
Abstract
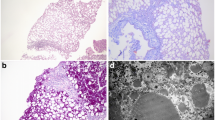
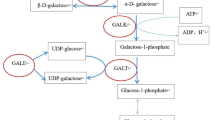
Transient infantile hypertriglyceridemia (HTGT1; OMIM #614480) is a rare autosomal recessive disorder, which manifests in early infancy with transient hypertriglyceridemia, hepatomegaly, elevated liver enzymes, persistent fatty liver and hepatic fibrosis. This rare clinical entity is caused by inactivating mutations in the GPD1 gene, which encodes the cytosolic isoform of glycerol-3-phosphate dehydrogenase. Here we report on four patients from three unrelated families of diverse ethnic origins, who presented with hepatomegaly, liver steatosis, hypertriglyceridemia, with or without fasting ketotic hypoglycemia. Whole exome sequencing revealed the affected individuals to harbor deleterious biallelic mutations in the GPD1 gene, including the previously undescribed c.806G > A (p.Arg269Gln) and c.640T > C (p.Cys214Arg) mutations. The clinical features in three of our patients showed several differences compared to the original reports. One subject presented with recurrent episodes of fasting hypoglycemia along with hepatomegaly, hypetriglyceridemia, and elevated liver enzymes; the second showed a severe liver disease, with intrahepatic cholestasis associated with kidney involvement; finally, the third presented persistent hypertriglyceridemia at the age of 30 years. These findings expand the current knowledge of this rare disorder, both with regard to the phenotype and molecular basis. The enlarged phenotypic spectrum of glycerol-3-phosphate dehydrogenase 1 deficiency can mimic other inborn errors of metabolism with liver involvement and should alert clinicians to recognize this entity by considering GPD1 mutations in appropriate clinical settings.

Similar content being viewed by others
References
Basel-Vanagaite L, Zevit N, Har Zahav A et al (2012) Transient infantile hypertriglyceridemia, fatty liver, and hepatic fibrosis caused by mutated GPD1, encoding glycerol-3-phosphate dehydrogenase 1. Am J Hum Genet 90(1):49–60
Brown LJ, Koza RA, Marshall L et al (2002) Lethal hypoglycemic ketosis and glyceroluria in mice lacking both the mitochondrial and the cytosolic glycerol phosphate dehydrogenases. J Biol Chem 277(36):32899–904
Cordeddu V, Redeker B, Stellacci E et al (2014) Mutations in ZBTB20 cause Primrose syndrome. Nat Genet 46(8):815–7
Dimmock D, Maranda B, Dionisi-Vici C et al (2009) Citrin deficiency, a perplexing global disorder. Mol Genet Metab 96(1):44–9
Drmanac R, Sparks AB, Callow MJ et al (2010) Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327(5961):78–81
Fiermonte G, Parisi G, Martinelli D et al (2011) A new Caucasian case of neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD): a clinical, molecular, and functional study. Mol Genet Metab 104(4):501–6
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14(1):27–8
Joshi M, Eagan J, Desai NK et al (2014) A compound heterozygous mutation in GPD1 causes hepatomegaly, steatohepatitis, and hypertriglyceridemia. Eur J Hum Genet 22(10):1229–32
Komatsu M, Yazaki M, Tanaka N et al (2008) Citrin deficiency as a cause of chronic liver disorder mimicking non-alcoholic fatty liver disease. J Hepatol 49(5):810–20
Kortum F, Caputo V, Bauer CK et al (2015) Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat Genet 47(6):661–7
Li MX, Gui HS, Kwan JS et al (2012) A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res 40(7):e53
MacDonald MJ, Marshall LK (2000) Mouse lacking NAD + -linked glycerol phosphate dehydrogenase has normal pancreatic beta cell function but abnormal metabolite pattern in skeletal muscle. Arch Biochem Biophys 384(1):143–53
Moriyama M, Fujimoto Y, Rikimaru S et al (2015) Mechanism for increased hepatic glycerol synthesis in the citrin/mitochondrial glycerol-3-phosphate dehydrogenase double-knockout mouse: urine glycerol and glycerol 3-phosphate as potential diagnostic markers of human citrin deficiency. Biochim Biophys Acta 1852(9):1787–95
Niceta M, Stellacci E, Gripp KW et al (2015) Mutations impairing GSK3-mediated MAF phosphorylation cause cataract, deafness, intellectual disability, seizures, and a down syndrome-like facies. Am J Hum Genet 96(5):816–25
Ou X, Ji C, Han X et al (2006) Crystal structures of human glycerol 3-phosphate dehydrogenase 1 (GPD1). J Mol Biol 357(3):858–69
Song YZ, Li BX, Chen FP et al (2009) Neonatal intrahepatic cholestasis caused by citrin deficiency: clinical and laboratory investigation of 13 subjects in mainland of China. Dig Liver Dis 41(9):683–9
Song YZ, Deng M, Chen FP et al (2011) Genotypic and phenotypic features of citrin deficiency: five-year experience in a Chinese pediatric center. Int J Mol Med 28(1):33–40
Wu JW, Yang H, Wang SP et al (2015) Inborn errors of cytoplasmic triglyceride metabolism. J Inherit Metab Dis 38(1):85–98
Acknowledgments
The authors wish to thank the patients and their families for their kind assistance. This study was supported in part by intramural funds provided by Ospedale Pediatrico Bambino Gesù (Ricerca corrente 2015 and 2016, Vite Coraggiose and GeneRare) and by the Association “La Vita e’ un Dono”.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
None.
Informed consent
All procedures followed were in accordance with the ethical standards of the responsible institutional committee on human experimentation and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study. Proof that informed consent was obtained must be available upon request.
Additional information
Communicated by: Jean-Marie Saudubray
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
Urinary organic acids profile in patient C at the age of 1.5 years showing massive elevation of glycerol excretion and dicarboxylic aciduria. Peaks are: 1. lactic acid; 2. 3-hydroxy-butyric acid; 3. glycerol; 4. succinic acid; 5. internal standard; 6. 3-hydroxyadipic lactone; 7. adipic acid; 8. 4-hydroxy-phenyllacetic acid; 9. cis-4-octenedioic acid; 10. 3-hydroxy-adipic acid; 11. suberic acid; 12. internal standard; 13. cis aconitic acid; 14. decenedioic acid; 15. 3-hydroxy-octenedioic acid; 16. 3-hydroxy-octanedioic acid; 17. sebacic acid; 18. 3-hydroxy-decenedioic acid; 19. 2-hydroxy-decanedioic acid; 20. 3-hydroxy-decanedioic acid; 21. 3-hydroxy-dodecanedienedioic acid; 22. 3-hydroxy-dodecenedioic acid; 23. 3-hydroxy-dodecanedioic acid; 24. tetradecenedioic acid. (PPTX 71 kb)
Supplementary Fig. 2
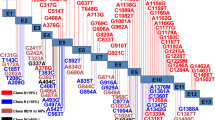
Structural impact of the Cys214Arg and Arg269Gln substitutions in GPD1. Panel A) The X-ray structure of the ternary GPD1/DHAP/NAD+ complex is reported with the N- and C-terminal domains shown in blue and green, respectively. NAD+ (pink) and DHAP (orange) are also shown. The lateral chains of the two affected residues, Cys214 and Arg269, are highlighted in purple. Panel B) Close up of the active site region. The lateral chain of Arg269 and those of other residues with role in catalysis are shown. Panel C) Dimeric form of the ternary GPD1/DHAP/NAD+ complex. Panel D) Close up of the inter-monomer interacting region. The hydrogen bonds highlighted by the X-ray structure, between Asn222 (monomer 1) and Tyr263 and Ile152 (monomer 2) are shown. Molecular graphics images were generated with VMD (Humphrey et al 1996). (PPTX 2289 kb)
Supplementary Table 1
Summary of demographic, clinical and molecular data of all known patients with transient infantile hypertriglyceridemia caused by biallelic mutations in GPD1 (DOCX 14 kb)
Rights and permissions
About this article

Cite this article
Dionisi-Vici, C., Shteyer, E., Niceta, M. et al. Expanding the molecular diversity and phenotypic spectrum of glycerol 3-phosphate dehydrogenase 1 deficiency. J Inherit Metab Dis 39, 689–695 (2016). https://doi.org/10.1007/s10545-016-9956-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-016-9956-7