödel C, Mirsch J, Harter PN, Tomicic MT, Mittelbronn M, Kaina B, Rödel F (2011) Survivin inhibition and DNA double-strand break repair: a molecular mechanism to overcome radioresistance in glioblastoma. Radiother Oncol 101:51–58
Turner N, Tutt A, Ashworth A (2004) Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer 4:814–819
Bah N, Maillet L, Ryan J, Dubreil S, Gautier F, Letai A, Juin P, Barillé-Nion S (2014) Bcl-xL controls a switch between cell death modes during mitotic arrest. Cell Death Dis 5:e1291
Nelson G, Buhmann M, von Zglinicki T (2009) DNA damage foci in mitosis are devoid of 53BP1. Cell Cycle 8(20):3379–3383
Laulier C, Barascu A, Guirouilh-Barbat J, Pennarun G, Le Chalony C, Chevalier F, Palierne G, Bertrand P, Verbavatz JM, Lopez BS (2011) Bcl-2 inhibits nuclear homologous recombination by localizing BRCA1 to the endomembranes. Cancer Res 71:3590–3602
Supiot S, Gouard S, Charrier J, Apostolidis C, Chatal JF, Barbet J, Davodeau F, Cherel M (2005) Mechanisms of cell sensitization to alpha radioimmunotherapy by doxorubicin or paclitaxel in multiple myeloma cell lines. Clin Cancer Res 11:7047s–7052s
Jézéquel P, Frénel JS, Campion L, Guérin-Charbonnel C, Gouraud W, Ricolleau G, Campone M (2013) bc-GenExMiner 3.0: new mining module computes breast cancer gene expression correlation analyses. Database (Oxford) bas060
Jézéquel P, Campone M, Gouraud W, Charbonnel C, Leux C, Ricolleau G, Campion L (2012) bc-GenExMiner: an easy-to-use online platform for gene prognostic analyses in breast cancer. Breast Cancer Res Treat 131:765–775
Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, Meyer T, Cimprich KA (2009) A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 35:228–239
Minocherhomji S, Hickson ID (2014) Structure-specific endonucleases: guardians of fragile site stability. Trends Cell Biol 24:321–327
Laguette N, Brégnard C, Hue P, Basbous J, Yatim A, Larroque M, Kirchhoff F, Constantinou A, Sobhian B, Benkirane M (2014) Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell 156:134–145
Naim V, Wilhelm T, Debatisse M, Rosselli F (2013) ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat Cell Biol 15:1008–1015
Kostrouchova M, Kostrouch Z, Saudek V, Piatigorsky J, Rall JE (2003) BIR-1, a Caenorhabditis elegans homologue of Survivin, regulates transcription and development. Proc Natl Acad Sci USA 100:5240–5245
Wang F, Dai J, Daum JR, Niedzialkowska E, Banerjee B, Stukenberg PT, Gorbsky GJ, Higgins JM (2010) Histone H3 Thr-3 phosphorylation by Haspin positions Aurora B at centromeres in mitosis. Science 330:231–235
Tang L, Ling X, Liu W, Das GM, Li F (2012) Transcriptional inhibition of p21WAF1/CIP1 gene (CDKN1) expression by Survivin is at least partially p53-dependent: evidence for Survivin acting as a transcription factor or co-factor. Biochem Biophys Res Commun 421:249–254
Li Y, Ma X, Wu X, Liu X, Liu L (2014) Prognostic significance of survivin in breast cancer: meta-analysis. Breast J 20(5):514–524
Faversani A, Vaira V, Moro GP, Tosi D, Lopergolo A, Schultz DC, Rivadeneira D, Altieri DC, Bosari S (2014) Survivin family proteins as novel molecular determinants of doxorubicin resistance in organotypic human breast tumors. Breast Cancer Res 16(3):R55
Hu Y, Xu K, Yagüe E (2015) miR-218 targets survivin and regulates resistance to chemotherapeutics in breast cancer. Breast Cancer Res Treat 151(2):269–280
Rödel C, Haas J, Groth A, Grabenbauer GG, Sauer R, Rödel F (2003) Spontaneous and radiation-induced apoptosis in colorectal carcinoma cells with different intrinsic radiosensitivities: survivin as a radioresistance factor. Int J Radiat Oncol Biol Phys 55:1341–1347
Lu B, Mu Y, Cao C, Zeng F, Schneider S, Tan J et al (2004) Survivin as a therapeutic target for radiation sensitization in lung cancer. Cancer Res 64:2840–2845
Rauch A, Hennig D, Schäfer C, Wirth M, Marx C, Heinzel T, Schneider G, Krämer OH (2014) Survivin and YM155: how faithful is the liaison? Biochim Biophys Acta 1845:202–220
Ling X, Cao S, Cheng Q, Keefe JT, Rustum YM, Li F (2012) A novel small molecule FL118 that selectively inhibits Survivin, Mcl-1, XIAP and cIAP2 in a p53-independent manner, shows superior antitumor activity. PLoS One 7:e45571
Iwasa T, Okamoto I, Suzuki M, Nakahara T, Yamanaka K, Hatashita E, Yamada Y, Fukuoka M, Ono K, Nakagawa K (2008) Radiosensitizing effect of YM155, a novel small-molecule Survivin suppressant, in non-small cell lung cancer cell lines. Clin Cancer Res 14:6496–6504
Véquaud E, Séveno C, Loussouarn D, Engelhart L, Campone M, Juin P, Barillé-Nion S (2015) YM155 potently triggers cell death in breast cancer cells through an autophagy-NF-kB network. Oncotarget 6(15):13476–13486
Acknowledgments
We thank Dr Dario Altieri and Dr Thomas Von Zglinicki for providing us plasmids coding for human Survivin and for GFP-fused 53BP1, respectively. We thank Bernard Lopez (UMR8200, IGR, Paris) for fruitful discussions.
Authors’ contributions
EV performed and analyzed most of the molecular studies and has been involved in the design and the interpretation of data. GD performed the comet assays. PJezequel carried out the transcriptomic analysis. PJuin participated in the design of the study and helped to draft the manuscript. SBN conceived the study and drafted the manuscript. All authors read and approved the final manuscript.
Grant Support
This work was supported by the Ligue interrégionale contre le Cancer (comités Charentes, Loire-Atlantique and Vendée).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Véquaud, E., Desplanques, G., Jézéquel, P. et al. Survivin contributes to DNA repair by homologous recombination in breast cancer cells. Breast Cancer Res Treat 155, 53–63 (2016). https://doi.org/10.1007/s10549-015-3657-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-015-3657-z