
Abstract
Background
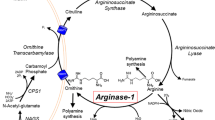
Arginase 1 (ARG1) deficiency is a rare urea cycle disorder (UCD). This hypothesis-generating study explored clinical phenotypes, metabolic profiles, molecular genetics, and treatment approaches in a cohort of children and adults with ARG1 deficiency to add to our understanding of the underlying pathophysiology.
Methods
Clinical data were retrieved retrospectively from physicians using a questionnaire survey. Plasma aminoacids, guanidinoacetate (GAA), parameters indicating oxidative stress and nitric oxide (NO) synthesis as well as asymmetric dimethylarginine (ADMA) were measured at a single study site.
Results
Nineteen individuals with ARG1 deficiency and 19 matched controls were included in the study. In patients, paraparesis, cognitive impairment, and seizures were significantly associated suggesting a shared underlying pathophysiology. In patients plasma GAA exceeded normal ranges and plasma ADMA was significantly elevated. Compared to controls, nitrate was significantly higher, and the nitrite:nitrate ratio significantly lower in subjects with ARG1 deficiency suggesting an advantage for NO synthesis by inducible NO synthase (iNOS) over endothelial NOS (eNOS). Logistic regression revealed no significant impact of any of the biochemical parameters (including arginine, nitrates, ADMA, GAA, oxidative stress) or protein restriction on long-term outcome.
Conclusion
Three main hypotheses which must be evaluated in a hypothesis driven confirmatory study are delineated from this study: 1) clinical manifestations in ARG1 deficiency are not correlated with arginine, protein intake, ADMA, nitrates or oxidative stress. 2) GAA is elevated and may be a marker or an active part of the pathophysiology of ARG1 deficiency. 3) Perturbations of NO metabolism merit future attention in ARG1 deficiency.
Similar content being viewed by others


References
Adam S, Champion H, Daly A et al (2012) Dietary management of urea cycle disorders: UK practice. J Hum Nutr Diet 25:398–404
Amayreh W, Meyer U, Das AM (2014) Treatment of arginase deficiency revisited: guanidinoacetate as a therapeutic target and biomarker for therapeutic monitoring. Dev Med Child Neurol 56:1021–1024
Anderssohn M, Schwedhelm E, Lüneburg N, Vasan RS, Böger RH (2010) Asymmetric dimethylarginine as a mediator of vascular dysfunction and a marker of cardiovascular disease and mortality: an intriguing interaction with diabetes mellitus. Diab Vasc Dis Res 7:105–118
Andrade F, Llarena M, Lage S, Aldámiz-Echevarría L (2015) Quantification of arginine and its methylated derivatives in healthy children by liquid chromatography-tandem mass spectrometry. J Chromatogr Sci 53:787–792
Arias A, Ormazabal A, Moreno J et al (2006) Methods for the diagnosis of creatine deficiency syndromes: a comparative study. J Neurosci Methods 156:305–309
Carvalho DR, Brum JM, Speck-Martins CE et al (2012) Clinical features and neurologic progression of hyperargininemia. Pediatr Neurol 46:369–374
Crombez EA, Cederbaum SD (2005) Hyperargininemia due to liver arginase deficiency. Mol Gen Metab 84:243–251
Deignan JL, Marescau B, Livesay JC et al (2008) Increased plasma and tissue guanidino compounds in a mouse model of hyperargininemia. Mol Gen Metab 93:172–178
Deignan JL, DeDeyn PP, Cederbaum SD et al (2010) Guanidino compound levels in blood, cerebrospinal fluid, and post-mortem brain material of patients with argininemia. Mol Gen Metab 100:S31–S36
Delwing de Lima D, Delwing F, da Cruz JG, Wyse AT, Delwing-Dal Magro D (2012) Protective effect of antioxidants on blood oxidative stress caused by arginine. Fundam Clin Pharmacol 26:250–258
Delwing D, Delwing D, Bavaresco CS, Wyse AT (2008) Protective effect of nitric oxide synthase inhibition or antioxidants on brain oxidative damage caused by intracerebroventricular arginine administration. Brain Res 1193:120–127
Grioni D, Furlan F, Canonico F, Parini R (2014) Epilepsia partialis continua and generalized nonconvulsive status epilepticus during the course of argininemia: a report on two cases. Neuropediatr 45:123–128
Joncquel-Chevalier Curt M, Cheillan D, Briand G et al (2013) Creatine and guanidinoacetate reference values in a French population. Mol Genet Metab 110:263–267
Kayacelebi AA, Langen J, Weigt-Usinger K et al (2015) Biosynthesis of homoarginine (hArg) and asymmetric dimethylarginine (ADMA) from acutely and chronically administered free L-arginine in humans. Amino Acids 47(9):1893–1908
Kolling J, Wyse AT (2010) Creatine prevents the inhibition of energy metabolism and lipid peroxidation in rats subjected to GAA administration. Metab Brain Dis 25:331–338
Loscalzo J (2003) Adverse effects of supplemental L-arginine in atherosclerosis. Consequences of methylation stress in a complex catabolism? Arterioscler Thromb Vasc Biol 23:3–5
Lüneburg N, Xanthakis V, Schwedhelm E (2011) Reference intervals for plasma L-arginine and the L-arginine:asymmetric dimethylarginine ratio in the Framingham Offspring Cohort. J Nutr 141:2186–2190
Mc Guire PJ, Parikh A, Diaz GA (2009) Profiling of oxidative stress in patients with inborn errors of metabolism. Mol Genet Metab 98:173–180
Nagasaka H, Tsukahara H, Yorifuji T et al (2009) Evaluation of endogenous nitric oxide synthesis in congenital urea cycle enzyme defects. Metabolism 58:278–282
Olsen RK, Cornelius N, Gregersen N (2015) Redox signalling and mitochondrial stress responses; lessons from inborn errors of metabolism. J Inherit Metab Dis doi: 10.1007/s10545-015-9861-5
Pastore A, Martinelli D, Piemonte F et al (2014) Glutathione metabolism in cobalamin deficiency type C (cblC). J Inherit Metab Dis 37:125–129
Ribas GS, Vargas CR, Wajner M (2014) L-carnitine supplementation as a potential antioxidant therapy for inherited neurometabolic disorders. Gene 533:469–476
Rüegger CM, Lindner M, Ballhausen D et al (2014) Cross-sectional observational study of 208 patients with non-classical urea cycle disorders. J Inherit Metab Dis 37:21–30
Schlune A, Ensenauer R, Häussinger D, vom Dahl S, Mayatepek E (2015) Hyperarginiemia due to arginase I deficiency: the original patients and their natural history, and a review of the literature. Amino Acids. doi:10.1007/s00726-015-2032-z
Scholl-Bürgi S, Sigl SB, Häberle J et al (2008) Amino acids in CSF and plasma in hyperammonaemic coma due to arginase1 deficiency. J Inherit Metab Dis 31:S323–S328
Schulze A, Battini R (2008) Pre-symptomatic treatment of creatine biosynthesis defects. In: Salomons GS, Wyss M (eds) Creatine and creatine kinase in health and disease, Subcellular Biochemistry 46:167–181
Schulze F, Wesemann R, Schwedhelm E et al (2004) Determination of asymmetric dimethylarginine (ADMA) using a novel ELISA assay. Clin Chem Lab Med 42:1377–1383
Schulze F, Lenzen H, Hanefeld C et al (2006) Asymmetric dimethylarginine is an independent risk factor for coronary heart disease: results from the multicenter coronary artery risk determination investigating the influence of ADMA concentration (CARDIAC) study. Am Heart J 152:e1–e8
Snyderman SE, Sansaricq C, Norton PM, Goldstein F (1979) Argininemia treated from birth. J Pediatr 95:61–63
Tsikas D (2005) Methods of quantitative analysis of the nitric oxide metabolites nitrite and nitrate in human biological fluids. Free Rad Res 39:797–815
Vockley JG, Tabor DE, Kern RM et al (1994) Identification of mutations (D128G, H141L) in the liver arginase gene of patients with hyperargininemia. Hum Mutat 4:150–154
Wijnands KA, Hoeksema MA, Meesters DM et al (2014) Arginase-1 deficiency regulates arginine concentrations and NOS2-mediated NO production during endotoxemia. PLoS One 9:e86135
Zugno AI, Stefanello FM, Scherer EB et al (2008) Guanidinoacetate decreases antioxidant defenses and total protein sulfhydryl content in striatum of rats. Neurochem Res 33:1804–1810
Acknowledgments
We gratefully acknowledge the contribution of Veronique Rüfenacht and Martin Volleberg, University Children’s Hospital Zürich, Switzerland; Andreas Kurringer, Christian Kerle, Fulya Zimmerer, Martin Fleger, Maximilian Obwegeser, Evelyn Gamper and Klaus Ludescher, LKH Bregenz, Austria and Luca Fierro and George A. Diaz, Icahn School of Medicine at Mount Sinai, New York, USA. The Government of Vorarlberg, Austria and Nutricia Metabolics, Friedrichsdorf, Germany have financially supported the study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All procedures followed were in accordance with the ethical standards of the responsible local committees on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained by the physicians from their patients or their caregivers for being included in the study.
Conflict of interest
None.
Additional information
Communicated by: Johan Lodewijk Karel Van Hove
Rights and permissions
About this article

Cite this article
Huemer, M., Carvalho, D.R., Brum, J.M. et al. Clinical phenotype, biochemical profile, and treatment in 19 patients with arginase 1 deficiency. J Inherit Metab Dis 39, 331–340 (2016). https://doi.org/10.1007/s10545-016-9928-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-016-9928-y