
Abstract
The assessment of urinary α-aminoadipic semialdehyde (α-AASA) has become the diagnostic laboratory test for pyridoxine dependent seizures (PDS). α-AASA is in spontaneous equilibrium with its cyclic form Δ1-piperideine-6-carboxylate (P6C); a molecule with a heterocyclic ring structure. Ongoing diagnostic screening and monitoring revealed that in some individuals with milder ALDH7A1 variants, and patients co-treated with a lysine restricted diet, α-AASA was only modestly increased. This prompted us to investigate the diagnostic power and added value of the assessment of urinary P6C compared to α-AASA. Urine samples were diluted to a creatinine content of 0.1 mmol/L, followed by the addition of 0.01 nmol [2H9]pipecolic acid as internal standard (IS) and 5 μL was injected onto a Waters C18 T3 HPLC column. Chromatography was performed using water/methanol 97/3 (v/v) including 0.03 % formic acid by volume with a flow rate of 150 μL/min and detection was accomplished in the multiple reaction monitoring mode: P6C m/z 128.1 > 82.1; [2H9]pipecolic acid m/z 139.1 > 93.1. Due to the dualistic nature of α-AASA/P6C, and the lack of a proper internal standard, the method is semi quantitative. The intra-assay CVs (n = 10) for two urine samples of proven PDS patients with only modest P6C increases were 4.7% and 8.1%, whereas their inter-assay CVs (n = 10) were 16 and 18% respectively. In all 40 urine samples from 35 individuals with proven PDS, we detected increased levels of P6C. Therefore, we conclude that the diagnostic power of the assessments of urinary P6C and α-AASA is comparable.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The finding that α-aminoadipic semialdehyde dehydrogenase (α-AASA DH/Antiquitin) deficiency is the underlying defect in the vast majority of individuals affected with pyridoxine dependent seizures (PDS), has provided the metabolic field with a specific biomarker: α-aminoadipic semialdehyde (α-AASA) (Mills et al. 2006; Plecko et al. 2007; Struys & Jakobs 2007). Moreover, it has paved the way for molecular diagnosis by investigations of the corresponding gene, i.e., ALDH7A1. Previously, the assessment of pipecolic acid in plasma and cerebrospinal fluid was the only biochemical tool for the metabolic laboratories (Plecko et al. 2000; Willemsen et al. 2005). A drawback of the plasma pipecolic acid assessment is that pipecolic acid can also be increased secondary to peroxisome biogenesis disorders or liver disease (Steinberg et al. 2006). In our laboratory, we started the measurement of α-AASA in body fluids in 2005 (Mills et al. 2006) (with urine being the preferred matrix), and we have detected in more than 100 individuals increases of α-AASA. In all cases, when DNA was subsequently sent to our laboratory, mutations were detected in the ALDH7A1 gene, illustrating the specificity of the α-AASA assessment. Chemically, α-AASA is an interesting molecule, and has not been picked up in the 50 years following the first description of PDS (Hunt et al. 1954). α-AASA is in spontaneous equilibrium with its cyclic form Δ1-piperideine-6-carboxylate (P6C); a molecule with a heterocyclic ring structure (Fig. 1). We have prepared α-AASA out of commercially available allysine ethylene acetal (Mills et al. 2006), and the obtained product is indeed a mixture of both forms: α-AASA and P6C. This dualistic nature hampers absolute quantification; however the increase in the concentrations of α-AASA, even when patients were on pyridoxine supplementation, were several fold the upper limits of the age-matched control population.

Spontaneous equilibrium of P6C and α-AASA
Ongoing diagnostic screening and monitoring in our laboratory has revealed that some individuals with milder ALDH7A1 variants, and patients co-treated with a lysine restricted diet, displayed only modest increases.
This prompted us to investigate the diagnostic power and added value of the assessment of urinary P6C compared to α-AASA. We have developed a sensitive LC-MS/MS method which allowed us to detect P6C in non affected individuals. Urine samples were upfront diluted to a creatinine content of 0.1 mmol/L and aliquots were directly injected onto the LC-MS/MS without the need for derivatisation. Reference values were established and 40 urine samples of individuals in which α-AASA was previously found to be increased were used for retrospective P6C quantification.
Material and methods
Urine specimens
Reference urine samples
The study was approved by the human medical ethics committee of Máxima Medical Centre Veldhoven. Reference urine samples (n = 91) were obtained from neonates admitted to a level IIIa neonatology intensive care unit (NICU) in Veldhoven, the Netherlands. When urine was sampled for standard neonatal care, left over material was stored at -20°C; no additional urine samples were collected for the sole purpose of this study. From the patient’s medical records, gestational age, birth weight, actual weight at urine sampling, Apgar score, protein intake, medication including anti-epileptic drugs and hypothermia treatment were documented. In all these samples α-AASA was assessed and found to be within the appropriate reference range. Reference values beyond the neonatal period were obtained by analyzing P6C levels in α-AASA negative urine samples which had been sent to our laboratory for diagnostic studies.
Urine samples from PDS patients
In 40 urines, stored at -20°C, of genetically proven and urinary α-AASA positive PDS patients, we have retrospectively determined the concentration of P6C. The vast majority of these children were on pyridoxine treatment at the time of urine sampling, and in some individual cases, children were co-treated with a lysine restricted diet.
Materials
[2H9]pipecolic acid was purchased from CDN isotopes, Pointe-Clair, Canada. All other reagents and solvents were of analytical grade. P6C was prepared out of allysine ethylene acetal (Chiralix, The Netherlands) as described before (Mills et al. 2006). 1H-NMR studies of the obtained solution showed small amounts of allysine ethylene acetal, in the presence of a complicated spectrum of multiplets in the region of 1.5 to 2.5 ppm. The actual concentration of α-AASA relative to P6C could not be concluded by the obtained 1H-NMR data. Tandem mass spectrometer Q1 scans, in both the negative and positive electrospray ionization mode, showed signals corresponding to α-AASA and P6C, and a less abundant signal to allysine ethylene acetal (detectable in positive mode). Product ion scans (positive mode) of m/z 128.1 (corresponding to the M + 1 signal of P6C) yielded two main fragmentation products with m/z 82 and m/z 55. Product ion scans (positive mode) of a standard solution of pipecolic acid, which can be regarded as the dihydro-analogue of P6C, showed two main fragmentation products with m/z 84 and m/z 56. All these combined data confirmed the identity of P6C, and we have optimized the LC-MS/MS using the obtained α-AASA/P6C reference solution.
P6C sample preparation and LC-MS/MS determination
Urine samples were diluted to a creatinine content of 0.1 mmol/L with solvent A (see below), followed by the addition of 0.01 nmol [2H9]pipecolic acid (10 μL of aqueous 0.001 mmol/L solution) serving as internal standard (IS), resulting in a final sample volume of 400 μL. Prepared samples were stable for more than 1 week at 4°C. 5 μL was injected onto a Waters C18 T3 HPLC column (150 × 2.1 mm, bead size 3.5 μm). Chromatography was performed using solvent A (water/methanol 97/3 (v/v) including 0.03 % formic acid by volume) with a flow rate of 150 μL/min. After elution of P6C and the IS, the column was rinsed with solvent B (water/methanol 50/50 (v/v) including 0.03 % formic acid by volume), and was subsequently allowed to regenerate with 100% solvent A. Total runtime including regeneration of the column was 7 minutes. The column was directly connected to an AB Sciex 4000 QTrap tandem mass spectrometer operating in the positive electrospray ionization mode and at optimized settings, and detection was accomplished in the multiple reaction monitoring mode. The MRM transitions for P6C were: m/z 128.1 > 82.1 (quantifier) and m/z 128.1 > 55.1 (qualifier); the MRM transition for [2H9]pipecolic acid was m/z 139.1 > 93.1 . The P6C m/z 128.1 > 82.1 transition and the [2H9]pipecolic acid m/z 139.1 > 93.1 transitions correspond to the loss of the carboxyl moiety from the molecules. In addition, also the MRM transition for endogenous pipecolic acid, i.e., m/z 130.1 > 84 was monitored. Results were calculated by directly using the obtained P6C/IS and pipecolic acid/IS peak-area ratios.
Urine spots were prepared by applying 5 μL of urine onto a Guthrie card, after which the urine was allowed to dry at room temperature and the spots were stored in the dark at room temperature. The edges of the spots were marked with a pencil, cut out by scissors, put in a small tube and extracted in 500 μL of double distilled water. The extraction was stimulated by continuous motion of the tube for 30 minutes at room temperature, after which the tube were centrifuged at 14000 g and the supernatant was used for P6C quantification. The amount of reconstituted urine sample taken into preparations was determined by the dilution of the original urine spot relative to the volume of the extraction solvent. α-AASA has been assessed by LC-MS/MS as described before (Mills et al. 2006). Briefly, 1 nmol [15 N]-α-aminoadipic acid (10 μL of aqueous 0.1 mmol/L solution) serving as internal standard, was added to 10 μL of urine. Subsequently, 100 μL of borate buffer (0.1 mmol/L, pH 10) and 100 μL of Fmoc-Cl solution (1,5 mg/mL acetone) were added, samples were mixed vigorously, and allowed to react for 15 minutes at room temperature. 5 uL of the sample was injected onto an LC-MS/MS (as for P6C). Chromatography was performed using a Waters X-Terra C18 column (150 mm × 4.6 mm ID; 5 μm particle size) applying a binary linear gradient starting with 100% solvent A (H2O/ACN 90/10 v/v, containing 125 mg ammonium formate) changing to 100% solvent B (H2O/ACN 90/10 v/v, containing 125 mg ammonium formate) in 10 minutes. Analytes were measured in negative mode using the following transitions: m/z -382.1 > -186.1 (α-aminoadipic acid); m/z -383.1 > -187.1 ([15 N]-α-aminoadipic acid); m/z -366.1 > -170.1 (α-AASA quantifier); m/z -366.1 > -144.1 (α-AASA qualifier).
Stability studies
Multiple aliquots of two urine samples of known PDS patients were stored under 3 different conditions: 1. at room temperature, 2. in the refrigerator at 4°C, and 3. in the freezer at -20°C. The samples were prepared and analyzed over a period of 40 days. Multiple Guthrie card spots from urine samples of five known PDS patients were prepared, stored in the dark at room temperature, and analyzed over a period of 30 days.
Results
Analytical performance of the LC-MS/MS method
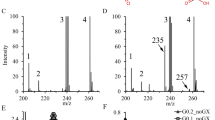
The mass fragmentograms of the LC-MS/MS measurement in urine samples from a PDS positive and PDS negative individual are depicted in fig. 2. P6C elutes at 2.25 minutes and [2H9]pipecolic acid at 2.50 minutes. It is important to note that due to the dualistic nature of α-AASA/P6C absolute quantification of one or the other is virtually impossible. Therefore all quantitative results presented in this study, represent estimations of the P6C concentration. For all experiments, we have chosen to use a single point calibration for the quantification of P6C, using the known concentration of the internal standard as direct reference. We made the assumption that pipecolic acid and P6C have identical mass spectrometric characteristics, although we are currently not able to substantiate this.

Mass fragmentograms of the LC-MS/MS measurement of P6C; in the left panel: control urine sample (concentration 0.02 mmol/mol creatinine), and in the right panel: PDS patient urine sample (concentration 2.05 mmol/mol creatinine). IS represents the signal of [2H9]pipecolic acid serving as internal standard
Assay characteristics
The intra day coefficient of variations (CVs) (n = 10) for two urine samples of two proven PDS patients with modest P6C increases were 4.7% (average ± sd: 0.38 ± 0.018 mmol/mol creatinine) and 8.1% (average ± sd: 0.68 ± 0.055 mmol/mol creatinine); the inter day CVs (n = 10) for these two urine samples were 16% (average ± sd: 0.35 ± 0.056 mmol/mol creatinine) and 18% (average ± sd: 0.69 ± 0.13 mmol/mol creatinine) respectively. Linearity of the method was examined by multiple dilutions of a positive PDS urine sample with a negative urine sample with an identical creatinine concentration. The quantification of P6C was linear over the range of 1 to 30-fold dilutions, with absolute P6C concentrations ranging from 0.3 μM to 9.3 μM. The limit of detection (S/N ratio =10) was estimated by verifying the P6C peak height relatively to the observed noise at the same chromatographic region and was found to be approximately 0.02 mmol/mol creatinine.
Stability studies
Multiple aliquots of two urines samples have been stored under three conditions. P6C was stable in both urines when samples were stored at -20°C for 40 days. Samples stored at 4°C showed clearly that P6C is not stable in this condition with a decrease at 40 days of 25% in urine sample A and 50% in urine sample B. P6C levels in urines stored at room temperature show a dramatic decrease of 75% for urine A to almost 100% in urine B. This decline is already notable after 5 days of storage with decreases of approximately 40%. P6C levels in urine spots on filter paper, stored in the dark at room temperature showed a decline to 50% of the original value after 10 days of storage.
Urinary P6C and α-AASA reference values
Since PDS often clinically manifest in the neonatal period, 91 urine samples were obtained from neonates admitted to a level IIIa NICU in Veldhoven, the Netherlands. Samples were directly stored at -20°C until further analysis. Additionally, 100 urine samples from individuals, in which α-AASA was normal (see reference ranges below), were used to obtain P6C reference values for all age groups. P6C and α-AASA values in newborns correlated positively with protein intake, and negatively with gestational age and body weight, but there was no correlation of P6C and α-AASA values with any studied neonatal IC condition, treatment, or gender. The Spearman correlation between P6C and birth weight was -0.18; P = 0.09; between P6C and gestational age -0.21; P = 0.05, and between P6C and protein intake 0.33; P = 0.003 (Fig. 3a). The Spearman correlation between α-AASA and birth weight was -0.20; P = 0.04; between α-AASA and gestational age -0.21; P = 0.05, and between α-AASA and protein intake 0.33; P = 0.004 (Fig. 3b). For the whole studied control population, the levels of urinary P6C showed a clear age-dependency with P6C levels up to 0.37 mmol/mol creatinine in the first months of life, decreasing to ≤ 0.05 mmol/mol creatinine for children older than 1 year (Fig. 4). We define the following urinary P6C reference ranges: ≤ 6 months: P6C <0.37 mmol/mol creatinine; >6 months ≤1 year: P6C ≤ 0.1 mmol/mol creatinine; > 1 year: P6C ≤ 0.05 mmol/mol creatinine. The reference ranges for urinary α-AASA are: ≤ 6 months: α-AASA <2 mmol/mol creatinine; >6 months ≤1 year: α-AASA ≤ 1 mmol/mol creatinine; > 1 year α-AASA ≤ 0.5 mmol/mol creatinine. An overview of the used methodologies for the assessment of both P6C and α-AASA are depicted in Table 1. As our reference population consists of an overrepresentation of premature newborns, we recommend other laboratories to establish in-house reference ranges for α-AASA and P6C.


P6C levels in reference population; the insert displays P6C levels in children from 0 to 12 months of age
Urinary P6C levels in proven PDS patients
In a unique collection of 40 urine samples from 35 individuals with proven PDS, P6C was retrospectively assessed. In all urine samples in which previously α-AASA was found to be increased, we detected increased levels of P6C. The observed concentrations of P6C appear to be age-dependent. We have depicted all the positive urine samples in Fig. 5, illustrating the diagnostic performance of the measurement of urinary P6C. There is a moderate correlation between the urinary levels of α-AASA and P6C in this cohort of patient samples, as is illustrated in Fig. 6. It is of note that in all these patient samples pipecolic acid concentrations were within the appropriate reference ranges.

P6C levels in patients with proven PDS using a logarithmic Y-axis presentation

Relationship between α-AASA and P6C levels in urine samples from PDS patients; spearman correlation is 0.78; p < 0.001
Discussion
The differential diagnosis in neonatal and infantile seizures includes PDS. Although relatively rare, with an estimated incidence up to 1:276.000 births in The Netherlands (Bok et al. 2007), this condition is treatable and early diagnosis is of utmost importance. The current knowledge about the primary defect in PDS has provided new diagnostic biomarkers, i.e., α-AASA and P6C. Recent studies revealed that the clinical phenotype of PDS varies from classical PDS presenting as therapy resistant neonatal seizures to partly treatable seizures with the first presentation in early childhood (Mills et al. 2010). The focus of our group, as well as others has been on the urinary levels of α-AASA, which can be detected by either LC-MS/MS or GC-MS, following a derivatisation procedure (Mills et al. 2006; Pitt & Eggington 2007). α-AASA is known to be in spontaneous equilibrium with its cyclic form P6C, however the mechanism of this equilibrium is poorly understood. Ongoing diagnostics and treatment monitoring with respect to PDS revealed that in individual cases the α-AASA increase was only modest compared to the appropriate reference range. These observations prompted us to focus on urinary P6C. P6C has previously been assessed in plasma of PDS patients and indeed, increases were detected (Sadilkova et al. 2009). We have chosen to investigate P6C levels in urine of patients as urinary samples can be obtained non-invasively, which is advantageous in subsequent therapy monitoring. Moreover, we have noticed that α-AASA has a strong tendency to bind with protein, and it can be expected that the same is true for P6C.
Our proposed LC-MS/MS method for the urinary P6C quantification shows sufficient sensitivity, is reproducible, and is relatively easy to perform. In an acute situation, results can be obtained within 15 minutes after urine sampling. The stability studies clearly revealed the importance of proper sample storage and shipment. The strong decline in P6C concentrations in Guthrie cards excludes this sampling procedure since there is a potential risk of false negative results.
The observed positive correlation of both P6C and α-AASA and the protein intake by neonates may argue for a lysine restricted diet therapy, thereby lowering the flux through the lysine degradation pathway, resulting in lower levels of P6C and α-AASA. Currently, we are aware that such studies are underway, however no official results have been published.
The diagnostic strength of urinary P6C and α-AASA assessments is comparable, implying that both markers can be applied in a diagnostic setting. In those cases where there is doubt about the identity of P6C or α-AASA, the alternative method can be used as a second tier test. The availability of P6C and α-AASA as biomarkers for PDS allows and encourages low-threshold screening for this treatable disorder, also in those cases where the clinical suspicion of PDS is low according to the classical criteria (Baxter 2001).
References
Baxter P (2001) Pyridoxine dependent/responsive seizures. In: Baxter P (ed) Vitamin responsive conditions in paediatric neurology. Mac Keith, London, pp 109–165
Bok LA, Struys EA, Willemsen MA et al (2007) Pyridoxine-dependent seizures in Dutch patients: diagnosis by elevated urinary alpha-aminoadipic semialdehyde levels. Arch Dis Child 92:687–689
Hunt AD, Stokes J, McCrory WW et al (1954) Pyridoxine dependency: report of a case of intractable convulsions in an infant controlled by pyridoxine. Pediatrics 13:140–145
Mills PB, Struys E, Jakobs C et al (2006) Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med 12:307–309
Mills PB, Footitt EJ, Mills KA et al (2010) Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency). Brain 133:2148–2159
Pitt JJ, Eggington M (2007) Urine screening strategies for 2-aminoadipic semialdehyde dehydrogenase deficiency. J Inherit Metab Dis 30 (Suppl1)
Plecko B, Stöckler-Ipsiroglu S, Paschke E et al (2000) Pipecolic acid elevation in plasma and cerebrospinal fluid of two patients with pyridoxine-dependent epilepsy. Ann Neurol 48:121–125
Plecko B, Paul K, Paschke E et al (2007) Biochemical and molecular characterization of 18 patients with pyridoxine-dependent epilepsy and mutations of the antiquitin (ALDH7A1) gene. Hum Mutat 28:19–26
Sadilkova K, Gospe SM Jr, Hahn SH (2009) Simultaneous determination of alpha-aminoadipic semialdehyde, piperideine-6-carboxylate and pipecolic acid by LC-MS/MS for pyridoxine-dependent seizures and folinic acid-responsive seizures. J Neurosci Methods 30:136–141
Steinberg SJ, Dodt G, Raymond GV et al (2006) Peroxisome biogenesis disorders. Biochim Biophys Acta 1763:1733–1748
Struys EA, Jakobs C (2007) Alpha-aminoadipic semialdehyde is the biomarker for pyridoxine dependent epilepsy caused by alpha-aminoadipic semialdehyde dehydrogenase deficiency. Mol Genet Metab 91:405
Willemsen MA, Mavinkurve-Groothuis AM, Wevers RA et al (2005) Pipecolic acid: a diagnostic marker in pyridoxine-dependent epilepsy. Ann Neurol 58:653
Acknowledgements
We would like to thank Erwin Jansen for his technical assistance.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Sylvia Stoeckler-Ipsiroglu
Competing interest: None declared.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Struys, E.A., Bok, L.A., Emal, D. et al. The measurement of urinary Δ1-piperideine-6-carboxylate, the alter ego of α-aminoadipic semialdehyde, in Antiquitin deficiency. J Inherit Metab Dis 35, 909–916 (2012). https://doi.org/10.1007/s10545-011-9443-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-011-9443-0