
Abstract
Amino acids are involved in various metabolic pathways and some of them also act as neurotransmitters. Since biosynthesis of l-glutamate and γ-aminobutyric acid (GABA) requires 2-oxoglutarate while 3-phosphoglycerate is the precursor of l-glycine and d-serine, evolutionary selection of these amino acid neurotransmitters might have been driven by their capacity to provide important information about the glycolytic pathway and Krebs cycle. Synthesis and recycling of amino acid neurotransmitters as well as composition and function of their receptors are often compromised in inherited metabolic diseases. For instance, increased plasma l-phenylalanine concentrations impair cerebral biosynthesis of protein and bioamines in phenylketonuria, while elevated cerebral l-phenylalanine directly acts via ionotropic glutamate receptors. In succinic semialdehyde dehydrogenase deficiency, the neurotransmitter GABA and neuromodulatory γ-hydroxybutyric acid are elevated. Chronic hyperGABAergic state results in progressive downregulation of GABAA and GABAB receptors and impaired mitophagy. In glycine encephalopathy, the neurological phenotype is precipitated by l-glycine acting both via cortical NMDA receptors and glycine receptors in spinal cord and brain stem neurons. Serine deficiency syndromes are biochemically characterized by decreased biosynthesis of l-serine, an important neurotrophic factor, and the neurotransmitters d-serine and l-glycine. Supplementation with l-serine and l-glycine has a positive effect on seizure frequency and spasticity, while neurocognitive development can only be improved if treatment starts in utero or immediately postnatally. With novel techniques, the study of synaptic dysfunction in inherited metabolic diseases has become an emerging research field. More and better therapies are needed for these difficult-to-treat diseases.

Similar content being viewed by others

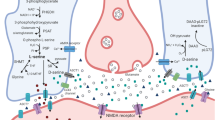
Abbreviations
- GABA:
-
γ-Aminobutyric acid
- GE:
-
Glycine encephalopathy (synonym, non-ketotic hyperglycinemia)
- GHB:
-
γ-Hydroxybutyric acid
- LNAA:
-
Large neutral amino acids
- PKU:
-
Phenylketonuria
- SSADH:
-
Succinic semialdehyde dehydrogenase
References
Absalom N, Eghorn LF, Villumsen IS et al (2012) α4βδ GABA(A) receptors are high-affinity targets for γ-hydroxybutyric acid (GHB). Proc Natl Acad Sci U S A 109:13404–13409
Alfadhel M, Nashabat M, Al Qahtani H et al (2016) Mutation in SLC6A9 encoding a glycine transporter causes a novel form of non-ketotic hyperglycinemia in humans. Hum Genet 135:1263–1268
Applegarth DA, Toone JR (2004) Workshop report. Glycine encephalopathy (nonketotic hyperglycinemia): review and update. J Inherit Metab Dis 27:417–422
Bak LK, Schousboe A, Waagepetersen HS (2006) The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem 98:641–653
Baker PR, Friederich MW, Swanson MA et al (2014) Variant non-ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX2. Brain 137:366–379
Bickel H, Gerrard J, Hickmans EM (1953) Influence of phenylalanine intake on phenylketonuria. Lancet 265:812–813
Bjoraker KJ, Swanson MA, Coughlin CR 2nd et al (2016) Neurodevelopmental outcome and treatment efficacy of benzoate and dextromethorphan in siblings with attenuated nonketotic hyperglycinemia. J Pediatr 17:234–239
Braissant O (2010) Current concepts in the pathogenesis of urea cycle disorders. Mol Genet Metab 100:S3–S12
Brassier A, Valayannopoulos V, Bahi-Busson N et al (2016) Two new cases of serine deficiency disorders treated with L-serine. Eur J Paediatr Neurol 20:53–60
Burgard P, Schmidt E, Rupp A, Schneider W, Bremer HJ (1996) Development of the patients of the German Collaborative Study of children treated for phenylketonuria. Eur J Pediatr 155(Suppl 1):S33–S38
Burgard P, Ullrich K, Ballhausen D et al (2017) Issues with European guidelines for phenylketonuria. Lancet Diabetes Endocrinol 5:681–683
Cusmai R, Martinelli D, Moavero R et al (2012) Ketogenic diet in early myoclonic encephalopathy due to nonketotic hyperglycinemia. Eur J Paediatr Neurol 16:509–513
De Koning TJ, Duran M, van Maldegem L et al (2002) Congenital microcephaly and seizures due to 3-phosphoglycerate dehydrogenase deficiency: outcome of treatment with amino acids. J Inherit Metab Dis 25:119–125
De Koning TJ, Klomp LW, van Oppen AC, Beemer FA, Dorland L, van den Berg I, Berger R (2004) Prenatal and early postnatal treatment in 3-phosphoglycerate-dehydrogenase deficiency. Lancet 364:2221–2222
De Koning TJ (2017) Amino acid synthesis deficiencies. J Inherit Metab Dis 40:609–620
Donarum EA, Stephan DA, Larkin K et al (2006) Expression profiling reveals multiple myelin alterations in murine succinate semialdehyde dehydrogenase deficiency. J Inherit Metab Dis 29:143–156
Dwyer CA, Scudder SL, Lin Y, Dozier LE, Phan D, Allen NJ, Patrick GN, Esko JD (2017) Neurodevelopmental changes in excitatory synaptic structure and function in the central cortex of sanfilippo syndrome IIIA mice. Sci Rep 7:46576
Elliott GR, Leys SP (2010) Evidence for glutamate, GABA and NO in coordinating behavior in the sponge, Ephydatia muelleri (Demospongiae, Spongillidae). J Exp Biol 213:2310–2321
Flusser H, Korman SH, Sato K, Matsubara Y, Galil A, Kure S (2005) Mild glycine encephalopathy (NKH) in a large kindred due to a silent exonic GLDC splice mutation. Neurology 64:1426–1430
Furuya S, Tabata T, Mitoma J et al (2000) L-Serine and glycine serve as major astroglia-derived trophic factors for cerebellar purkinje neurons. Proc Natl Acad Sci U S A 97:11528–11533
Furuya S (2008) An essential role for de novo biosynthesis of L-serine in CNS development. Asia Pac J Clin Nutr 17(S1):312–315
Glushakov AV, Dennis DM, Sumners C, Seubert CN, Martynyuk AE (2003) L-phenylalanine selectively depresses currents at glutamatergic excitatory synapses. J Neurosci Res 72:116–124
Glushakov AV, Glushakova O, Varshney M et al (2005) Long-term changes in glutamatergic synaptic transmission in phenylketonuria. Brain 128:300–307
Gupta M, Hogema BM, Grompe M et al (2003) Murine succinate semialdehyde dehydrogenase deficiency. Ann Neurol 54(Suppl 6):S81–S90
Guthrie R, Susi A (1963) A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 32:338–343
Häberle J, Börg B, Rutsch F et al (2005) Congenital glutamine deficiency with glutamine synthetase mutations. N Engl J Med 353:1926–1933
Harper D, Maynard-Smith J (2003) Animal signals. Oxford University Press
Harris KD, Zahavi A (2013) The evolution of ACh and GABA as neurotransmitters: a hypothesis. Med Hypotheses 81:760–762
Hart CE, Race V, Achouri Y et al (2007) Phosphoserine aminotransferase deficiency: a novel disorder of serine biosynthesis pathway. Am J Hum Genet 80:931–937
Hennermann JB, Berger JM, Grieben U, Scharer G, van Hove JLK (2012) Prediction of long-term outcome in glycine encephalopathy: a clinical survey. J Inherit Metab Dis 35:253–261
Hogema BM, Gupta M, Senephansiri H et al (2001) Pharmacologic rescue of lethal seizures in mice deficient in succinate semialdehyde dehydrogenase deficiency. Nat Genet 29:212–216
Hoover-Fong JE, Shah S, van Hove JLK, Applegarth D, Toone J, Hamosh A (2004) Natural history of nonketotic hyperglycinemia in 65 patients. Neurology 63:1847–1853
Huttenlocher PR (2000) The neuropathology of phenylketonuria: human and animal studies. Eur J Pediatr 159:S102–S106
Ikonomidou C, Bosch F, Miksa M et al (1999) Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283:70–74
Ikonomidou C, Bittigau P, Ishimaru MJ et al (2000) Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 287:1056–1060
Jakobs C, Bojasch M, Mönch E, Rating D, Siemes H, Hanefeld F (1981) Urinary excretion of gamma-hydroxybutyric acid in a patient with neurological abnormalities. The probability of a new inborn error of metabolism. Clin Chim Acta 111:169–178
Keller MA, Turchyn AV, Ralser M (2014) Non-enzymatic glycolysis and pentose phosphate pathway-like reactions in a plausible Archean ocean. Mol Syst Biol 10:725
Kikuchi G, Motokawa Y, Yoshida T, Hiraga K (2008) Glycine cleavage system: reaction mechanism, physiological significance, and hyperglycinemia. Proc Jpn Acad Ser B Physiol Biol Sci 84:246–263
Koenig MK, Hodgemans R, Riviello JJ et al (2017) Phenotype of GABA-transaminase deficiency. Neurology 88:1919–1924
Kristan WB Jr (2016) Early evolution of neurons. Curr Biol 26:R949–R954
Kumar A, Dejanovic B, Hetsch F, et al (2017) S-sulfocysteine/NMDA receptor-dependent signaling underlies neurodegeneration in molybdenum cofactor deficiency. J Clin Invest https://doi.org/10.1172/JCI89885
Lakhani R, Vogel KR, Till A et al (2014) Defects in GABA metabolism affect selective autophagy pathways and are alleviated by mTOR inhibition. EMBO Mol Med 6:551–566
Martin W, Baross J, Kelley D, Russell MJ (2008) Hydrothermal vents and the origin of life. Nat Rev Microbiol 6:805–814
Matalon R, Surendran S, Michals Matalon K et al (2003) Future role of large neutral amino acids in transport of phenylalanine into the brain. Pediatrics 112:1570–1574
McDermot KD, Nelson W, Reichert CM, Schulman JD (1980) Attempts at use of strychnine sulfate in the treatment of nonketotic hyperglycinemia. Pediatrics 65:61–64
Miller SL (1953) A production of amino acids under possible primitive earth conditions. Lancet 117:528–529
Mills PB, Struys E, Jakobs C et al (2006) Mutations in antiquitin in individuals with pyridoxine-dependent epilepsy. Nat Med 12:307–309
Morrison PF, Sankar R, Shields WD (2006) Valproate-induced chorea and encephalopathy in atypical nonketotic hyperglycinemia. Pediatr Neurol 35:356–358
Muntau AC, Röschinger W, Habich M, Demmelmair H, Hoffmann B, Sommerhoff CP, Roscher AA (2002) Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. N Engl J Med 347:2122–2132
Novarino G, El-Fishawy P, Kayserili H et al (2012) Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science 338:394–397
Olney JW, Wozniak DF, Jevtovic-Todorovic V, Ikonomidou C (2001) Glutamate signaling and the fetal alcohol syndrome. Ment Retard Dev Disabil Res Rev 7:267–275
Oparin AI (1968) The origin of life (The origin and development of life). Moscow: Moscow Worker publisher, 1924 (in Russian). English translation: Oparin AI. (NASA TTF-488). Washington: D.C.L GPO
Ortez C, Jou C, Cortès-Saladefont E et al (2013) Infantile parkinsonism and GABAergic hypotransmission in a patient with pyruvate carboxylase deficiency. Gene 532:302–306
Pardridge WM (1998) Blood-brain barrier carrier-mediated transport and brain metabolism of amino acids. Neurochem Res 23:635–644
Parker ET, Cleaves HJ, Dworkin JP et al (2011) Primordial synthesis of amines and amino acids in a 1958 Miller H2S-rich spark discharge experiment. Proc Natl Acad Sci U S A 108:5526–5531
Pearl PL, Gibson KM, Acosta MT et al (2003) Clinical spectrum of succinic semialdehyde dehydrogenase deficiency. Neurology 60:1413–1417
Pearl PL, Gibson KM, Quezado Z et al (2009) Decreased GABA-A binding on FMZ-PET in succinic semialdehyde dehydrogenase deficiency. Neurology 73:423–429
Pearl PL, Shukla L, Theodore WH, Jakobs C, Gibson KM (2011) Epilepsy in succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism. Brain and Development 33:796–805
Pietz J, Kreis R, Rupp A, Mayatepek E, Rating D, Boesch C, Bremer HJ (1999) Large neutral amino acids block phenylalanine transport into brain tissue in patients with phenylketonuria. J Clin Invest 103:1169–1178
Poddar R, Chen A, Winter L, Rajagopal S, Paul S (2017) Role of AMPA receptors in homocysteine-NMDA receptor-induced crosstalk between ERK and p38 MAK. J Neurochem 142:560–573
Schell MJ, Brady RO Jr, Molliver ME, Snyder SH (1997) D-Serine as a neuromodulator: regional and developmental localizations in rat brain glia resemble NMDA receptors. J Neurosci 17:1604–1615
Schousboe A, Westergaard N, Sonnewald U et al (1993) Glutamate and glutamine metabolism and compartmentation in astrocytes. Dev Neurosci 15:359–366
Schousboe A, Westergaard N, Waagepetersen HS, Larsson OM, Barken IJ, Sonnewald U (1997) Trafficking between glia and neurons of TCA cycle intermediates and related metabolites. Glia 21:99–105
Schulze A, Tran C, Levandovsky V, Patel V, Cortez MA (2016) Systemic availability of guanidinoacetate affects GABAA receptor function and seizure threshold in GAMT deficient mice. Amino Acids 48:2041–2047
Sibarov DA, Abushik PA, Ginitullin R, Antonov SM (2016) GluN2A subunit-containing NMDA receptors are the preferential targets of homocysteine. Front Cell Neurosci 10:246
Snead OC, Gibson KM (2005) γ-Hydroxybutyric acid. N Engl J Med 352:2721–2732
Soto D, Olivella M, Grau C et al (2018) Rett-like encephalopathy caused by a de novo GRIN2B mutation is attenuated by D-serine dietary supplement. Biol Psychiatry 83:160–172
Srour M, Hamdan FF, Gan-Or Z et al (2015) A homozygous mutation in SLC1A4 in siblings with severe intellectual disability and microcephaly. Clin Genet 88:e1–e4
Tabatabaie L, Klomp LW, Berger R, de Koning TJ (2010) L-Serine synthesis in the central nervous system: a review on serine deficiency disorders. Mol Genet Metab 99:256–262
Tada K, Kure S (1993) Non-ketotic hyperglycinaemia: molecular lesion, diagnosis and pathophysiology. J Inherit Metab Dis 16:691–703
Urey HC (1952) On the early chemical history of the Earth and the origin of life. Proc Natl Acad Sci U S A 38:351–363
Van Spronsen FJ, van Wegberg AM, Ahring K et al (2017) Key European guidelines for the diagnosis and management of patients with phenylketonuria. Lancet Diabetes Endocrinol 5:743–756
Venzi M, di Giovanni G, Crunelli V (2015) A critical evaluation of the gamma-hydroxybutyrate (GHB) model of absence epilepsy. CNS Neurosci Ther 21:123–140
Vogel KR, Ainslie GR, Gibson KM (2016) mTOR inhibitors rescue premature lethality and attenuate dysregulation of GABAergic/glutamatergic transcription in murine succinate semialdehyde dehydrogenase deficiency (SSADHD), a disorder of GABA metabolism. J Inherit Metab Dis 39:877–886
Wu Y, Buzzi A, Frantseva M et al (2006) Status epilepticus in mice deficient for succinate semialdehyde dehydrogenase: GABAA receptor-mediated mechanisms. Ann Neurol 59:42–52
Wächtershäuser G (1990) Evolution of the first metabolic cycles. Proc Natl Acad Sci U S A 87:200–204
Westergaard N, Sonnewald U, Schousboe A (1994) Release of alpha-ketoglutarate, malate and succinate from cultured astrocytes: possible role in amino acid neurotransmitter homeostasis. Neurosci Lett 176:105–109
Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann HJ, Fukuda A (2004) Cl-uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J Physiol 557:829–841
Yoshida K, Furuya S, Osuka S et al (2004) Targeted disruption of the mouse 3-phosphoglycerate dehydrogenase gene causes severe neurodevelopmental defects and results in embryonic lethality. J Biol Chem 279:3573–3577
Zahavi A, Zahavi A (2012) The logic of analog signaling and the theory of signal selection. Isr J Ecol Evol 58:269–278
Zhang XV, Martin ST (2006) Driving parts of Krebs cycle in reverse through mineral photochemistry. J Am Chem 128:16032–16033
Zinnanti WJ, Lazovic J, Griffin K et al (2009) Dual mechanism of brain injury and novel treatment strategy in maple syrup urine disease. Brain 132:903–918
Acknowledgements
I thank Dr. Angeles Garcia-Cazorla for the fruitful discussion and valuable input.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Stefan Kölker declares that he has no conflict of interest.
Informed consent
Not applicable.
Animal rights
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Communicated by: Robert Steiner
Rights and permissions
About this article

Cite this article
Kölker, S. Metabolism of amino acid neurotransmitters: the synaptic disorder underlying inherited metabolic diseases. J Inherit Metab Dis 41, 1055–1063 (2018). https://doi.org/10.1007/s10545-018-0201-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-018-0201-4