
Abstract
Inherited neurotransmitter diseases are a subset of rare neurometabolic disorders characterized by hereditary deficiencies in neurotransmitter metabolism or transport. Non-ketotic hyperglycinaemia (NKH), called glycine encephalopathy, is an autosomal recessive glycine metabolism disorder characterized by an abnormal accumulation of glycine in all bodily tissues, including the CNS. The SLC6A9 gene, which codes for the GLYT1 protein, a biochemical abnormality in the GCS, and dihydrolipoamide dehydrogenase enzymes, which function as a GCS component, are responsible for the neonatal form’s symptoms, which include progressive encephalopathy, hypotonia, seizures, and occasionally mortality in the first few days of life. By changing the MAPK signalling pathways, glycine deprivation in the brain damages neurons by increasing NMDA receptor activation, increasing intracellular Ca levels, and leading to DNA breakage and cell death in the neuron region. In addition to the previously mentioned clinical diagnosis, NKH or GE would be determined by MLPA and 13C glycine breath tests. Pediatricians, surgeons, neurologists, and geneticists treat NKH and GE at the newborn period; there is no cure for either condition.
Similar content being viewed by others


Introduction
Endogenous chemical messengers known as neurotransmitters improve the synaptic connection between neurons that are primarily engaged in a variety of physiological and psychological processes. Rare neurometabolic conditions known as inherited neurotransmitter disorders (NTDs) include enzymes involved in the synthesis of neurotransmitters or their required co-factors [1]. In these NTDs, one of the rare NTDs is glycine encephalopathy, called non-ketotic hyperglycinaemia. It is an autosomal recessive disorder marked by impaired glycine cleavage enzyme activity [2]. Mabry and colleagues originally described non-ketotic hyperglycinaemia in two newborns from the same sibship in 1963, presenting with severe neurological discomfort, hypotonia, frequent convulsions, and obtundation.
The syndrome is unique from Childs and colleagues [3] previously documented hyperglycinaemia with acidosis (1961). Glycine concentrations in plasma are elevated in a range of metabolic diseases in infants and children. In the ketotic-hyperglycinaemia condition, elevated blood levels of glycine seem to be involved in the accumulation of one or more unusual organic acids produced by the breakdown of amino acids chain. Propionic acidaemia [4] and methylmalonic acidaemia [5] are two conditions that are often linked to hyperglycinaemia; however, patients with isovaleric acidaemia [6] and beta-ketothiolase deficiency have also been reported to have it [7]. The majority of children with hyperglycinaemia, however, are classified as having non-ketotic hyperglycinaemia since there are often no detectable abnormalities in organic acid metabolism.
Many people with this diverse collection of illnesses suffer a life-threatening sickness in their infants, characterized by intractable seizures, stiffness, severe mental retardation, and early mortality. The non-ketotic hyperglycinaemia condition has been treated in a variety of ways, but none of them have worked [8].
According to Rees and colleagues in 2003, the spinal cord circuit is where the glycine receptor (GlyR) performs its most well-known function. They are often located in the spinal cord, help with synaptic transmission [9,10,11], and are essential for both motor control and pain perception [12,13,14]. They first occur during the early stages of spinal cord development, and the growth of their subunit composition is controlled [15]. It’s interesting to note that GlyR has been demonstrated to influence the differentiation of interneurons and synaptogenesis in the spinal cord [11].
The glycine cleavage system is made up of the glycine decarboxylase gene, which codes for the P-protein, the amino methyltransferase gene, which codes for the T-protein, and the glycine cleavage system H-gene, which codes for the H-protein [16]. While people with variation NKH have changes in the genes that make up lipoate and its cofactor. In addition, the disruption of GlyRs activity results in disease of the brain.
In individuals with side epilepsy, alternative splice variants have been identified [17]. Additionally, a mutation in the gene encoding the amino methyltransferase enzyme (AMT), also known as the Glyr-2 subunit [18], is present in autistic patients and is a significant contributor to glycine breakdown [19]. Five symmetrically organized subunits of the trans-membrane protein complexes known as GlyRs are positioned around a central pore. So far, five kinds of GlyR subunits have been identified: four alphas and one beta [8, 20]. For some alpha subunits, alternate splicing can result in novel variants [21,22,23,24]. Alpha subunits can produce homomeric or heteromeric receptors when they are coupled with the beta subunit [25, 26].
Glycine may be investigated as an amino acid that can aid COVID-19 patients reduce tissue damage and cytokine storm, according to Chuan Yuan Li., 2020 suggestion [27]. Glycine protects cells from pyroptosis and the production of proinflammatory cytokines by binding to its receptor GlyR, inducing a chloride influx that results in cellular membrane hyperpolarization.
If glycine is successful in clinical trials, it can be given to COVID-19 patients everywhere in the globe right now.
Aetiology
Glycine builds up in the brain as a result of defects in the glycine cleavage pathway in NKH, an autosomal recessive disorder, as was previously mentioned [28]. Glycine accumulation in the brain changes the MAPK signalling pathways, which injures neurons [29]. Glycine activates N-methyl-d-aspartate receptors in the cerebral hemispheres and cerebellum, but it inhibits N-methyl-d-aspartate receptors in the brainstem and spinal cord [30, 31]. 5, 10-Methylenetetrahydrofolate, a byproduct of glycine hydrolysis, is also necessary for DNA synthesis. Since neural stem cell proliferation depends on the glycine cleavage pathway, it is highly expressed throughout development [32].
In NKH, a wide range of neurological dysfunctions, including severe infant hypotonia, failure to thrive, minor mental impairment, and learning challenges. The most severe and frequent form of NKH is called a classic form, which is characterized by infant encephalopathy, respiratory problems, and multifocal myoclonic convulsions. In those with classical NKH, GLDC gene mutations can occur in up to 75% of cases [33]. Less than 1% and 20% of instances of NKH, respectively, are caused by mutations in the GCSH and AMT genes [33, 34]. Medical symptoms are significantly influenced by the quantity of remaining glycine cleavage system hobby [28, 35]. The infantile shape typically includes seizures after 6 months of life.
Patients with late-onset NKH commonly show typical intellectual traits, along with spastic diplegia and optic atrophy [36]. The literature has also identified a transitory form of NKH, in which biochemical and electroencephalogram (EEG) abnormalities resolve by 2 months of age [37, 38]. In situations where the hepatic and cerebral glycine cleavage systems did not mature in a timely manner, full medical judgments are made. There have been reports of anatomical brain deformities, aberrant white matter signalling, and progressive brain atrophy. Patients that have NKH [39, 40]. The function of glycine in neural stem cells and synaptic transmission proliferation [41]. Callosal corpus, one of the characteristics of NCH-related deformities is dysgenesis. Involvement of the substantia Nigra, red nucleus, thalamus, deep grey matter nucleus, and ventral brainstem tract has also been reported.
Pathophysiology
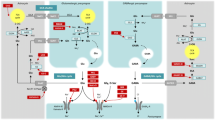
A significant increase in glycine concentration in cerebrospinal fluid was associated with NKH. It exhibits that glycine build-up in the brain is caused by a glycine cleavage device failure. A proportion of coronary artery disease (CAD) patients have been shown to have excessive amounts of glycine in their brains [42, 43]. The GCS is a mitochondrial multi-enzyme complex found in the brain, liver, kidney, and testis that catalyses the breakdown of glycine to carbon dioxide, ammonia, and one carbon unit [44], as shown in Fig. 1 [45].

Enzymatic reaction of GCS. Glycine breaks down into carbon dioxide, ammonia, and one carbon atom
Kure and colleagues in 2011 [46] say that GCS has four proteins: glycine dehydrogenase, amino methyltransferase, GCS H-protein, and dihydrolipoamide dehydrogenase which are encoded genes for each of these proteins, GLDC, AMT, GCSH, and DLD, respectively. The enzyme that is a part of other complex enzyme systems is called dihydrolipoamide dehydrogenase (DLD gene) (pyruvate dehydrogenases complex and the branched-chain ketoacid dehydrogenase complex).
Although NKH's neurological impairment is assumed to be caused by a high glycine level in the brain, its exact pathophysiology is yet unknown. However, several studies have helped us understand the relationship between glycine and neuroexcitotoxicity. Glycine is now believed to act as an inhibitory neurotransmitter at a strychnine-touchy receptor [47,48,49,50,51]. According to Johnson and Ascher and colleagues in 1987 [52], the glutaminergic receptor N-methyl-d-aspartate (NMDA) is potentiated by the excitatory property of glycine.
Based on experimental findings with cultured mouse brain neurons, it has been determined that glycine increases NMDA-mediated responses at a location roughly connected to the NMDA receptor. The NMDA receptor possesses excitatory amino acid receptors that are hyperactive, according to McDonald and Johnston and colleagues in 1992 [53], which have been linked to the pathophysiology of neuronal damage in a number of neurological conditions such hypoxia, ischaemia, hyperglycinaemia, or physical brain trauma. Growing neurophysiological evidence is in favour of this.
Sato and colleagues in 1991 [54] studied GCS studied rat brains utilizing P-protein antibody immunostaining, and discovered that the GCS was restricted to the astrocytes. The brain stem and spinal cord exhibited the least amount of astrocyte staining, whereas the hippocampus and cerebellar cortex had the most. In different parts of the brain, astrocyte staining varied. The fact that astrocytes in the CNS contain GCS indicates that glycine breakdown occurs mostly in these cells.
Because there is a link between the locations of the GCS and the NMDA receptors, it is seen that astrocytes of the GCS are associated with NMDA receptors. According to Tada K and colleagues in 1993 [55], the addition of glutamate causes the DNA chromosomal of the neurons to disintegrate into nucleosomal size DNA fragments, which results in glutamate-induced neuronal death (Fig. 2). Endonuclease inhibitors reduced DNA fragmentation and neuronal death, indicating that activating endonuclease is what causes DNA fragmentation. Instead of glutamate, this might cause an increase in intracellular Ca2 + concentration, which would then cause DNA fragmentation. The subcellular mechanism of neuroexcitotoxicity may thus be DNA fragmentation by glutamate, according to their studies.

Pathophysiology of NKH
Genetics
According to Irene Mademont-Soler and colleagues in 2020 [56], a high concentration of glycine in the brain causes glycine encephalopathy, also known as glycine transporter 1 encephalopathy (GLYT1 encephalopathy). The protein GLYT1, which includes 14 exons, an intercellular N- and C-terminal, as well as 12 trans-membrane domains, is encoded by SLC6A9, according to Rayan Alfallaj and colleagues in 2019 [16]. When two Na and Cl ions bind to glycine, GLYT1 activity, or glycine transport at glial cells, is stimulated. In each of their case studies including GLYT1, Rayan Alfallaj and colleagues in 2019 [16] proposed an autosomal recessive inheritance pattern. A homozygous missense mutation in the SLC6A9 gene causes GLYT1, a deadly disease.
Diagnosis
Glycine encephalopathy, according to Rayan Alfallaj and colleagues in 2019 [16], and Kure and colleagues in 2011 [46], can be diagnosed in neonates or babies who have seizures, muscle hypotonia, and lethargy that are not easily explained by an infection, trauma, hypoxia, or other frequent paediatric issues. Based on the clinical symptoms, high levels of glycine in the CSF (greater than 0.09), normal plasma, enzymes, and genetic tests for SLC6A9 mutations. Less than 0.04 is seen in both normal and ketotic-hyperglycinaemia. GE is characterized by an EEG finding of a burst suppression pattern during the first month of life.
The 13C glycine breath test and the multiplex ligation-dependent probe amplification (MLPA) are the two methods for the identification of significant deletions in DLD which were introduced by Kure and colleagues in 2011 [46] as a result of recent reports on improvements in GE diagnosis. Both approaches would make it easier for patients with hyperglycinaemia to confirm the diagnosis of GE.
Treatment
Only supportive care is now provided as a treatment for the illness. The therapy of GLYT1 involves the involvement of paediatricians, neurologists, geneticists, genetic counsellors, dietitians, physiotherapists, occupational therapists, and orthopaedic surgeons. Children with glycine transporter 1 encephalopathy (GLYT1) are at risk for aspiration, malnutrition, and stunting because of issues with stomach incoordination and swallowing. A feeding tube, such as a gastrostomy tube, may be a successful therapy for many patients in order to guarantee that they consume enough calories and prevent recurrent aspirations. It's also a good idea to get your hearing and eyesight checked often. Last but not least, it is essential to consult orthopaedic surgeons, physiotherapists, and occupational therapists on a regular basis [46].
Conclusion
NKH, also known as GE, inherited neurotransmitter diseases that are a subset of rare neurometabolic disorders characterized by hereditary deficiencies in neurotransmitter metabolism or transport on GCS. There is no cure for NKH or GE; it is treated in the neonatal stage by paediatricians, surgeons, neurologists, and geneticists. Glycine deficiency in the brain causes neuronal damage by altering MAPK signalling pathways, resulting in increased NMDA receptor activation and intracellular Ca buildup, as well as cell death in the neuron area due to DNA fragmentation. GE can be diagnosed using MLPA and 13C glycine breath testing, in addition to the previously mentioned clinical diagnosis.
Availability of data and materials
Not applicable.
Abbreviations
- NKH:
-
Non-ketotic hyperglycinaemia
- GE:
-
Glycine encephalopathy
- GCS:
-
Glycine cleavage system
- NTD:
-
Neurotransmitter disorder
- GlyR:
-
Glycine receptor
- GLYT1:
-
Glycine transporter 1 encephalopathy
- AMT:
-
Amino methyl transferase enzyme
- NMDA:
-
N-Methyl-d-aspartate receptor
- GCSH:
-
Glycine cleavage system protein H
- DLD:
-
Dihydrolipoamide dehydrogenase
- GLDC:
-
Glycine dehydrogenase
- MLPA:
-
Multiplex ligation dependent probe amplification
- CAD:
-
Coronary artery disease
- EEG:
-
Electroencephalogram
- CSF:
-
Cerebrospinal fluid
References
Lim Y, Mankad K, Kinali M, Tan A. Neuroimaging spectrum of inherited neurotransmitter disorders. Neuropediatrics. 2019;51(01):006–21. https://doi.org/10.1055/s-0039-1698422.
Stence N, Fenton L, Levek C, Tong S, Coughlin C, Hennermann J, et al. Brain imaging in classic nonketotic hyperglycinemia: quantitative analysis and relation to phenotype. J Inherit Metab Dis. 2019;42(3):438–50. https://doi.org/10.1002/jimd.12072.
Childs B, Nyhan W, Borden M, Bard L, Cooke R. Idiopathic hyperglycinemia and hyperglycinuria: a new disorder of amino acid metabolism. I. Pediatrics. 1961;27(4):522–38. https://doi.org/10.1542/peds.27.4.522.
Morel C, Watkins D, Scott P, Rinaldo P, Rosenblatt D. Prenatal diagnosis for methylmalonic acidemia and inborn errors of vitamin B12 metabolism and transport. Mol Genet Metab. 2005;86(1–2):160–71. https://doi.org/10.1016/j.ymgme.2005.07.018.
Khan A, Zahid B, Khan S, Ahmad S. Isovaleric Acidemia: a rare case of an inborn error of metabolism. Cureus. 2020. https://doi.org/10.7759/cureus.7150.
Hillman RE, Keating JP. Beta-ketothiolase deficiency as a cause of the “ketotic hyperglycinemia syndrome.” Pediatrics. 1974;53(2):221–5. https://doi.org/10.1542/peds.53.2.221.
Nyhan WL, Borden M, Childs B. Idiopathic hyperglycinemia: a new disorder of amino acid metabolism. Pediatrics. 1961;27(4):539–50. https://doi.org/10.1542/peds.27.4.539.
Matzenbach B, Maulet Y, Sefton L, Courtier B, Avner P, Guénet JL, et al. Structural analysis of mouse glycine receptor alpha subunit genes identification and chromosomal localization of a novel variant. J Bio Chem. 1994;269(4):2607–12. https://doi.org/10.1016/s0021-9258(17)41987-9.
Colin I, Rostaing P, Augustin A, Triller A. Localization of components of glycinergic synapses during rat spinal cord development. J Comp Neurol. 1998;398(3):359–72. https://doi.org/10.1002/(sici)1096-9861(19980831)398:3%3c359::aid-cne5%3e3.0.co;2-z.
Moss S, Smart T. Constructing inhibitory synapses. Nat Rev Neurosci. 2001;2(4):240–50. https://doi.org/10.1038/35067500.
Scain A, Le Corronc H, Allain A, Muller E, Rigo J, Meyrand P, et al. Glycine release from radial cells modulates the spontaneous activity and its propagation during early spinal cord development. J Neurosci Res. 2010;30(1):390–403. https://doi.org/10.1523/jneurosci.2115-09.2010.
Harvey R, Depner U, Wässle H, Ahmadi S, Heindl C, Reinold H, et al. GlyR α3: an essential target for spinal PGE 2 mediated inflammatory pain sensitization. Science. 2004;304(5672):884–7. https://doi.org/10.1126/science.1094925.
Zeilhofer H. The glycinergic control of spinal pain processing. Cell Mol Life Sci. 2005;62(18):2027–35. https://doi.org/10.1007/s00018-005-5107-2.
Lynch JW, Callister RJ. Glycine receptors: a new therapeutic target in pain pathways. Curr Opin Invest Drugs. 2006;7:48–53.
Watanabe E, Akagi H. Distribution patterns of mRNAs encoding glycine receptor channels in the developing rat spinal cord. Neurosci Res. 1995;23(4):377–82. https://doi.org/10.1016/0168-0102(95)00972-v.
Alfallaj R, Alfadhel M. Glycine transporter 1 encephalopathy from biochemical pathway to clinical disease: review. Child Neurol Open. 2019. https://doi.org/10.1177/2329048x19831486.
Eichler S, Förstera B, Smolinsky B, Jüttner R, Lehmann T, Fähling M, et al. Splice-specific roles of glycine receptor α3 in the hippocampus. Eur J Neurosci. 2009;30(6):1077–91. https://doi.org/10.1111/j.1460-9568.2009.06903.x.
Piton A, Gauthier J, Hamdan F, Lafrenière R, Yang Y, Henrion E, et al. Systematic resequencing of X-chromosome synaptic genes in autism spectrum disorder and schizophrenia. Mol Psychiatry. 2010;16(8):867–80. https://doi.org/10.1038/mp.2010.54.
Yu T, Chahrour M, Coulter M, Jiralerspong S, Okamura-Ikeda K, Ataman B, et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron. 2013;77(2):259–73. https://doi.org/10.1016/j.neuron.2012.11.002.
Lynch JW. Native glycine receptor subtypes and their physiological roles. Neuropharmacology. 2009;56(1):303–9. https://doi.org/10.1016/j.neuropharm.2008.07.034.
Kuhse J, Kuryatov A, Maulet Y, Malosio ML, Schmieden V, Betz H. Alternative splicing generates two isoforms of the α2 subunit of the inhibitory glycine receptor. FEBS Lett. 1991;283(1):73–7. https://doi.org/10.1016/0014-5793(91)80557-j.
Malosio ML, Grenningloh G, Kuhse J, Schmieden V, Schmitt B, Prior P, et al. Alternative splicing generates two variants of the alpha 1 subunit of the inhibitory glycine receptor. J Bio Chem. 1991;266(4):2048–53. https://doi.org/10.1016/s0021-9258(18)52207-9.
Nikolic Z, Laube B, Weber RG, Lichter P, Kioschis P, Poustka A, et al. The human glycine receptor subunit α3. J Bio Chem. 1998;273(31):19708–14. https://doi.org/10.1074/jbc.273.31.19708.
Legendre P. Glycine receptors caught between genome and proteome—functional implications of RNA editing and splicing. Front Mol Neurosci. 2009. https://doi.org/10.3389/neuro.02.023.2009.
Pribilla I, Takagi T, Langosch D, Bormann J, Betz H. The atypical M2 segment of the beta subunit confers picrotoxinin resistance to inhibitory glycine receptor channels. EMBO J. 1992;11(12):4305–11. https://doi.org/10.1002/j.1460-2075.1992.tb05529.x.
Oertel J, Villmann C, Kettenmann H, Kirchhoff F, Becker C-M. A novel glycine receptor β subunit splice variant predicts an unorthodox transmembrane topology. J Bio Chem. 2007;282(5):2798–807. https://doi.org/10.1074/jbc.m608941200.
Li C-Y. Can glycine mitigate COVID-19 associated tissue damage and cytokine storm? Radiat Res. 2020;194(3):199.
Hennermann JB, Van Hove JL, Coughlin CR II. Glycine encephalopathy and epilepsy. J Inherit Metab. 2017. https://doi.org/10.1891/9780826168641.0027.
Moura AP, Parmeggiani B, Gasparotto J, Grings M, Fernandez Cardoso GM, Seminotti B, et al. Glycine administration alters MAPK signalling pathways and causes neuronal damage in rat brain: putative mechanisms involved in the neurological dysfunction in nonketotic hyperglycinemia. Mol Neurobiol. 2017;55(1):741–50. https://doi.org/10.1007/s12035-016-0319-z.
Belcastro V, Barbarini M, Barca S, Mauro I. A novel amt gene mutation in a newborn with nonketotic hyperglycinemia and early myoclonic encephalopathy, European. J Pediatric Neurol. 2016;20(1):192–5. https://doi.org/10.1016/j.ejpn.2015.08.008.
Korman SH, Gutman A. Pitfalls in the diagnosis of glycine encephalopathy (non-ketotic hyperglycinemia). Dev Med Child Neurol. 2007;44(10):712–20. https://doi.org/10.1111/j.1469-8749.2002.tb00275.x.
Ichinohe A, Kure S, Mikawa S, Ueki T, Kojima K, Fujiwara K, et al. Glycine cleavage system in neurogenic regions. Eur J Neurosci. 2004;19(9):2365–70. https://doi.org/10.1111/j.0953-816x.2004.03345.x.
Van Hove J, Hennermann J, Coughlin II C. Nonketotic hyperglycinemia (Glycine Encephalopathy) and lipoate deficiency disorders. Inborn Metabolic Diseases. 2016; pp. 349-356. https://doi.org/10.1007/978-3-662-49771-5_23.
Krawiec C, Anastasopoulou C. Nonketotic Hyperglycinemia. [Updated 2022 Jul 24]. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2022. https://www.ncbi.nlm.nih.gov/books/NBK556140/.
Dinopoulos A, Matsubara Y, Kure S. Atypical variants of nonketotic hyperglycinemia. Mol Genet Metab. 2005;86(1–2):61–9. https://doi.org/10.1016/j.ymgme.2005.07.016.
Prasad M, Iqbal M, Mordekar SR. Nonketotic hyperglycinemia case series. J Pediatr Neurosci. 2015;10(4):355. https://doi.org/10.4103/1817-1745.174445.
Aliefendioğlu D, Tana Aslan A, Coşkun T, Dursun A, Çakmak FN, Kesimer M. Transient nonketotic hyperglycinemia: two case reports and literature review. Pediatr Neurol. 2003;28(2):151–5. https://doi.org/10.1016/s0887-8994(02)00501-5.
Zammarchi E, Donati M, Ciani F. Transient neonatal nonketotic hyperglycinemia: a 13-year follow-up. Neuropediatrics. 1995;26(06):328–30. https://doi.org/10.1055/s-2007-979785.
Mourmans J, Majoie CBLM, Barth PG, Duran M, Akkerman EM, Poll-The BT. Sequential MR imaging changes in nonketotic hyperglycinemia. AJNR Am J Neuroradiol. 2006;27(01):208–11.
Raghavendra S, Ashalatha R, Thomas SV, Kesavadas C. Focal neuronal loss, reversible subcortical focal T2 hypointensity in seizures with a nonketotic hyperglycemic hyperosmolar state. Neuroradiology. 2007;49(4):299–305. https://doi.org/10.1007/s00234-006-0189-6.
Langan TJ, Pueschel SM. Nonketotic hyperglycemia: clinical, biochemical, and therapeutic considerations. Curr Probl Pediatr. 1983;13(3):8–30. https://doi.org/10.1016/0045-9380(83)90016-6.
Perry TL, Urquhart N, MacLean J, Evans ME, Hansen S, Davidson AG, et al. Nonketotic hyperglycinemia. N Engl J Med. 1975;292(24):1269–73. https://doi.org/10.1056/nejm197506122922404.
Hayasaka K, Tada K, Fueki N, Aikawa J. Prenatal diagnosis of nonketotic hyperglycinemia: enzymatic analysis of the glycine cleavage system in chorionic villi. J Pediatr. 1990;116(3):444–5. https://doi.org/10.1016/s0022-3476(05)82841-0.
Kikuchi G. The glycine cleavage system: composition, reaction mechanism, and physiological significance. Mol Cell Biochem. 1973;1(2):169–87. https://doi.org/10.1007/bf01659328.
Sakata Y, Owada Y, Sato K, Kojima K, Hisanaga K, Shinka T, et al. Structure and expression of the glycine cleavage system in rat central nervous system. Brain Res Mol. 2001;94(1–2):119–30. https://doi.org/10.1016/s0169-328x(01)00225-x.
Kure S. Two novel laboratory tests facilitating diagnosis of glycine encephalopathy (nonketotic hyperglycinemia). Brain Dev. 2011;33(9):753–7. https://doi.org/10.1016/j.braindev.2011.03.001.
McDearmid J, Liao M, Drapeau P. Glycine receptors regulate interneuron differentiation during spinal network development. Proc Natl Acad Sci. 2006;103(25):9679–84. https://doi.org/10.1073/pnas.0504871103.
Ando T, Nyhan W, Connor J, Rasmussen K, Donnell G, Barnes N, et al. The oxidation of glycine and propionic acid in propionic acidemia with ketotic hyperglycinemia. Pediatr Res. 1972;6(6):576–83. https://doi.org/10.1203/00006450-197206000-00006.
Mohammad SA, Abdelkhalek HS. Nonketotic hyperglycinemia: spectrum of imaging findings with emphasis on diffusion-weighted imaging. Neuroradiology. 2017;59(11):1155–63. https://doi.org/10.1007/s00234-017-1913-0.
Khong P-L, Lam BCC, Chung BHY, Wong K-Y, Ooi G-C. Diffusion weighted MR imaging in neonatal nonketotic hyperglycinemia. AJNR Am J Neuroradiol. 2003;24(06):1181–2118.
Kanekar S, Byler D. Characteristic MRI findings in neonatal nonketotic hyperglycinemia due to sequence changes in GLDC gene encoding the enzyme glycine decarboxylase. Metab Brain Dis. 2013;28(4):717–20. https://doi.org/10.1007/s11011-013-9415-1.
Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325(6104):529–31. https://doi.org/10.1038/325529a0.
Johnston MV. Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Fetal and Perinatal Neurology. 1992:19–30. https://doi.org/10.1159/000420416.
Sato K, Yoshida S, Fujiwara K, Tada K, Tohyama M. Glycine cleavage system in astrocytes. Brain Res J. 1991;567(1):64–70. https://doi.org/10.1016/0006-8993(91)91436-5.
Tada K, Kure S. Non-ketotic hyperglycinaemia: molecular lesion, diagnosis and pathophysiology. J Inherit Metab Dis. 1993;16(4):691–703. https://doi.org/10.1007/bf00711901.
Mademont-Soler I, Casellas-Vidal D, Trujillo A, Espuña-Capote N, Maroto A, del García-González M, et al. glyt1 encephalopathy: further delineation of disease phenotype and discussion of pathophysiological mechanisms. Am J Med Genet A. 2020;185(2):476–85. https://doi.org/10.1002/ajmg.a.61996.
Acknowledgements
The authors would like to acknowledge the management of JSS Academy of Higher Education & Research, Mysuru, Karnataka, for supporting the basic research ideas and also for the resources provided.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
BS searched, collected papers and contributed in writing the manuscript. BMK, SB and TS revised review and manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No ethical approval was obtained or required for the purposes of this review.
Consent for publication
Dr. Kanthesh M Basalingappa, Associate Professor & Coordinator, Division of Molecular Biology, School of Life Science, JSS Academy of Higher Education & Research, Mysuru-570015. Email: kantheshmb@jssuni.edu.in.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bhumika, S., Basalingappa, K.M., Gopenath, T.S. et al. Glycine encephalopathy. Egypt J Neurol Psychiatry Neurosurg 58, 132 (2022). https://doi.org/10.1186/s41983-022-00567-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41983-022-00567-6