
Abstract
While biosimilars of low molecular-weight biologics such as G-CSF have been available in Europe since 2006, biosimilars of monoclonal antibodies (mAbs) have only become available in the last year. Unlike G-CSF, mAbs are large and complex and often play a direct role in the survival of patients with life-threatening illnesses such as breast cancer. Several biosimilars are currently under development for the treatment of breast cancer, and the use of biosimilars in a setting that directly impacts patient survival raises a number of questions. In this review, we discuss the biosimilar mAbs currently in development for the treatment of breast cancer. We provide an overview of the European Medicine Agency guidelines and historic data on the development of biosimilars in order to discuss the development of biosimilar mAbs for breast cancer. Biosimilars offer a highly attractive path toward reducing the cost of medical care and should be pursued with great interest. However, for agents used to treat life-threatening diseases such as cancer, a cautious approach must be taken to ensure that there is no negative impact on patient care. Clinical trials for biosimilar mAbs must be carried out in an appropriately sensitive patient population using endpoints that can accurately demonstrate both the similarity of the biosimilar and its efficacy in the indication. Due to the abbreviated approval pathway, rigorous pharmacovigilance must be in place once a biosimilar mAb is approved in order to ensure its long-term safety and efficacy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The emergence of biosimilar antibodies in oncology
The discovery of the HER2 proto-oncogene and the development of the HER2-targeted antibody trastuzumab (Herceptin®, Genentech) more than two decades ago represent landmark achievements in the treatment of breast cancer. Prior to trastuzumab, women with HER2-positive breast cancer had few treatment options and progressed rapidly. The introduction of trastuzumab in previously untreated patients with metastatic disease resulted in a 4.8 month increase in median overall survival (OS) [1]. Women with HER2-positive metastatic breast cancer now have survival rates similar to patients with hormone receptor–positive breast cancer, a disease that historically had a more favorable prognosis. Trastuzumab has since been approved for use in many indications, including neoadjuvant and adjuvant breast cancer. In early breast cancer, one year of treatment with adjuvant trastuzumab with chemotherapy results in a statistically significant reduction in the risk of disease recurrence by as much as 48 % in some trials [2, 3]. Since its approval, trastuzumab has become the standard of care for patients with HER2-positive breast cancer.
In 2014, the patent exclusivity rights for trastuzumab will expire in Europe, opening the door for the creation of copy versions. Unlike small-molecule drugs such as aspirin and tyrosine kinase inhibitors, which are produced via chemical synthesis, trastuzumab belongs to a unique class of agents known as biologics. Biologics are complex drugs that are derived from living organisms such as bacterial and eukaryotic cells [4]. Because of the size and complexity of biologics and the variability introduced during production, it is impossible to make an identical copy, or generic version, of a biologic. Instead, copies of biological medicines are known as “biosimilars,” a term that highlights the fact that they are similar to the reference products but not entirely identical. Importantly, only copies of biologics that have undergone a comparability exercise and have been approved by a regulatory body can be called “biosimilar” [5].
The first biosimilars introduced in Europe were biosimilar somatropins in 2006. These were followed by biosimilar erythropoietins in 2007 and biosimilar filgrastims starting in 2008 [6]. Until recently, only biosimilars of these lower molecular-weight biologics were available in Europe. This changed in September 2013 when the European Commission granted marketing authorization for two biosimilars of the anti-tumor necrosis factor alpha (TNF-α) antibody infliximab [7]. This represents the first time a biosimilar of a monoclonal antibody (mAb) has been approved by a regulatory body. Currently, several biosimilar versions of trastuzumab are under development. It is expected that approval of a biosimilar trastuzumab may come as early as 2014. If so, biosimilar trastuzumab will be the first biosimilar mAb available for the treatment of cancer. The development of biosimilar trastuzumab represents a unique and exciting opportunity in the field of breast cancer. In this review, we will explore the development and approval of biosimilar trastuzumab and discuss in detail a number of issues relevant to breast oncologists when considering biosimilar trastuzumab for their patients.
Pathway to biosimilar trastuzumab approval
Guidelines for biosimilar mAb development
Biosimilars are approved on the basis of a regulatory pathway different from both generics and originators. Because biosimilars are the copies of molecules that have already been approved through a rigorous clinical trial program, the dossiers for their approval are reduced compared to those of the originators. However, because they are not identical to the reference products, biosimilars require more thorough testing than generics. The European Medicines Agency (EMA) has issued several guidelines regulating the development of biosimilars, including guidelines for the development and testing of biosimilar mAbs [8, 9]. Prior to being approved, a biosimilar must demonstrate comparability to a reference product in terms of quality characteristics, biological activity, safety, and efficacy. This is achieved through a stepwise comparability exercise that includes in vitro analytical testing, non-clinical comparative testing, and one or more clinical trials [8].
A key component of the biosimilarity exercise is an accelerated clinical trial program in which the pharmacokinetics, clinical efficacy, clinical safety, and immunogenicity of the biosimilar are compared to that of the originator. According to the EMA, the goal of the clinical trial program is to “demonstrate similar efficacy and safety compared to the reference medicinal product, not patient benefit per se, which has already been established by the reference medicinal product” [8]. Throughout the clinical trial program, all testing must be done in a sensitive and homogenous patient population so that any differences between the biosimilar and the originator can be easily detected. Likewise, the clinical endpoint for these trials should be sensitive to the detection of product-related differences. The EMA recommends overall response rate (ORR) or complete response (CR) rate as endpoints for clinical efficacy studies of biosimilar mAbs in oncology, as these endpoints are less likely than survival endpoints to be influenced by factors such as previous lines of therapy and tumor burden [8]. Very often biosimilar clinical trials are carried out for only one or two of the reference drugs’ indications. The expectation is that if data from these trials are robust and there is adequate justification, the biosimilar may be approved for indications of the reference medicinal product in which it has not been tested. Extrapolation of indications is essential to the biosimilar concept and has led to much controversy, as we will discuss in greater detail.
Clinical trials of biosimilar trastuzumab
The first biosimilar mAbs were approved in Europe in 2013, and several biosimilar trastuzumab candidates are currently in development. Of these, CT-P6, from the Korean manufacturer Celltrion, is the furthest along in the clinical development pathway. CT-P6 in combination with paclitaxel has demonstrated comparability to Herceptin plus paclitaxel in a pooled analysis of data from a phase I/IIb pharmacokinetics study [10] and a phase III efficacy and safety study [11] in women with HER2-positive metastatic breast cancer. CT-P6 demonstrated equivalent pharmacokinetics and a similar safety profile to Herceptin in the phase I/IIb study [10]. In the phase III clinical efficacy and safety trial, 475 women with previously untreated HER2-positive metastatic breast cancer received either CT-P6 or Herceptin, both in combination with paclitaxel. The primary endpoint was ORR by independent review with a predefined equivalence margin of 15 %. Time to treatment failure (TTF) and cardiotoxicity as measured by LVEF were secondary endpoints. In the pooled analysis of the phase I/IIb and phase III studies (Table 1) [11], the primary endpoint of equivalent ORR at cycle 8 was met, with 56.6 % of patients responding to CT-P6 compared with 61.9 % of patients receiving Herceptin. Time to treatment failure in responding patients was 11.07 versus 12.52 months (P = 0.0978). Time to treatment failure data were not presented for the intent-to-treat population. CT-P6 had a toxicity profile similar to that of Herceptin with no statistically significant differences. To date, no data on immunogenicity have been released. On the basis of these data, Celltrion has submitted CT-P6 for approval in Korea [12] and is expected to do so in Europe in 2014.
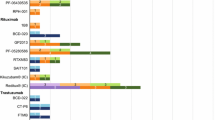
Though CT-P6 will likely be the first biosimilar trastuzumab approved in Europe, there are several other biosimilar candidates close behind in development. Of particular interest is the Pfizer biosimilar candidate PF-05280014, which was compared to Herceptin in a phase I pharmacokinetics study in healthy male volunteers. In this trial, PF-05280014 demonstrated comparability to Herceptin on the basis of serum concentration, area under the serum concentration–time curve and secondary pharmacokinetic parameters. There were similar incidences of adverse events in each treatment arm and no unexpected immunogenicity [13]. A phase III trial comparing PF-05280014 to Herceptin, both in combination with paclitaxel, in women with HER2-positive metastatic breast cancer is planned [14]. Several other biosimilar trastuzumab candidates are also in phase III trials (Table 2) [11, 14–18].
Considerations for biosimilar trastuzumab development
Patient population
According to the EMA guidelines for biosimilar mAbs, clinical trials must be carried out in a sufficiently sensitive and homogenous population [8]. For trastuzumab biosimilars tested in breast cancer, the metastatic setting may not be a sufficiently sensitive and homogenous population. Metastatic disease is a highly heterogeneous state that can vary based on prior treatment, line of therapy, disease burden, comorbidities, location of metastasis, and molecular phenotype of metastatic cells. Because common breast cancer treatments such as chemotherapy and radiotherapy are associated with an immunosuppressive effect [19, 20], women with metastatic disease are more likely to be immunologically impaired. These women also have a greater risk of developing secondary cancers as a result of previous therapy [21, 22].
While breast cancer is by nature a highly heterogeneous disease, early breast cancer represents a far more sensitive and homogeneous population in which to carry out clinical trials of a biosimilar trastuzumab. At this stage patients have received the same treatments, have a reduced disease burden, and do not suffer the adverse events associated with treatments received in later lines of therapy. Because the goal of a biosimilar clinical trial is to detect any differences between the biosimilar and the originator, the heterogenous nature of metastatic disease, the risk for secondary tumors, and the potential for immune impairment make patients with metastatic breast cancer a poor population for biosimilar clinical trials. Clinical testing of biosimilar trastuzumab in patients with early breast cancer will allow for a more careful and thorough biosimilarity assessment.
Clinical trial endpoints
A second challenge for clinical trials of biosimilar trastuzumab is the selection of clinical trial endpoints. While survival is generally a preferred endpoint in oncology clinical trials, survival endpoints may not be appropriate for a biosimilar comparability trial as they can be influenced by confounding factors such as tumor burden, disease status, and previous lines of therapy. The EMA suggests response as an endpoint for biosimilar trials [8]. Because trastuzumab directly impacts patient survival, many oncologists may feel uncomfortable using a biosimilar version that has not demonstrated a survival benefit in clinical trial. While ORR may be a preferable endpoint for detecting differences between products, it is not always associated with long-term improvements in patient outcome [23, 24]. Pathologic complete response (pCR), however, has been shown to correlate closely with improvements in disease-free survival (DFS) and OS in patients with early breast cancer [25]. Use of pCR as a primary endpoint in early breast cancer follows EMA recommendations both for a response endpoint and for clinical testing in a sensitive and homogenous population. A good practice would be to carry out biosimilar trastuzumab trials using these parameters with long-term survival as a secondary endpoint.
Considerations for clinical practice
Extrapolation
Extrapolation of a biosimilar to indications for which it was not tested during the clinical trial program is common practice in Europe. Many of the currently available biosimilars were approved in every indication of the originator after testing in a single indication. For the recently approved biosimilar mAbs Remsima™ (Celltrion) and Inflectra™ (Hospira), approval was granted for all indications of the originator Remicade® (infliximab, Janssen) after a phase I trial in ankylosing spondylitis and a phase III trial in rheumatoid arthritis. Extrapolated indications include ulcerative colitis, Crohn’s disease, psoriatic arthritis, and plaque psoriasis [26, 27]. According to the EMA guidelines, extrapolation of biosimilar data to other indications of the originator is allowed, provided the mechanism of action is the same in each indication and/or adequate scientific justification can be provided based on the totality of evidence, including experience gained with the reference product and good clinical efficacy and safety data for the biosimilar [8]. The precise mechanism of action for trastuzumab is unknown. Several putative mechanisms of action for trastuzumab have been identified, and it may be that any combination of these mechanisms results in the antibody’s efficacy. It is also likely that these mechanisms may contribute differently for each indication of trastuzumab [28].
Indication extrapolation forms the basis of the biosimilar concept, and without it there would be minimal financial benefit associated with biosimilars [29]. Current opinion regarding extrapolation of indications for biosimilar mAbs holds that if the clinical efficacy and immunogenicity testing are done in the most sensitive patient population, it would be appropriate to extrapolate to other indications of the reference antibody [30]. For HER2-positive breast cancer, this would mean clinical testing of a biosimilar trastuzumab in the adjuvant or neoadjuvant setting, with extrapolation to metastatic breast cancer. The converse, a biosimilar tested in the metastatic setting extrapolated to early breast cancer, would not be acceptable. As discussed above, the metastatic setting does not allow for appropriate evaluation of immunogenicity signals because many patients with metastatic breast cancer are immune compromised. Extrapolating a biosimilar trastuzumab tested in metastatic breast cancer to early breast cancer means an increased risk of unpredictable immune responses that could reduce efficacy and increase adverse events. A trial in the neoadjuvant setting is highly recommended in order to justify the use of biosimilar trastuzumab in the adjuvant setting.
Labeling
All biologic manufacturers must submit a pharmacovigilance plan as part of the marketing authorization application. This plan comprises pre- and post-authorization immunogenicity testing, a risk management plan based on safety issues identified during the clinical trials, and post-marketing safety commitments such as targeted questionnaires, phase IV studies, and specialized follow-up for long-term use [9, 31]. The goal of this plan is to identify any product-associated safety risks not observed during clinical testing and provide a framework to rapidly report and manage such incidences.
Central to the pharmacovigilance plan is the need to be able to accurately trace which medicines a patient is given. Many medicines are prescribed by international nonproprietary name (INN), which provides information regarding the composition and type of drug. For biologics and biosimilars, the situation is more complicated. Like generics, biosimilars are given the same INN as the originator [32]. While generics are identical to their originators, and in many cases can be used interchangeably, biosimilars are not. When tracking the adverse events and other safety concerns associated with a biosimilar or biologic, it is of utmost importance that the appropriate drug is identified. Thus, prescription by brand name is recommended by several regulatory agencies within Europe [33, 34] and by the EMA. Because of this, in 2013, the summary of product characteristics, or label, of two biologic products was altered to include the statement “in order to improve the traceability of biological medicinal products, the trade name of the administered product should be clearly recorded (or stated) in the patient file” [35, 36]. It is expected that this statement will be added to other biologic labels in the future, including Herceptin’s.
The contents of the Herceptin label are of interest with regard to biosimilar trastuzumab, because biosimilars currently receive very similar labels to those of their originators [37]. The label for a biosimilar product may not identify that it is a biosimilar, what clinical studies have been done to validate its comparability, or which indications are extrapolated. This is worrisome, as it is essential that, when administering any drug to a patient, a clinician have as much information as possible about that. Information on comparability trial data and extrapolation can be found in a biosimilar medicine’s European Public Assessment Report. However, this is difficult to access and interpret, and many community physicians are not fully aware of the information contained in this document. The label for each drug is the primary source of information for the practicing clinician and should be as clear and complete as possible. Because of this, we recommend the label for any approved biosimilar trastuzumab be altered to identify that the product is a biosimilar. Importantly, the clinical trial data to justify comparability and extrapolation should be included. The document should also be updated with results of the post-approval pharmacovigilance activities once this information is available [30].
Interchangeability, substitution, and switching
Interchangeability has been an issue of serious concern for biosimilars since the approval of the first biosimilar medicines nearly a decade ago. Because generic medicines are therapeutically equivalent to their originators, they are often considered interchangeable with the originators. Because biosimilars are not therapeutically equivalent to their originators, many physicians feel strongly that biosimilars should not be considered interchangeable. Others argue that interchangeability is essential to the incorporation of biosimilars into clinical practice and is an obvious consequence of the biosimilarity exercise [38]. Currently, each country in the European Union (EU) is allowed to decide individually which medicines are interchangeable.
Interchangeability becomes an issue when the traceability of a biologic product is compromised through automatic substitution or switching. When products are interchangeable, the patient may receive either the biosimilar or the originator, regardless of what the physician has prescribed and recorded. If the patient later develops an adverse reaction to the drug, the physician may incorrectly attribute the source because of this automatic substitution. It is also possible that the patient may be switched back and forth between the biosimilar and the originator, either due to poor record keeping as a result of automatic substitution or because the drugs are considered interchangeable. Switching between two similar biologic drugs increases the risk of anti-drug antibodies, which can lead to adverse immunologic reactions and decreased drug efficacy. Because the patient has received multiple drugs, the origin of these adverse events cannot be traced.
There is no EU-wide policy on automatic substitution. Automatic substitution is regulated at the national level and varies by country [39]. Currently no country has passed legislation allowing this practice with biologics, and many have specifically prohibited it. We agree with this action, as we feel that automatic substitution and switching with biosimilars are risky behaviors that preclude successful pharmacovigilance activities. A biosimilar trastuzumab will never be therapeutically equivalent to the originator, and so these medicines should not be considered interchangeable. Because trastuzumab is used to prolong survival in patients with a fatal disease, it is irresponsible to take risks that might impair the efficacy and safety of the patients’ treatment.
The future of biosimilar trastuzumab in Europe
The eventual approval of a biosimilar trastuzumab in Europe is certain and is not an outcome to be feared or prevented. Less expensive treatments for patients and increased competition for innovators will ultimately improve the state of breast cancer treatment. However, before this eventuality can occur, it is our responsibility as caregivers to ensure we are protecting our patients’ interest and well-being. Here, we have outlined several recommendations for the development of biosimilar trastuzumab and suggestions for its use in clinical practice. It is our hope that these recommendations will serve as a guide for clinicians, pharmacists, regulators, and biosimilar manufacturers as biosimilar trastuzumab is incorporated into the European marketplace.
To summarize, it is our opinion that biosimilar trastuzumab candidates have many challenges to overcome before approval. The majority of clinical trials involve patients with metastatic breast cancer, which is not a sensitive and homogenous population, and use endpoints that do not always correlate with survival. Using endpoints, such as pCR in early breast cancer, which predict survival would be preferable. When a biosimilar trastuzumab that has been tested in early breast cancer is approved in Europe, extrapolation of the data to metastatic breast cancer might be appropriate, assuming this is supported by the marketing authorization application. However, if a biosimilar trastuzumab were approved based on testing in metastatic breast cancer, it would not be appropriate to extrapolate to a potentially curable patient population such as early breast cancer; this would require additional testing. As with all biopharmaceuticals, biosimilar trastuzumab will require thorough pharmacovigilance following authorization. In order to facilitate this, physicians should prescribe all biologics by brand name. This will avoid confusion between an originator drug and a biosimilar, and will also aid in preventing accidental substitution or switching of medicines. Because biosimilars are not identical copies of their originator drugs, they should not be considered interchangeable, and switching between these medicines with or without the physician’s consent should be avoided. We feel that a combination of good clinical trial design and good post-approval practices will allow biosimilar trastuzumab to become an important and trusted component of breast cancer care.
Biosimilar trastuzumab may be the first biosimilar mAb approved for oncology, but many others will soon follow. Patents for several therapeutic antibodies expire in the next 5 years, and biosimilars of rituximab and bevacizumab are already in development. All of the considerations for biosimilar trastuzumab discussed here will apply for many of these upcoming biosimilars. With that in mind, our final and most important recommendation is for thorough education of all parties involved in the biosimilar decision-making process. Even as we enter our eighth year of biosimilars in Europe, many key parties lack a basic understanding of what a biosimilar is, how it differs from a generic, why these differences may or may not be important, and how to minimize any perceived risk associated with biosimilars. As we move toward a future where biosimilars will be part of the treatment landscape for many diseases, it is essential that we ensure our colleagues and patients are properly educated on the subject. Adequate education for all involved parties allows everyone involved to make informed decisions based on knowledge, comfort, and transparency rather than fear and confusion. This is the keystone to ensure successful integration of biosimilar mAbs into the treatment of cancer.
References
Slamon DJ, Leyland-Jones B, Shak S et al (2009) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344:783–792
Perez EA, Romond EH, Suman VJ et al (2011) Four-year follow-up of trastuzumab plus adjuvant chemotherapy for operable human epidermal growth factor receptor 2–positive breast cancer: joint analysis of data from NCCTG N9831 and NSABP B-31. J Clin Oncol 29:3366–3373
Goldhirsch A, Gelber RD, Piccart-Gebhart MJ, Herceptin Adjuvant (HERA) Trial Study Team et al (2013) 2 years versus 1 year of adjuvant trastuzumab for HER2-positive breast cancer (HERA): an open-label, randomized controlled trial. Lancet 382:1021–1028
Revers L, Furczon E (2010) An introduction to biologics and biosimilars. Part II: Subsequent entry biologics: biosame or biodifferent? Can Pharm J 143:184–191
Weise M, Bielsky MC, De Smet K et al (2011) Biosimilars—why terminology matters. Nat Biotechnol 29:691–693
Carey K (2011) Biosimilars encircle Rituxan, US debates innovator exclusivity. Nat Biotechnol 29:177–178
European Medicines Agency (2013) European public assessment reports: Biosimilars. http://www.ema.europa.eu/ema/index.jsp?curl=pages%2Fmedicines%2Flanding%2Fepar_search.jsp&mid=WC0b01ac058001d125&searchTab=searchByAuthType&alreadyLoaded=true&isNewQuery=true&status=Authorised&status=Withdrawn&status=Suspended&status=Refused&keyword=Enter+keywords&searchType=name&taxonomyPath=&treeNumber=&searchGenericType=biosimilars&genericsKeywordSearch=Submit. Accessed Nov 2013
Guideline on similar biological medicinal products containing monoclonal antibodies—non-clinical and clinical issues. London, United Kingdom: Committee for Medicinal Products for Human Use, European Medicines Agency 2012
Guideline on immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use. London, United Kingdom: Committee for Medicinal Products for Human Use, European Medicines Agency 2012
Im Y-H, Krasnozhon D, Bondarenko I, et al. (2013) Phase I/IIb clinical trial comparing PK and safety of trastuzumab and its biosimilar candidate CT-P6. Presented at the St Gallen Breast Cancer Conference 13–16 March 2013 in St. Gallen, Switzerland. Abstract 268
Im Y-H, Odarchenko P, Grecea D, et al. (2013) Double-blind, randomized, parallel group, phase III study to demonstrate equivalent efficacy and comparable safety of CT-P6 and trastuzumab, both in combination with paclitaxel, in patients with metastatic breast cancer as first-line treatment. J Clin Oncol 31: Poster 629
Celltrion files for approval of CT-P6, breast cancer mAb biosimilar, in Korea. Incheon, Korea: Celltrion, Inc; 4 June 2013. http://www.celltrion.com/en/COMPANY/notice_view.asp?idx=348&code=ennews. Accessed Nov 2013
Yin D, Barker KB, Ruifeng L, et al. (2013) A phase I pharmacokinetics trial comparing PF-05280014 (a potential biosimilar) and trastuzumab in healthy volunteers (REFLECTIONS B327-01). J Clin Oncol 31: Abstract 612
Ewesuedo R, Barker KB, Taylor CT, Jacobs I (2013) A phase 3 randomized, double-blind trial comparing PF-05280014 + paclitaxel vs trastuzumab + paclitaxel for treatment of HER2 + metastatic breast cancer. Cancer Res 73 (24 Suppl): Abstract OT1-1-03
Clinical Trials.gov (Internet). Bethesda (MD): National Library of Medicine (US). 2000 Feb 29—Identifier NCT01084876, Demonstrate efficacy and safety of metastatic breast cancer (COMPARE); 9 March 2010. http://clinicaltrials.gov/ct2/show/NCT01084876. Accessed Dec 2013
PBR (Web site). Biocon may launch Roche’s Herceptin biosimilar version by March 2014. http://drugdiscovery.pharmaceutical-business-review.com/news/biocon-may-launch-roches-herceptin-biosimilar-version-by-March-2014-220813. Accessed Nov 2013
Clinical Trials.gov (Internet). Bethesda (MD): National Library of Medicine (US). 2000 Feb 29—Identifier NCT01764022, A safety and efficacy study of BCD-022 with paclitaxel compared to herceptin with paclitaxel in HER2-positive metastatic breast cancer patients; 7 January 2013. http://clinicaltrials.gov/ct2/show/NCT01764022. Accessed Dec 2013
Clinical Trials.gov (Internet). Bethesda (MD): National Library of Medicine (US). 2000 Feb 29—Identifier NCT01901146, Efficacy and Safety Study of ABP 980 Compared With Trastuzumab in Subjects With HER2 Positive Early Breast Cancer (Lilac); 20 May 2013. http://clinicaltrials.gov/ct2/show/NCT01901146. Accessed Dec 2013
Tsavaris N, Kosmas C, Vadiaka M et al (2002) Immune changes in patients with advanced breast cancer undergoing chemotherapy with taxanes. Br J Cancer 87:21–27
Standish LJ, Torkelson C, Hamill FA et al (2008) Immune defects in breast cancer patients after radiotherapy. J Soc Integr Oncol 6:110–121
Saso R, Kulkarni S, Mitchell P et al (2000) Secondary myelodysplastic syndrome/acute myeloid leukaemia following mitoxantrone-based therapy for breast carcinoma. Br J Cancer 83:91–94
Smith RE (2003) Risk for the development of treatment-related acute myelocytic leukemia and myelodysplastic syndrome among patients with breast cancer: review of the literature and the National Surgical Adjuvant Breast and Bowel Project experience. Clin Breast Cancer 4:273–279
Hotta K, Kiura K, Fujiwara Y et al (2009) Association between incremental gains in the objective response rate and survival improvement in phase III trials of first-line chemotherapy for extensive disease small-cell lung cancer. Ann Oncol 20:829–834
Lee L, Wang L, Crump M (2011) Identification of potential surrogate end points in randomized clinical trials of aggressive and indolent non-Hodgkin’s lymphoma: correlation of complete response, time-to-event and overall survival end points. Ann Oncol 22:1392–1403
Von Minckwitz G, Untch M, Blohmer JU et al (2012) Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J Clin Oncol 30:1796–1804
European Medicines Agency (2013) European public assessment reports: Remsima. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002576/human_med_001682.jsp. Accessed Nov 2013
European Medicines Agency. European public assessment reports: Inflectra. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002778/human_med_001677.jsp. Accessed Nov 2013
Vu T, Claret FX (2012) Trastuzumab: updated mechanisms of action and resistance in breast cancer. Front Oncol 2:62
Simoens S (2011) Biosimilar medicines and cost-effectiveness. Clinicoecon Outcomes Res 3:29–36
Schneider CK, Vleminckx C, Gravanis I et al (2012) Setting the stage for biosimilar monoclonal antibodies. Nat Biotechnol 30:1179–1185
Guideline on good pharmacovigilance practices (GVP): module V—risk management systems. London, United Kingdom: Heads of Medicines Agencies, European Medicines Agency, 2012
World Health Organization (2006) WHO informal consultation on international nonproprietary names (INN) policy for biosimilar products. World Health Organization, Geneva
Medicines and Healthcare Products Regulatory Agency (2008) Biosimilar products. Drug Saf Updat 1:8
The Association of the British Pharmaceuticals Industry (2013) ABPI position on biosimilar medicines. The Association of the British Pharmaceuticals Industry, London
MabThera® (rituximab) [package insert] (2013) Roche Registration Limited, Welwyn Garden
Avastin® (bevacizumab) [package insert] (2013) Roche Registration Limited, Welwyn Garden
Mellstedt H, Niederwieser D, Ludwig H (2008) The challenge of biosimilars. Ann Oncol 19:411–419
Ebbers HC, Crow SA, Vulto AG, Schellekens H (2012) Interchangeability, immunogenicity, and biosimilars. Nat Biotechnol 30:1186–1190
Niederwieser D, Schmitz S (2011) Biosimilar agents in oncology/haematology: from approval to practice. Eur J Haematol 86:277–288
Acknowledgments
The authors would like to thank Chelsey Goins, PhD, for her assistance with writing the manuscript, and Heather Tomlinson (ELS) for her editorial assistance and assistance preparing the manuscript for publication. This work was supported by F. Hoffman-La Roche Ltd.
Conflict of interest
Dr Cortés has disclosed that he has performed in a consultant or advisory role for Celgene, Roche, and Novartis, and has received honoraria from Celgene, Roche, Novartis, and Eisai. Dr Diéras has disclosed that she has participated in advisory boards for Roche, GSK, and Pfizer. Dr Curigliano has declared no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Cortés, J., Curigliano, G. & Diéras, V. Expert perspectives on biosimilar monoclonal antibodies in breast cancer. Breast Cancer Res Treat 144, 233–239 (2014). https://doi.org/10.1007/s10549-014-2879-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-014-2879-9