
Abstract
Primary or premature ovarian insufficiency (POI) is the most common long-term complication experienced by girls and women with classic galactosemia; more than 80% and perhaps more than 90% are affected despite neonatal diagnosis and careful lifelong dietary restriction of galactose. In this review we explore the complexities of timing and detection of galactosemia-associated POI and discuss potential underlying mechanisms. Finally, we offer recommendations for follow-up care with current options for intervention.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Galactosemia is an inborn error of metabolism that results from impaired activity of any of the three enzymes of the Leloir pathway: galactokinase (GALK, EC 2.7.1.6), galactose-1-phosphate uridylyltransferase (GALT, EC 2.7.7.12), or UDP-galactose 4′-epimerase (GALE, EC 5.1.3.2) (reviewed in Fridovich-Keil and Walter 2008). Classic galactosemia (OMIM 230400), the most common clinically severe form of the disorder, results from profound impairment of GALT. Classic galactosemia impacts about 1/60,000 live births, although prevalence differs substantially among populations (reviewed in Fridovich-Keil and Walter 2008). Affected newborns appear normal, but following exposure to milk, which contains abundant galactose, develop symptoms that can escalate rapidly from vomiting and diarrhea to jaundice, failure to thrive, hepatomegaly, and E. coli sepsis, which can be lethal. Neonatal diagnosis with immediate dietary galactose restriction prevents or resolves the acute symptoms of classic galactosemia. However, despite even presymptomatic diagnosis and strict lifelong dietary intervention, a majority of patients go on to experience a constellation of troubling long-term complications. Sequelae include cognitive and/or behavioral impairment in close to half of all patients, speech difficulties in at least half of all patients, low bone mineral density in many patients, ataxia or tremor in some patients, and primary or premature ovarian insufficiency (POI) in at least 80% of all girls and women (reviewed in Fridovich-Keil and Walter 2008).
What is primary or premature ovarian insufficiency (POI)?
Primary or premature ovarian insufficiency is a spectrum disorder of ovarian dysfunction that differs considerably among women. In its mildest form, POI may present as diminished ovarian reserve with subfertility and often an elevated FSH level in a woman’s fourth decade of life. However, more severe forms of POI will present with primary amenorrhea in the young adolescent with absent sex steroid production and a complete lack of secondary sexual characteristics. Many women with POI have an intermediate phenotype, with normal progression through puberty and later onset of irregular to absent menstrual cycles and/or infertility. Proposed clinical criteria for the diagnosis of intermediate POI include secondary amenorrhea for at least 4 consecutive months or more, prior to the age of 40, in the setting of low estradiol and elevated FSH (Goswami and Conway 2005; Welt 2008).
The term “primary ovarian insufficiency” was first used in the literature more than 60 years ago (Albright 1942) and was re-introduced recently as a more appropriate term to replace less encompassing, compassionate, and descriptive terms such as “premature ovarian failure” or “premature menopause” that had come into use (Welt 2008). POI is not, in fact, the early onset of natural menopause. Many women with POI experience irregular ovarian function after their diagnosis, with months of regular menses and ovulation alternating with intervals of amenorrhea and hypoestrogenism. Antral follicle counts, determined by ultrasound, and serum FSH levels may vary considerably. This fluctuating course of ovarian function is seldom seen once a woman has experienced natural menopause and amenorrhea at a more typical age. Of note, 5–10% of women given a diagnosis of POI may still spontaneously conceive, and there is very little evidence that any current treatment increases this rate (Van Kasteren and Schoemaker 1999).
The prevalence of POI in the general population is not entirely known. An often quoted incidence of 1% is based largely on a 1986 study of female residents of Rochester, MN (Coulam et al. 1986). Of women in this cohort between the ages of 15 and 29, the incidence of POI was 1 in 10,000; for women between the ages of 30 and 39, it was 7.6 in 10,000. In contrast, the majority of reported information about the prevalence of diminished ovarian reserve comes from data on infertility patients. In 2007, 13% of women who underwent in vitro fertilization for infertility were diagnosed with diminished ovarian reserve (Centers for Disease Control and Prevention 2007). However, the prevalence of diminished ovarian reserve in the general population is unclear, in part because many cases may remain subclinical.
While there are many recognized causes of POI, in the majority of cases the etiology remains unknown. Approximately 23% of cases are caused by Turner’s syndrome or mosaicism for monosomy X (Conway 2000); this is the single most frequently recognized genetic cause of POI. Approximately 20% of women with POI show evidence of autoimmune disease (LaBarbera et al. 1988). Ovarian insufficiency has been associated with autoimmunity in a variety of organs including autoimmune Addison’s disease, Hashimoto’s thyroiditis, type 1 diabetes, rheumatoid arthritis, pernicious anemia, celiac sprue, vitiligo, lupus, Sjörgren’s syndrome, and autoimmune polyendocrine syndromes I and II (Wheatcroft and Weetman 1997). Abnormal karyotypes other than monosomy X represent a small contribution (Persani et al. 2009), and many single gene defects, other than galactosemia, have also been identified in families with POI; the most significant of these is the fragile X premutation, which accounts for roughly 5% of women with POI (Bussani et al. 2004; Conway et al. 1998; Gersak et al. 2003; Murray et al. 1998). Other notable mutations associated with POI involve the genes folliculogenesis growth factor GDF-9, BMP-15, and the sex determining factor FOXL2. (Crisponi et al. 2001; Di Pasquale et al. 2004; Laissue et al. 2006). Mutations impairing the expression or function of FSH (B subunit) or FSH receptor may also contribute to apparent ovarian dysfunction, although the former results in low levels of FSH, which would be atypical for POI (reviewed in Rubio-Gozalbo et al. 2010).
POI in galactosemia
The most common long-term complication reported for girls and women with profound GALT deficiency is POI, with an incidence of >80% and perhaps >90% (e.g., Kaufman et al. 1981; Waggoner et al. 1990; reviewed in Berry 2008; Fridovich-Keil and Walter 2008; Rubio-Gozalbo et al. 2010). Of note, patients with trace levels of residual GALT activity may demonstrate a milder phenotype (Berry 2008; Guerrero et al. 2000; Kaufman et al. 1988), leading to confusion or controversy in some instances. There have been no reports of ovarian dysfunction among women with Duarte galactosemia (who demonstrate about 25% normal GALT activity), and a cohort of galactosemia carrier women demonstrated apparently normal ovarian reserve and age at menopause (Knauff et al. 2007), although many of the women in that study were ascertained on the basis of their children, meaning there may have been a bias in the cohort toward women who were (or had been) fertile.
POI detection in prepubertal girls with classic galactosemia
Because of its prevalence in the patient population, there has been considerable interest in presymptomatic diagnosis of POI among prepubertal girls with classic galactosemia. Until recently, the only minimally invasive approach involved measurement of blood FSH values and estradiol values. However, FSH is an indirect measure, reflecting the hypothalamic response to diminished ovarian function, and this response may be absent in prepubertal girls. Ovarian imaging studies in prepubertal girls might also be informative, but these are more complex and are not routinely performed, especially on very young girls.
Recently we reported data suggesting that anti-Müllerian hormone (AMH), which is produced by granulosa cells of primary through late preantral and preovulatory follicles (La Marca and Volpe 2006), may provide a meaningful predictor of ovarian function in prepubertal girls with classic galactosemia (Sanders et al. 2009). In nongalactosemic women, AMH levels correlate with antral follicle counts visible by ultrasound and also with histologically determined primordial follicle counts (Hansen et al. 2010). While the reported results with galactosemic girls were promising, longitudinal studies will be needed to test the true predictive value of serum or plasma AMH levels measured in this population.
Timing of the onset of POI in galactosemia
POI in galactosemia may manifest as primary amenorrhea, secondary amenorrhea, or oligomenorrhea. Explanations for any of these outcomes could include a reduced initial oocyte pool, increased follicular atresia during development, or perhaps diminished maturation of primordial follicles. Determining the point in development at which POI begins has been difficult; whether the fundamental defect arises prenatally, in infancy, or later in childhood, ovarian insufficiency is not clinically apparent until puberty. Furthermore, a GALT-deficient animal model has not yet been reported that recapitulates the reproductive phenotype. However, results from numerous studies suggest that galactosemia-associated ovarian dysfunction has its roots in early development.
Newborn screening for galactosemia and prenatal diagnosis have allowed for early initiation of dietary galactose restriction; unfortunately, these advances have not led to a reduced incidence of POI in galactosemia (Rubio-Gozalbo et al. 2006; Schweitzer et al. 1993; Waggoner et al. 1990). The failure of dietary galactose restriction to improve reproductive outcomes suggests that GALT deficiency may first affect ovarian tissue in the pre- or perinatal period, prior to diagnosis and intervention. Those animal studies that have been reported support this hypothesis. In genetically wild-type rats, prenatal exposure to a high level of galactose has been shown to interfere with the migration of primordial germ cells to the developing gonad (Bandyopadhyay et al. 2003), and female pups demonstrate reduced initial oocyte pools (Chen et al. 1981).
Galactosemic females are undoubtedly exposed to galactose prenatally: galactose, galactitol, and gal-1-P levels have all been detected at abnormally high levels in the tissues of galactosemic fetuses (Holton 1995). This accumulation in utero is most likely due to self-intoxication from de novo galactose synthesis. Leloir enzyme activity is detectable in the human fetal liver by the 10th week of gestation (Holton 1995), and maternal adherence to a lactose-restricted diet does not prevent accumulation of galactose metabolites in the cord blood (Irons et al. 1985) or amniotic fluid (Jakobs et al. 1988) of galactosemic newborns.
Studies of serum biomarkers used to assess ovarian status in galactosemic girls also suggest that galactosemia-associated POI begins in early life. Serum AMH levels, used clinically as an indicator of follicular function and ovarian reserve (La Marca and Volpe 2006), are abnormally low in galactosemic girls relative to age-matched controls, even in girls younger than 2 years (Sanders et al. 2009). Elevated FSH level, an indirect measure of ovarian function, has been observed in galactosemic girls as early as 10 months of age and may remain high throughout the prepubertal years (Beauvais and Guilhaume 1984; Gitzelmann and Steinmann 1984; Irons et al. 1986; Kaufman et al. 1986; Schwarz et al. 1986; Steinmann et al. 1981; reviewed in Berry 2008; Rubio-Gozalbo et al. 2006, 2010). FSH production in response to GnRH stimulation is also abnormally high in classic galactosemics (reviewed in Berry 2008).
Combined, these findings suggest that the ovaries of most galactosemic girls are already functionally different in neonatal life with possible toxicity starting in fetal life. This may lead to fewer follicles at birth resulting in accelerated depletion of follicles in this population of girls and women. However, FSH measurements in prepubertal girls may be misleading. For example, prior to the age of 2 years an elevated FSH level may reflect factors independent of ovarian function of the child, and even a normal FSH level in a prepubertal girl is not fully reassuring since the hypothalamic-pituitary-ovarian (HPO) axis is thought to be quiescent after 1–2 years of age and not fully reactivated until just prior to puberty (Conte et al. 1975). Whether timing of the HPO axis is altered in galactosemic girls remains unclear. Thus, depending on whether the HPO axis is active in a given prepubertal girl, her FSH levels may not start to rise until early puberty even if her ovaries are dysfunctional.
Morphologic and histologic studies of ovaries from galactosemic girls might provide more insight into the timing of onset of ovarian dysfunction, but the limited studies that have been published are mostly confined to women in whom POI had already been diagnosed. In two case reports concerning prepubertal girls, the ovaries appeared normal; postmortem examination of a galactosemic newborn who died of E. coli sepsis revealed morphologically normal ovaries containing abundant oocytes (Levy et al. 1984), and a 7-year-old girl undergoing ultrasound for appendicitis was also found to have ovaries that appeared normal. Intriguingly, this same girl was later diagnosed with POI and underwent laparoscopy at age 17, which revealed streak ovaries (Kaufman et al. 1981). In imaging studies of girls with POI examined at pubertal age or later, the ovaries are invariably abnormal and usually described as hypoplastic or “streak-like.” Histological examination most often shows few if any follicles; in the cases where follicles have been observed they did not appear to have matured beyond the primordial stage (reviewed in Rubio-Gozalbo et al. 2010).
Lack of information on ovarian morphology and histology in a majority of galactosemic girls in infancy and early childhood makes it impossible to determine whether the diminished ovarian tissue seen in older girls and women reflects a failure of development, a regression of ovarian tissue over time, or a combination of both. The one documented case of apparently normal ovaries later becoming streak-like (Kaufman et al. 1981) suggested that the mechanism may be related to accumulated galactose ootoxicity over time.
Animal studies of galactose exposure reinforce the notion that the galactosemic ovary remains vulnerable to damage throughout life. The ovaries of genetically wild-type rats fed a high galactose diet exhibit decreased follicular development (Liu et al. 2006) and increased apoptosis of maturing follicles (Lai et al. 2003). In mice, preferential activation of the prolactin short-form receptor in the ovary, which represses FOXO3 and subsequently inhibits GALT expression, results in accelerated follicular depletion and consequent ovarian failure (Halperin et al. 2008). These studies indicate that even in the absence of developmental damage to the ovary, later exposure to galactose or its metabolites may contribute to POI, both by suppressing normal follicular maturation and by increasing follicular death.
Recent reports that, at least in some galactosemic women, POI follows a fluctuating course (Gubbels et al. 2008, 2009) are also suggestive of multiple layers of galactose ootoxicity. These observations are also consistent with the apparently fluctuating course of POI in many women who do not have galactosemia (Van Kasteren and Schoemaker 1999). For these women, prohibition of primordial follicle maturation may be intermittently relieved by an as yet unknown mechanism, allowing for intervals of spontaneous ovulation, normal menstrual cycles, and fertility. Of note, one study of a small cohort of galactosemic women looked at spontaneous fertility outcomes (Gubbels et al. 2008) and reported that galactosemia patients with a diagnosis of POI may be more likely to conceive than women who have POI due to other causes. This possibility needs to be studied in a larger cohort because it may alter how galactosemic patients are counseled about their probability of conceiving.
Possible mechanisms underlying POI in galactosemia
The question remains: What makes the human ovary so vulnerable to profound GALT deficiency? Though the answer remains elusive, a number of logical scenarios have been postulated (reviewed in Berry 2008); these include increased apoptosis of oocytes and/or ovarian stromal cells due to the individual or combined effects of excess gal-1P, galactitol, and/or other galactose metabolites, or defective glycosylation of ovarian or other proteins or lipids leading to direct and/or indirect ovarian dysfunction. Of course, the formal possibility also remains that GALT may have "moonlighting" functions independent of the Leloir pathway that are essential for ovarian function. However, reports of a correlation between milder ovarian dysfunction and the presence of residual GALT activity (Kaufman et al. 1988; Waggoner et al. 1990) or residual ability to metabolize galactose as measured by whole body galactose oxidation (Guerrero et al. 2000) argue against this possibility. Historically, women with GALK deficiency have not been considered at increased risk of POI, yet they accumulate galactose and/or galactitol in their blood and tissues; the individual roles of galactose and/or galactitol in ovarian toxicity must, therefore, be viewed with some caution. That said, profound GALK deficiency is extraordinarily rare, so that all studies of patient outcome are, by definition, studies of small cohorts (reviewed in Fridovich-Keil and Walter 2008). The long-term natural history of GALK-deficiency may therefore be imperfectly understood, leaving open the possibility that some subtle ovarian dysfunction impacts at least a subset of these patients.
Direct ovarian damage?
Direct ovarian damage could occur via a number of possible routes. Galactose may cause deposition of methyglyoxal by hampering the glutathione redox cycle and enhancing apoptosis (Liu et al. 2000). Similarly, galactitol, which is metabolized poorly, may accumulate in ovarian cells causing osmotic stress, swelling, and cell dysfunction. Glutathione levels may decrease, allowing increased hydrogen peroxide accumulation and apoptosis (Meyer et al. 1992). Finally, gal-1P may inhibit key enzymes that normally metabolize glucose-1P, impairing cellular metabolism (Gitzelmann 1995), and/or leading to increased cell stress (Slepak et al. 2005).
Studies on the effects of excess galactose in rat invariably show ovarian damage. Pregnant rats fed a 50% galactose diet showed a striking reduction in oocyte number in female pups; the most prominent effects were noted after exposure to galactose during the premeiotic stages of oogenesis (Chen et al. 1981). Female mice fed a 50% galactose diet for 2, 4, or 6 weeks demonstrated a decrease in the normal ovulatory response, as evidenced by a reduction in the number of corpora lutea when compared with controls (Swartz and Mattison 1988). Further, exposure of galactose-treated mice to a superovulatory regimen of pregnant mare’s serum gonadotropin (PMSG) and human chorionic gonadotropin (hCG) also failed to induce the anticipated increase in ovulatory response (Swartz and Mattison 1988); this effect was reversible upon cessation of galactose exposure (Swartz and Mattison 1988). In another study, pregnant rats were fed pellets with or without a 35% galactose supplement from day 3 of conception through weaning of their pups (Bandyopadhyay et al. 2003). Female pups were evaluated for serum levels of galactose and galactose-1-phosphate, growth rate, onset of puberty, reproductive cyclicity, ovarian complement of follicles, hypothalamo-pituitary-ovarian function, and follicular response to gonadotropins. Galactose in the maternal diet delayed the onset of puberty and also increased the frequency of hypergonadotropic hypoestrogenism in the female pups (Bandyopadhyay et al. 2003).
Indirect ovarian damage?
Another potential mechanism underlying POI in classic galactosemia involves indirect damage resulting from aberrant glycosylation. For example, appropriate levels and ratios of UDP-galactose and UDP-glucose are needed for proper synthesis of ovarian glycoproteins and glycolipids and also for support of germ cells, follicle maturation, and steroidogenesis. Numerous reports have demonstrated that GALT deficiency leads to aberrant glycosylation, especially prior to dietary galactose restriction (Charlwood et al. 1998; Dobbie et al. 1990; Haberland et al. 1971; Jaeken et al. 1992; Ornstein et al. 1992; Petry et al. 1991; Prestoz et al. 1997; Stibler et al. 1997; Sturiale et al. 2005). The structures of the truncated glycans detected in patient samples are consistent with a decreased capacity to galactosylate glycoproteins. In some cases the truncated N-glycans observed persist, albeit in small proportion, even after dietary treatment (Charlwood et al. 1998), however, the clinical relevance of these glycosylation abnormalities is not clear. For example, although Prestoz and colleagues (1997) reported finding a subpopulation of ostensibly abnormally glycosylated FSH in the blood of galactosemic women relative to controls, two direct tests of FSH bioactivity conducted more than 25 years apart using different sample sets and different methods both demonstrated no statistically significant difference between patients and controls (Kaufman et al. 1981; Sanders et al. 2009).
Gonadotropins and their receptors undergo extensive posttranslational modifications, including glycosylation, that are crucial for their longevity and function in vivo (Ulloa-Aguirre et al. 1995). For example, less acidic isoforms of recombinant FSH that carry incomplete or aberrant glycan additions have been found to have a shorter half-life (de Leeuw et al. 1996). In the FSH receptor, N-glycosylation occurs co-translationally and plays a role in the maturation and processing of the receptor (Menon et al. 2005). Interestingly, patients with primary congenital disorders of glycosylation type 1, due to mutations that affect the glycosylation machinery itself, also may have ovarian insufficiency based on hypergonadotropic hypogonadism (Kristiansson et al. 1995). Similar to classic galactosemics, these patients tend to have high concentrations of FSH in infancy, perhaps suggesting a similar mechanism of pathophysiology (Kristiansson et al. 1995).
An epigenetic mechanism?
Another possible mechanism of POI in classic galactosemia that needs to be explored involves prenatal or neonatal disturbance of the expression of genes involved in follicle development (Coman et al. 2010; Lai et al. 2008). Recently, the human tumor suppressor gene aplysia ras homolog 1 (ARHI), which is involved in ovarian folliculogenesis, was proposed as a possible target of toxicity in classic galactosemia (Lai et al. 2008). This gene is missing in rodents (Fitzgerald and Bateman 2004), and re-expression of this gene in mice causes failure of ovarian follicular maturation, poor growth, and impaired Purkinje cell development (Xu et al. 2000). ARHI is expressed consistently in normal ovarian cells (Yu et al. 1999).
Another gene that may be involved is NR5A1 (Lourenço et al. 2009). As confirmed by studies in mice, NR5A1 encodes a transcriptional regulator of genes that mediate the hypothalamic-pituitary-steroidogenic axis in both males and females. Recent studies of 25 women with sporadic ovarian insufficiency revealed that two carried mutations in NR5A1 that were not found in >700 control alleles, and functional studies using a tissue culture system demonstrated that these mutations impaired NR5A1 transactivational activity (Lourenço et al. 2009). Whether the ovarian insufficiency experienced by girls and women with classic galactosemia results from impairment of a pathway that involves NR5A1 remains unclear.
Recommendations for follow-up of girls and women with classic galactosemia, and potential intervention for POI in this population
Treatment of delayed or absent puberty
Currently, the treatment of galactosemic girls and women with ovarian insufficiency focuses mainly on estrogen supplementation to avoid secondary effects of hypoestrogenism. Unless the patient does not have a uterus, progesterone must be added at some point to reduce the risk of endometrial hyperplasia or cancer. Some physicians test for hypergonadotropism beginning after the age of 8 years, and then induce pubertal development at an age-appropriate time by estrogen supplementation if FSH and LH values are increased. However, high gonadotropin values may be found in these girls even at a very early age (Beauvais and Guilhaume 1984; Gitzelmann and Steinmann 1984; Irons et al. 1986; Kaufman et al. 1986; Schwarz et al. 1986; Steinmann et al. 1981; reviewed in Berry 2008; Rubio-Gozalbo et al. 2006, 2010) perhaps indicating early ovarian insufficiency resulting in a disturbed hypothalamic-pituary-ovarian axis. Therefore, the absence of developing secondary sex characteristics may be a more important benchmark of when to initiate therapy.
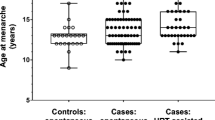
Previously, most physicians waited until a patient was at least 16 years of age to induce pubertal development with exogenous hormones if it had not occurred spontaneously (Gubbels et al. 2008); this practice led to a higher percentage of spontaneous menarche in the older cohort. However, postponing the induction of pubertal development to age 16 can have psychological disadvantages if classmates have pubertal development earlier. There may be physiological detriments as well, although this point remains unclear.
In the U.S., most clinicians currently initiate hormone replacement therapy (HRT) in girls with ostensible ovarian insufficiency between 12 and 14 years of age. Timing may be influenced by bone age, parental request, and growth velocity (Drobac et al. 2006). Initiating hormone replacement too early in girls with Turner’s syndrome has been shown to decrease maximum height (Chernausek et al. 2000), demonstrating the risk of premature intervention. In Europe, a study of clinical practice demonstrated that 40.4% of clinicians initiate treatment by age 11, 47.8% wait until age 13, and 7.5% wait until age 15 (Kiess et al. 2002). Since very low doses of estrogen may stimulate growth velocity in an adolescent with delayed puberty and higher doses may result in premature closure of the epiphysis, initial replacement at a relatively low dose should be considered with slow titration over several years (Sas et al. 1999; van Teunenbroek et al. 1996) to enable affected girls to reach higher adult stature.
There are many formulations and routes of administration for HRT. The most commonly prescribed are oral conjugated equine estrogens or oral 17β-estradiol (Drobac et al. 2006; Kiess et al. 2002). Transdermal and transvaginal administration of 17β-estradiol is becoming more popular for postmenopausal hormone therapy because it may be associated with less risk of venothromboembolism (Canonico et al. 2010). Further research is needed in young women with POI to determine which route of administration is best.
Risks of HRT in women with POI
Specific information on risks of HRT in POI remains scarce, as most studies have been performed in postmenopausal women, and these results should not be extrapolated to younger women (Armitage et al. 2003). One aspect that deserves further investigation is whether HRT in young women with ovarian insufficiency should continue until the average age at menopause. Women with POI have a two-fold increased risk of mortality, and while large randomized trials are lacking, HRT may ameliorate endothelial dysfunction in young women (Kalantaridou et al. 1998, 2004) and delay the onset of osteoporosis, a prevalent complication of POI (Anasti et al. 1998). In the absence of contraindications, HRT remains the best treatment option known for menopausal symptoms (hot flashes, vaginal dryness, etc.) in women with hypoestrogenism from POI.
Fertility treatment options
A general treatment strategy for women experiencing hypogonadotropic or normogonadotropic anovulation is ovulation induction with the administration of urinary or recombinant gonadotropins. However, for success this treatment requires the presence of responsive follicles, which may be a problem for women with classic galactosemia. If the ovarian dysfunction seen in galactosemia results from a follicular maturation arrest (Liu et al. 2006; Xu et al. 2000), then longer-term stimulation (for example, for several months) might, in theory, be beneficial (Gubbels et al., personal observation ). However, one might question the likelihood of benefit of administering exogenous FSH to a woman whose endogenous FSH is already elevated, unless, of course, the endogenous FSH is functionally impaired, which appears not to be the case (Kaufman et al. 1981; Sanders et al. 2009). Anecdotal evidence from a case report (Menezo et al. 2004) suggests there may be some role for exogenous gonadotropins; this possibility needs to be explored in a larger, placebo-controlled trial.
For those women in whom ovarian stimulation fails or cannot be performed, oocyte donation is an option. Pregnancy after this procedure has been described in galactosemia (Sauer et al. 1991). Egg donation is not readily available in all countries; some impose restrictions on the use of anonymous and compensated donors (e.g., Schenker 1997; Uroz and Guerra 2009). Furthermore, the treatment is costly and prior psychological counseling is recommended. However, this treatment is effective; compared to the chances of spontaneous pregnancy, the probability of conception with egg donation is significantly higher. In the U.S., egg donation IVF cycles have an approximate 55% chance of live birth per cycle (Centers for Disease Control and Prevention 2007).
Fertility preservation options
Options to preserve endogenous fertility in galactosemia need to be explored further. For postpubertal women, freezing embryos after in vitro fertilization is an option that is currently available, with good results. The probability of pregnancy resulting from frozen embryo transfers in the U.S. is approximately 30% per transfer (Centers for Disease Control and Prevention 2007). This success rate requires that the egg be fertilized with either the partner’s sperm or with donor sperm prior to cryopreservation, however, so this technology may not be a preferred option for every woman. Furthermore, this technique requires that the woman seeking treatment produce a relatively large number of oocytes with controlled ovarian hyperstimulation. If galactosemia does cause a diminished oocyte pool or impaired maturation of oocytes, as animal studies suggest (Liu et al. 2006; Xu et al. 2000), response to hormonal stimulation may be insufficient to justify the procedure. Reports to date involving galactosemic women are extremely limited.
In the prepubertal girl, ovarian tissue cryopreservation has been named as a possible mode to preserve fertility, and in Denmark, ovaries of galactosemic girls have already been frozen (Danish Galactosemia Society, personal communication). There are, however, several problems with this strategy worth considering. The current state of ovarian cryopreservation technology is not reliably established and pregnancies resulting from these procedures are rare; recommending this procedure may therefore offer false hope to galactosemic patients and their families. Indeed, both the American College of Obstetrics and Gynecology (ACOG) and the American Society of Reproductive Medicine (ASRM) currently recommend that women only undergo oocyte or ovarian cryopreservation as part of a research study as the technology is not yet ready for clinical use (ACOG 2008). Improvements in this technique, however, may be of great benefit. Trials are currently ongoing for (nongalactosemic) women trying to preserve fertility before needed chemotherapy and radiation treatment that may yield valuable information and improved options for galactosemic girls (Backhus et al. 2007; Grifo and Noyes 2010; Noyes et al. 2010). Unfortunately, if ovarian insufficiency has already occurred in early childhood, cryopreserving tissue after that point may be too late. In addition, whether these invasive procedures should even be considered for children is controversial.
Pregnancy in galactosemia
For those women with galactosemia who do achieve pregnancy, evidence from studies of small cohorts suggest that there are no adverse effects on the galactosemic mother or her infant (Gubbels et al. 2008; Schadewaldt et al. 2009). There is, however, a lack of long-tem prospective information on this subject. It is clear that pregnancy does not lead to metabolic crisis, but most patients have increased metabolite values. A prospective follow-up study of patients who try to conceive might provide useful information that is currently only available in anecdotal form.
Long-term follow-up of children born to galactosemic mothers would also be of interest. Current data from animal models are mixed. Offspring of rats fed a high galactose diet have a lower birth weight and placental weight compared to controls, although catch-up growth is seen as soon as the galactose intake is normalized (Spatz and Segal 1965). Rats born to mothers with high galactose intake also have larger livers with less liver-enzyme activity, but these abnormalities also resolve in the postnatal period after diet normalization (Spatz and Segal 1965). Offspring of rats fed a high galactose diet have smaller brains than rats born to mothers fed a normal diet (Haworth et al. 1969), indicating a possible effect on brain development. Cataracts in the offspring of rats fed a high galactose diet during pregnancy are also common (Segal and Bernstein 1963), but are completely reversible as soon as galactose intake is normalized. Of particular concern, a reduction in the number of oocytes at birth is seen in female pups born to rats fed a high galactose diet during pregnancy (Chen et al. 1981). Whether any of these observations from rodents foretell concerns for humans remains unclear, but they could and should be tested through long-term follow-up studies.
Challenges, uncertainties, and opportunities
Supplemental approaches to intervention?
That POI and other long-term complications are so common among treated patients with classic galactosemia reinforces the unavoidable conclusion that current intervention is inadequate. One approach to supplemental treatment that has been raised in the galactosemia literature proposes use of a small molecule inhibitor to block GALK function (Bosch 2006; Wierenga et al. 2008). The idea, in short, is to block accumulation of gal-1P in patient cells by blocking its synthesis, ostensibly converting the metabolic status and outcome of a GALT-deficient patient into that of a GALK-deficient patient.
This idea seems reasonable in theory, and some candidate inhibitors have already been reported (Wierenga et al. 2008); however, a number of challenges remain. First, are GALK-deficient patients truly unaffected beyond the well-described galactose-dependent cataracts? Second, how much GALK inhibition would be required to prevent gal-1P accumulation, and in what tissues? Studies in yeast (Mumma et al. 2008) suggest that GALK inhibition would need to be profound to impact gal-1P accumulation in a significant way. Logical consideration suggests another problem, namely that GALK inhibition is a cell-autonomous effect; inhibiting GALK in one cell might limit the accumulation of gal-1P in that cell, but it would do nothing to lower the accumulated galactose in the blood, or the accumulation of gal-1P in other cells. Drug delivery would therefore need to be truly systemic, including across the blood-brain barrier.
Other treatment options that deserve attention include tissue or organ transplantation, for example liver, and in vivo or ex vivo gene therapy. The possibility of restoring GALT activity by introducing a normal population of cells, or a functional copy of the GALT cDNA into a patient’s own genetic material, is intriguing. This approach would presumably mitigate or solve the metabolic problem, and not simply block it in some cells. In short, the treatment might not need to be truly systemic, because even partial correction, or correction of the defect in some cells but not others, should provide the body with a route to eliminate excess galactose from the bloodstream.
Timing of intervention
One of the key challenges to prevention of POI in classic galactosemia remains the uncertainty of when the fundamental damage occurs. Given that folliculogenesis occurs in utero, it remains possible that most if not all of the “ovarian damage” has already occurred prior to birth. If this is the case, strategies for intervention may need to target the prenatal period, though a host of additional concerns would accompany such early intervention. Given that postnatal intervention may also carry potentially serious risks, it will be important to know that there is a good chance for success to justify those risks. Alternatively, if the damage is not prenatal, or is reversible, it will be important to know when in the postnatal period any given form of intervention will have the greatest chance for success.
References
ACOG (2008) ACOG Committee opinion no. 405: ovarian tissue and oocyte cryopreservation. Obstet Gynecol 111:1255–1256
Albright F (1942) A syndrome characterized by primary ovarian insufficiency and decreased stature.Am J Med Sci 204:625–648
Anasti J, Kalantaridou S, Kimzey L, Defensor R, Nelson L (1998) Bone loss in young women with karyotypically normal spontaneous premature ovarian failure. Obstet Gynecol 91:12–15
Armitage M, Nooney J, Evans S (2003) Recent concerns surrounding HRT. Clin Endocrinol (Oxf) 59:145–155
Backhus L, Kondapalli L, Chang R, Coutifaris C, Kazer R, Woodruff T (2007) Oncofertility Consortium consensus statement: guidelines for ovarian tissue cryopreservation. Cancer Treat Res 138:235–239
Bandyopadhyay S, Chakrabarti J, Banerjee S, et al. (2003) Prenatal exposure to high galactose adversely affects initial gonadal pool of germ cells in rats. Hum Reprod 18:276–282
Beauvais P, Guilhaume A (1984) L’insuffisance ovarienne de la galactosémie congénitale. Presse Med 13:2685–2687
Berry G (2008) Galactosemia and amenorrhea in the adolescent. Ann NY Acad Sci 1135:112–117
Bosch A (2006) Classical galactosaemia revisited. J Inherit Metab Dis 29:516–525
Bussani C, Papi L, Sestini R et al. (2004) Premature ovarian failure and fragile X premutation: a study on 45 women. Eur J Obstet Gynecol Reprod Biol 112:189–191
Canonico M, Fournier A, Carcaillon L et al. (2010) Postmenopausal hormone therapy and risk of idiopathic venous thromboembolism: results from the E3N cohort study. Arterioscler Thromb Vasc Biol 30:340–345
Centers for Disease Control and Prevention (2007) Assisted reproductive technology report: national summary. http://www.cdc.gov/art/
Charlwood J, Clayton P, Keir G, Mian N, Winchester B (1998) Defective galactosylation of serum transferrin in galactosemia. Glycobiology 8:351–357
Chen Y, Mattison D, Feigenbaum L, Fukui H, Schulman J (1981) Reduction in oocyte number following prenatal exposure to a diet high in galactose. Science 214:1145–1147
Chernausek S, Attie K, Cara J, Rosenfeld R, Frane J, Genentech, Inc., Collaborative Study Group (2000) Growth hormone therapy of Turner syndrome: the impact of age of estrogen replacement on final height. J Clin Endocrinol Metab 85:2439–2445
Coman D, Murray D, Byrne J et al. (2010) Galactosemia, a single gene disorder with epigenetic consequences. Pediatr Res 67:286–292
Conte F, Grumbach M, Kaplan S (1975) A diphasic pattern of gonadotropin secretion in patients with the syndrome of gonadal dysgenesis. J Clin Endocrinol Metab 40:670–674
Conway G (2000) Premature ovarian failure. Br Med Bull 56:643–649
Conway G, Payne N, Webb J, Murray A, Jacobs P (1998) Fragile X premutation screening in women with premature ovarian failure. Hum Reprod Update 13:1184–1187
Coulam C, Adamson S, Annegers J (1986) Incidence of premature ovarian failure. Obstet Gynecol 67:604–606
Crisponi L et al. (2001) The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat Genet 27:159–166
de Leeuw R, Mulders J, Voortman G, Rombout F, Damm J, Kloosterboer L (1996) Structure-function relationship of recombinant follicle stimulating hormone (Puregon). Mol Hum Reprod 2:361–369
Di Pasquale E et al. (2004) Hypergonadtropic ovarian failure associated with an inherited mutation of human bone morphogenetic protein-15 (BMP-15) gene. Am J Hum Genet 75:106–111
Dobbie JA, Holton JB, Clamp JR (1990) Defective galactosylation of proteins in cultured skin fibroblasts from galactosaemic patients. Ann Clin Biochem 27:274–275
Drobac S, Rubin K, Rogol A, Rosenfield R (2006) A workshop on pubertal hormone replacement options in the US. J Pediatr Endocrinol Metab 19:55–64
Fitzgerald J, Bateman J (2004) Why mice have lost genes for COL21A1, STK17A, GPR145 and AHRI: evidence for gene deletion at evolutionary breakpoints in the rodent lineage. Trends Genet 20:408–412
Fridovich-Keil J, Walter J (2008) Galactosemia. In: Valle D, Beaudet A, Vogelstein B, Kinzler K, Antonarakis S, and Ballabio A (eds) The online metabolic and molecular bases of inherited disease – OMMBID, chap. 72. www.ommbid.com
Gersak K, Meden-Vrtovec H, Peterlin B (2003) Fragile X premutation in women with sporadic premature ovarian failure in Slovenia. Hum Reprod Update 18:1637–1640
Gitzelmann R (1995) Galactose-1-phosphate in the pathophysiology of galactosemia. Eur J Pediatr 154(Suppl 2):S45–S49
Gitzelmann R, Steinmann B (1984) Galactosemia: how does long-term treatment change the outcome? Enzyme 32:37–46
Goswami D, Conway G (2005) Premature ovarian failure. Hum Reprod Update 11:391–410
Grifo J, Noyes N (2010) Delivery rate using cryopreserved oocytes is comparable to conventional in vitro fertilization using fresh oocytes: potential fertility preservation for female cancer patients. Fertil Steril 93:391–396
Gubbels C, Land J, Rubio-Gozalbo M (2008) Fertility and impact of pregnancies on the mother and child in classic galactosemia. Obstet Gynecol Surv 63:334–343
Gubbels C, Kuppens S, Bakker J et al. (2009) Pregnancy in classic galactosemia despite undetectable anti-Müllerian hormone. Fertil Steril 91(1293):e13–e16
Guerrero NV, Singh RH, Manatunga A, Berry GT, Steiner RD, Elsas LJ (2000) Risk factors for premature ovarian failure in females with galactosemia. J Pediatr 137:833–841
Haberland C, Perou M, Brunngraber EG, Hof H (1971) The neuropathology of galactosemia: a histopathological and biochemical study. J Neuropathol Exp Neurol 30:431–447
Halperin J, Devi S, Elizur S et al. (2008) Prolactin signaling through the short form of its receptor represses forkhead transcription factor FOXO3 and its target gene galt causing a severe ovarian defect. Mol Endocrinol 22:513–522
Hansen K, Hodnett G, Knowlton N, Craig L (2010) Correlation of ovarian reserve tests with histologically determined primordial follicle number. Fertil Steril. doi:10.1016/j.fertnstert.2010.04.006
Haworth J, Ford J, Younoszai M (1969) Effect of galactose toxicity on growth of the rat fetus and brain. Pediatr Res 3:441–447
Holton J (1995) Effects of galactosemia in utero. Eur J Pediatr 154:S77–S81
Irons M, Levy H, Pueschel S, Castree K (1985) Accumulation of galactose-1-phosphate in the galactosemic fetus despite maternal milk avoidance. J Pediatr 107:261–263
Irons M, Levy H, Crowley W (1986) Gonadal function in galactosemia. Am J Hum Genet 39:A13
Jaeken J, Kint J, Spaapen L (1992) Serum lysosomal enzyme abnormalities in galactosaemia. Lancet 340:1472–1473
Jakobs C, Kleijer W, Bakker H, Gennip A, Przyrembel H, Niermeuer M (1988) Dietary restriction of maternal lactose intake does not prevent accumulation of galactitol in the amniotic fluid of fetuses affected with galactosaemia. Prenat Diagn 8:641–645
Kalantaridou S, Davis S, Nelson L (1998) Premature ovarian failure. Endocrinol Metab Clin North Am 27:989–1006
Kalantaridou S, Naka K, Papanikolaou E et al. (2004) Impaired endothelial function in young women with premature ovarian failure: normalization with hormone therapy. J Clin Endocrinol Metab 89:3907–3913
Kaufman F, Kogut M, Donnell G, Goebelsmann U, March C, Koch R (1981) Hypergonadotropic hypogonadism in female patients with galactosemia. N Engl J Med 304:994–998
Kaufman F, Donnell G, Roe T, Kogut M (1986) Gonadal function in patients with galactosaemia. J Inherit Metab Dis 9:140–146
Kaufman FR, Xu Y-K, Ng WG, Donnell GN (1988) Correlation of ovarian function with galactose-1-phosphate uridyl transferase levels in galactosemia. J Pediatr 112:754–756
Kiess W, Conway G, Ritzen M et al. (2002) Induction of puberty in the hypogonadal girl–practices and attitudes of pediatric endocrinologists in Europe. Horm Res 57:66–71
Knauff E, Richardus R, Eijkemans M et al. (2007) Heterozygosity for the classical galactosemia mutation does not affect ovarian reserve and menopausal age. Reprod Sci 14:780–785
Kristiansson B, Stibler H, Wide L (1995) Gonadal function and glycoprotein hormones in the carbohydrate-deficient glycoprotein (CDG) syndrome. Acta Paediatr 84:655–659
LaBarbera A, Miller M, Ober C, Rebar R (1988) Autoimmune etiology in premature ovarian failure. Am J Reprod Immunol Microbiol 16:115–122
Lai K, Cheng L, Cheung A, O W (2003) Inhibitor of apoptosis proteins and ovarian dysfunction in galactosemic rats. Cell Tissue Res 311: 417–425.
Lai K, Tang M, Yin X, Klapper H, Wierenga K, Elsas L (2008) ARHI: a new target of galactose toxicity in classic galactosemia. Biosci Hypotheses 1:263–271
Laissue P et al. (2006) Mutations and sequence varients in GDF9 and BMP15 in patients with premature ovarian failure. Eur J Endocrinol 154:739–744
La Marca A, Volpe A (2006) Anti-Mullerian hormone (AMH) in female reproduction: is measurement of circulating AMH a useful tool? Clin Endocrinol 64:603–610
Levy H, Driscoll S, Porensky R, Wender D (1984) Ovarian failure in galactosemia. N Engl J Med 310:50
Liu G, Hale G, Hughes C (2000) Galactose metabolism and ovarian toxicity. Reprod Toxicol 14:377–384
Liu G, Shi F, Blas-Machado U et al. (2006) Dietary galactose inhibits GDF-9 mediated follicular development in the rat ovary. Reprod Toxicol 21:26–33
Lourenço D, Brauner R, Lin L et al. (2009) Mutations in NR5A1 associated with ovarian insufficiency. N Engl J Med 360:1200–1210
Menezo Y, Lescaille M, Nicollet B, Servy E (2004) Pregnancy and delivery after stimulation with rFSH of a galatosemia patient suffering hypergonadotropic hypogonadism: case report. J Assist Reprod Genet 21:89–90
Menon K, Clouser C, Nair A (2005) Gonadotropin receptors: role of post-translational modifications and post-transcriptional regulation. Endocrine 26:249–257
Meyer W, Doyle M, Grifo J et al. (1992) Aldose reductase inhibition prevents galactose-induced ovarian dysfunction in the Sprague-Dawley rat. Am J Obstet Gynecol 167:1837–1843
Mumma JO, Chhay JS, Ross KL, Eaton JS, Newell-Litwa KA, Fridovich-Keil JL (2008) Distinct roles of galactose-1P in galactose-mediated growth arrest of yeast deficient in galactose-1P uridylyltransferase (GALT) and UDP-galactose 4′-epimerase (GALE). Mol Genet Metab 93:160–171
Murray A, Webb J, Grimley S, Conway G, Jacobs P (1998) Studies of FRAXA and FRAXE in women with premature ovarian failure. J Med Genet 35:637–640
Noyes N, Labella P, Grifo J, Knopman J (2010) Oocyte cryopreservation: a feasible fertility preservation option for reproductive age cancer survivors. J Assist Reprod Genet 27:495–499
Ornstein KS, McGuire EJ, Berry GT, Roth S, Segal S (1992) Abnormal galactosylation of complex carbohydrates in cultured fibroblasts from patients with galactose-1-phosphate uridyltransferase deficiency. Pediatr Res 31:508–511
Persani L, Rossetti R, Cacciatore C, Bonomi M (2009) Primary ovarian insufficiency: X chromosome defects and autoimmunity. J Autoimmun 33:35–41
Petry K, Greinix HT, Nudelman E et al. (1991) Characterization of a novel biochemical abnormality in galactosemia: deficiency of glycolipids containing galactose or N-acetylgalactosamine and accumulation of precursors in brain and lymphocytes. Biochem Med Metab Biol 46:93–104
Prestoz L, Couto A, Shin Y, Petry K (1997) Altered follicle stimulating hormone isoforms in female galactosaemia patients. Eur J Pediatr 156:116–120
Rubio-Gozalbo ME, Panis B, Zimmermann LJI, Spaapen LJ, Menheere PPCA (2006) The endocrine system in treated patients with classical galactosemia. Mol Genet Metab 89:316–322
Rubio-Gozalbo M, Gubbels C, Bakker J, Menheere P, Wodzig W, Land J (2010) Gonadal function in male and female patients with classic galactosemia. Hum Reprod Update 16:177–188
Sanders R, Spencer J, Epstein M et al. (2009) Biomarkers of ovarian function in girls and women with classic galactosemia. Fertil Steril 92:344–351
Sas T, de Muinck Keizer-Schrama S, Stijnen T et al. (1999) Final height in girls with Turner’s syndrome treated with once or twice daily growth hormone injections. Arch Dis Child 80:36–41
Sauer M, Kaufman F, Paulson R, Lobo R (1991) Pregnancy after oocyte donation to a woman with ovarian failure and classical galactosemia. Fertil Steril 55:1197–1199
Schadewaldt P, Hammen H, Kamalanathan L et al. (2009) Biochemical monitoring of pregnancy and breast feeding in five patients with classical galactosaemia–and review of the literature. Eur J Pediatr 168:721–729
Schenker J (1997) Assisted reproduction practice in Europe: legal and ethical aspects. Hum Reprod Update 3:173–184
Schwarz H, Zimmermann A, Carasso A, Zuppinger K (1986) Feminization in a galactosemic girl in the presence of hypergonadotropic hypogonadism. Acta Endocrinol Suppl (Copenh) 279:428–433
Schweitzer S, Shin Y, Jakobs C, Brodehl J (1993) Long-term outcome in 134 patients with galactosemia. Eur J Pediatr 152:36–43
Segal S, Bernstein H (1963) Observations on cataract formation in the newborn offspring of rats fed a high-galactose diet. J Pediatr 62:363–370
Slepak T, Tang M, Addo F, Lai K (2005) Intracellular galactose-1-phosphate accumulation leads to environmental stress response in yeast model. Mol Genet Metab 86:360–371
Spatz M, Segal S (1965) Transplacental galactose toxicity in rats. J Pediatr 67:438–446
Steinmann B, Gitzelmann R, Zachmann M (1981) Galactosemia: hypergonadotropic hypogonadism already found in prepubertal girls but only in adult males. Eur J Pediatr 135:337
Stibler H, von Dobeln U, Kristiansson B, Guthenberg C (1997) Carbohydrate-deficient transferrin in galactosaemia. Acta Paediatr 86:1377–1378
Sturiale L, Barone R, Fiumara A et al. (2005) Hypoglycosylation with increased fucosylation and branching of serum transferrin N-glycans in untreated galactosemia. Glycobiology 15:1268–1276
Swartz W, Mattison D (1988) Galactose inhibition of ovulation in mice. Fertil Steril 49:522–526
Ulloa-Aguirre A, Midgley A Jr, Beitins I, Padmanabhan V (1995) Follicle-stimulating isohormones: characterization and physiological relevance. Endocr Rev 16:765–787
Uroz V, Guerra L (2009) Donation of eggs in assisted reproduction and informed consent. Med Law 28:565–575
Van Kasteren Y, Schoemaker J (1999) Premature ovarian failure: a systematic review on therapeutic interventions to restore ovarian function and achieve pregnancy. Hum Reprod Update 5:483–492
van Teunenbroek A, de Muinck Keizer-Schrama S, Stijnen T et al. (1996) Yearly stepwise increments of the growth hormone dose results in a better growth response after four years in girls with Turner syndrome. J Clin Endocrinol Metab 81:4013–4021
Waggoner DD, Buist NRM, Donnell GN (1990) Long-term prognosis in galactosemia: results of a survey of 350 cases. J Inher Metab Dis 13:802–818
Welt C (2008) Primary ovarian insufficiency: a more accurate term for premature ovarian failure. Clin Endocrinol 68:499–509
Wheatcroft N, Weetman A (1997) Is premature ovarian failure an autoimmune disease? Autoimmunity 25:157–165
Wierenga K, Lai K, Buchwald P, Tang M (2008) High-throughput screening for human galactokinase inhibitors. J Biomol Screen 13:415–423
Xu F, Xia W, Luo R et al. (2000) The human ARHI tumor suppressor gene inhibits lactation and growth in transgenic mice. Cancer Res 60:4913–4920
Yu Y, Xu F, Peng H et al. (1999) NOEY2 (ARHI), an imprinted putative tumor suppressor gene in ovarian and breast carcinomas. Proc Natl Acad Sci USA 96:214–219
Acknowledgments
The authors gratefully acknowledge the many patients and families affected by galactosemia who have volunteered for studies over the past decades, and the many scientists and clinicians who have conducted those studies, generating the papers summarized in this article. We further thank an anonymous reviewer who made numerous detailed suggestions that substantially improved the quality of this document. This review article was supported in part by NIH grant DK059904 (to J.L.F.K.), and a Dutch Galactosemia Research fund grant (to E.R.).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Gerard T. Berry
Competing interest: None declared.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Fridovich-Keil, J.L., Gubbels, C.S., Spencer, J.B. et al. Ovarian function in girls and women with GALT-deficiency galactosemia. J Inherit Metab Dis 34, 357–366 (2011). https://doi.org/10.1007/s10545-010-9221-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-010-9221-4