
Abstract
Wilson disease (WD) is caused by mutations in the gene for ATP7B, a copper transport protein that regulates copper levels in cells. A large number of missense mutations have been reported to cause WD but genotype–phenotype correlations are not yet established. Since genetic screening for WD may become reality in the future, it is important to know how individual mutations affect ATP7B function, with the ultimate goal to predict pathophysiology of the disease. To begin to assess mechanisms of dysfunction, we investigated four proposed WD-causing missense mutations in metal-binding domains 5 and 6 of ATP7B. Three of the four variants showed reduced ATP7B copper transport ability in a traditional yeast assay. To probe mutation-induced structural dynamic effects at the atomic level, molecular dynamics simulations (1.5 μs simulation time for each variant) were employed. Upon comparing individual metal-binding domains with and without mutations, we identified distinct differences in structural dynamics via root-mean square fluctuation and secondary structure content analyses. Most mutations introduced distant effects resulting in increased dynamics in the copper-binding loop. Taken together, mutation-induced long-range alterations in structural dynamics provide a rationale for reduced copper transport ability.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Wilson disease (WD) is a rare autosomal recessively-inherited disorder of copper (Cu) metabolism caused by mutations in the ATP7B gene which codes for a transmembrane Cu transporting ATPase (Park et al. 1991; Mohr and Weiss 2019). The genetics of WD is complex with more than 450 disease-causing mutations confirmed (Mohr and Weiss 2019; Harada et al. 2001). Under physiological conditions, ATP7B has dual function, mediating excretion of Cu into the bile and Cu transport into the Golgi network for loading of Cu-dependent enzymes such as ceruloplasmin (Lo and Bandmann 2017). In patients, impaired Cu excretion leads to increased Cu accumulations, reaching toxic levels, primarily in the liver and eventually in other organs, particularly in the brain. The consequences are a wide range of symptoms such as progressive liver disease and liver failure, neurological syndrome as well as psychiatric symptoms (Mohr and Weiss 2019; Nagano et al. 1998; Rodriguez-Castro et al. 2015). The wide spectrum of phenotypic variations in WD patients often makes it hard to make an accurate and early clinical diagnosis (Jang et al. 2017; Dong et al. 2016; Braiterman et al. 2014).
WD is a rare disorder with an estimated prevalence of symptomatic disease of 1 in 30,000 and a heterozygous ATP7B mutation carrier frequency of 1 in 90 (Mohr and Weiss 2019). However, these numbers have been questioned as they are based on assumptions; it has been postulated that the number of people with disease and carriers are much higher (Mohr and Weiss 2019; Lo and Bandmann 2017). Because early diagnosis is essential to introduce treatment that will prevent future complications, there is an interest in genetic screening of infants for WD (Lo and Bandmann 2017). A number of pilot studies have investigated the possibility of large-scale genetic screening programs for WD (Lo and Bandmann 2017) and, despite technical hurdles, such programs may be reality in the future. Clearly, mutations causing WD should affect ATP7B function, but how this takes place at the molecular-mechanistic level is not known for most missense mutations. Whereas mutations that prematurely truncate the ATP7B polypeptide will intuitively abolish function, it is harder to depict how point mutations in a 1400-residue protein can cause dysfunction. To make best use of genetic screening efforts, it would be required to understand of how different mutations contribute to disease onset, progression and severity. Moreover, many functional aspects of ATP7B are not known and, therefore, laboratory studies of mutated ATP7B variants may also reveal fundamental insights on general mechanisms.
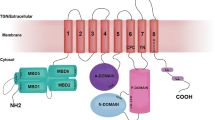
Most notably, ATP7B (and its human homolog ATP7A) (Banci et al. 2008) has six metal-binding domains (MBD) in the cytoplasmic N-terminal region (Fig. 1a) whereas yeast and bacterial homologs have two and one MBDs, respectively (Yu et al. 2017). All MBDs have a ferredoxin-like fold and a MXCXXC consensus Cu-binding motif (Yu et al. 2017, 2018; Boal and Rosenzweig 2009). The MBDs are thought to receive Cu from the cytoplasmic Cu chaperone Atox1 via direct protein–protein interactions, followed by intra-ATP7B transport of Cu through the membrane channel for release in the Golgi lumen. However, to explain that there are as many as six MBDs, regulatory roles via interactions with other ATP7B domains and/or with other proteins have been proposed (Yu et al. 2017; Gonzalez-Guerrero and Arguello 2008). The organization of the six MBDs during ATP7B’s ATP-dependent catalytic cycle is unknown due to lack of structural information on the full-length protein. Nonetheless, it has been proposed that Cu delivery to one of the MBDs will trigger conformation changes among the MBDs that in turn initiate the catalytic cycle (Niemiec et al. 2012, 2015; Mondol et al. 2016). Using Cu transport assays in vitro, it has been shown that several MBDs in ATP7B can be deleted, or Cu-binding sites mutated, without loss of Cu transport activity; the presence of only one MBD is sufficient for function (Forbes et al. 1999; Ponnandai Shanmugavel et al. 2017).

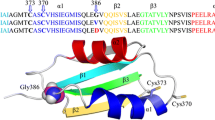
a Scheme of domain arrangement in ATP7B. On the cytoplasmic side, there are actuator (A), phosphorylation (P), ATP-binding (N) and six metal binding (MBD1-6) domains. b High-resolution NMR structures of MBD56 domains [PDB ID: 2EW9 (Achila et al. 2006)]. The GMX1CX2SC Cu(I)-binding motifs colored in orange and WD-causing missense mutations: L492S (green), A604P (yellow), R616W (blue), and G626A (pink). (Color figure online)
Despite ‘only’ regulatory roles proposed, and the redundancy of domains, it is interesting to note that at least 33 WD causing missense mutations are found in the MBDs (excluding linker regions) (Arioz et al. 2017). The MBDs closest to the membrane-spanning part of ATP7B, MBD5 and MBD6, appear to be a hotspot containing 23 of the 33 missense mutations in the MBDs (Arioz et al. 2017; Gourdon et al. 2012). Only two of the 33 mutations are localized in the Cu-binding motifs. One may thus speculate that most WD-causing mutations in the MBDs will affect MBD structural integrity, MBD–MBD interactions and/or interactions with other ATP7B domains. With the aim to explain dysfunction on a molecular level for WD missense mutations located in MBD5 and MBD6 at different positions within the ferredoxin fold, we here investigated Cu transport ability of four mutated ATP7B proteins. The variants (L492S in MBD5; A604P, R616W, and G626A in MBD6) have all been proposed to cause WD based on genetic analysis of WD patients in different families (Ljubic et al. 2016; Caca et al. 2001; Tuan Pham et al. 2017; Zong and Kong 2015), although patients homozygote for these mutations have not been discussed in the literature. The L492S mutation is situated in the first ß-strand of the ferredoxin-fold, the A604P mutation in the third ß-strand, and the R616W and G626A mutations in different ends of the second -helix of the ferredoxin fold (Fig. 1b). Thus, none of the mutations are near the Cu-binding site in the MBD. We found that three of the variants demonstrated reduced Cu transport efficiency in a yeast assay as compared to the wild type protein. Upon structural dynamics analysis using MD simulations, we found identifiable differences for the mutated domains as compared to the wild-type counterparts, which may be linked to the expected dysfunction of these ATP7B variants in vivo.
Materials and methods
Yeast strains and plasmid construction
High-stress resistance yeast S. cerevisiae CEN.PK 113-11C (MATa SUC2 MAL2-8 URA3-52 HIS3-∆1; provided by Dr. P. Kötter, Institute of Microbiology, Johann Wolfgang Goethe-University, Frankfurt, German) was used as a reference strain. The yeast strains were cultivated in liquid YEPD/YPD (Yeast Extract Peptone dextrose media) and grown at 30 °C. The yeast strain bearing CCC2 and ATX1 deletions was used in this study. For the expression of ATP7B and ATOX1, plasmids containing human ATP7B (p426GPD-ATP7B) and ATOX1 (p423GPD-ATOX1) were transformed to the yeast strain with CCC2 and ATX1 knockout (Δccc2Δatx1). The detailed construction of the yeast strain and plasmids p426GPD-ATP7B and p423GPD-ATOX1 were reported previously (Ponnandai Shanmugavel et al. 2017). The disease-causing ATP7B mutants namely L492S, A604P, R616 W, and G626A were constructed. The mutations were introduced into the full-length ATP7B by quickchange site-directed mutagenesis kit using the previously constructed p426GPD-ATP7B plasmid (Ponnandai Shanmugavel et al. 2017) as a template. All constructed plasmids were verified by sequencing (Eurofins). The DNA primers used for the plasmid construction are listed in the Supplementary Table S2. All plasmids were transformed into yeast cells by the standard lithium acetate method (Gietz and Woods 2006).
Yeast complementation assay
The Cu transport activity of the yeast strains was evaluated using growth curve analysis in iron limited medium. A single yeast colony from the plates was inoculated in Iron limited medium (SD medium containing 1.7 g L−1 yeast nitrogen base without Fe and Cu, 50 mM MES buffer pH 6.1, 20 g L−1 glucose, 5 g L−1 ammonium sulfate, complete supplement mixture CSM–Ura–His, 1 mM ferrozine (Fe chelator), 1 µM CuSO4 and 100 µM FeSO4) and incubated overnight at 30 °C and 200 rpm (Morin et al. 2009). Yeast cells from this culture were washed with ice cold deionized water and cultivated in the fresh iron-limited medium at an initial cell density of OD600 = 0.1. The growth of the cells was monitored and growth rates were determined from the exponential phase. All yeast growth experiments were carried out at identical conditions and in six biological replicates.
Protein extraction and western blotting
All yeast strains were grown in iron limited medium for 30 h at 30 °C. Cells were spun down by centrifugation at 2000×g at 4 °C for 10 min. Cells pellets were washed twice with ice-cold water and resuspended in lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 2.5 mM EDTA, 1% v/v Triton X100 and freshly added protease inhibitor). After disruption with glass beads, membranes were collected by centrifugation at 18,000×g (4 °C, 30 min). Samples were re-suspended in SDS loading buffer (0.5 M Tris–HCl, pH 6.8, 10% SDS, 0.5% (w/v) bromophenol blue, 87% glycerol, 100 mM DTT) and 50 mg of membranes were loaded on a 4–12% Bis–Tris gel (Invitrogen) and blotted onto PVDF membranes. ATP7B and Atox1 were detected with monoclonal rabbit ATP7B and Atox1 antibodies, respectively (Abcam, 1:1000 dilution), upon incubation overnight at 4 °C. Next, blots were incubated with horse radish peroxidase conjugated anti-rabbit IgG reagent (Thermo Scientific Pierce) for 15 min, 4 °C. Bands were detected by PierceTM Fast Western Blot Kits, SuperSignalTM West Femto, Rabbit (ThermoScientific Pierce) and visualized with a BioRad ChemiDoc XRSimage analyzer.
Cell localization of yeast-expressed ATP7B
Yeast cells were cultured to mid-log phase in the iron-limited medium. Harvested yeast cells were fixed in 5 ml of 50 mM KPO4 (pH 6.5), 1 mM MgCl2 and 4% formaldehyde for 2 h. After fixation, the cells were washed two times in 5 ml of PM buffer (100 mM KPO4 pH 7.5, 1 mM MgCl2) and followed by resuspension in PMST buffer (100 mM KPO4 pH 7.5, 1 mM MgCl2, 1 M sorbitol, 0.1% Triton X-100) to a final OD600 of 10. 100 µl yeast cells were incubated for 20 min in 0.6 µl of β-mercaptoethanol and 1 mg/ml zymolyase (Zymo Research). The spheroplasted cells were washed with PMST buffer and attached to polylysine-coated coverslips. Adherent cells were blocked in PMST–BSA buffer (0.5% BSA in PMST buffer) for 30 min. Next, the adherent cells were incubated overnight at 4 °C with primary antibody (1:500 rabbit monoclonal ATP7B antibody, Abcam) diluted in PMST–BSA buffer. After incubation, the cells were washed three times with PMST–BSA buffer and incubated with secondary antibody (1:1000 anti-rabbit Alexa 488, Abcam) for 3 h at room temperature, and with 0.4 mg/ml DAPI (staining nuclei) for 5 min. Cells were mounted in Vectashield mounting medium (Vector Laboratories). Images were acquired using a Leica DM 2000 inverted microscope and processed with the Leica application suite (LAS-AF lite) software.
In silico model building and molecular dynamics
All in silico systems were prepared based on the solution structure 2EW9 (a two-domain structure of the human ATP7B MBD56) (Achila et al. 2006). The first structure in the NMR structural ensemble were extracted as the starting coordinate for molecular dynamics (MD) simulations. The mutants L492S, A604P, G626A and R616W were in silico mutated based on the extracted wild-type structure (WT). Six single-domain systems were constructed by removing the linker between MBD5 and MBD 6. They are two wild-type proteins, i.e., MBD5WT and MBD6 WT, and four mutant protein systems, i.e., MBD5L492S, MBD6A604P, MBD6G626A, and MBD6R616W. The single-domain structure (wild-type system MBD5WT as the example) was solvated in a 60 Å rhombic dodecahedron water box. Na+Cl− ionic pairs (0.15 M) were added to neutralize the system and mimic the physiological conditions. The system was initially minimized for 10,000 steps under a series of position restraints and constraints, then heated to 300 K and equilibrated under NVT condition (constant volume and temperature) for 5 ns (ns) using the CHARMM program (version 42a1) (Brooks et al. 2009). The production MD simulation was carried out at NPT (constant pressure and temperature) condition, i.e., the pressure at 1 atm and the temperature at 340 K (Nilsson et al. 2013). The simulation lasted for 500 ns on each system using the NAMD program (version 2.12) and three replicas were performed (Phillips et al. 2005). MD snapshots were saved every 20 picoseconds along the trajectories for further analysis. Other details about the MD setting-up can be found elsewhere (Kumar et al. 2017). The trajectory analysis on all systems were done with CHARMM routines. All statistical figures were plotted by MATLAB (Version 2018a MathWorks, Inc.) and structural figures were generated with the PyMOL graphic software (Version 2.2 Schrödinger, LLC.).
Results
Probing ATP7B-mediated Cu-transport in a yeast assay
If an ATP7B mutation causes WD, the obvious speculation would be that the mutation hampers Cu transport by the mutated ATP7B protein. Therefore, to investigate Cu transport ability by the four mutated ATP7B proteins, we turned to a yeast assay that has been used by many to assess ATP7B dysfunction (Forbes et al. 1999; Ponnandai Shanmugavel et al. 2017) and that was improved by us recently to also include Cu delivery from human Atox1 (Ponnandai Shanmugavel et al. 2017; Shanmugavel and Wittung-Stafshede 2019). In this set up, the ∆ccc2∆atx1 double knockout yeast strain supplemented with the human ATOX1 gene acted as the background strain to which constructs with mutated ATP7B were introduced on high copy plasmids for the constitutive expression of mutant ATP7B proteins. The ∆ccc2∆atx1 yeast strain has inadequate growth on the iron-limited media due to lack of Cu transport that abolish Fet3p-mediated iron uptake (Ponnandai Shanmugavel et al. 2017). Thus, Cu transport by variants of ATP7B can be directly probed via yeast growth on iron limited media (Fig. S1). The growth rates of yeast expressing L492S ATP7B and R616W ATP7B matched the basal growth of yeast lacking ATP7B (Fig. 2a). This signifies an almost complete loss of Cu transport capability of these two mutated proteins. The A604P ATP7B expressing yeast showed increased growth as compared to L492S and R616W mutants, but substantially reduced growth compared to wild-type ATP7B (Fig. 2a). Surprisingly, the Cu transport efficiency was not reduced at all in yeast expressing G626A ATP7B. Thus, if Cu transport in yeast is a measure of WD severity, these results imply that L492S and R616W are worse mutations than A604P, and the G626A mutation is questioned with respect to if it really causes WD.

A Wilson disease mutants in ∆ccc2∆atx1 yeast. Growth rates (shown as bars, y-axis) of the ∆ccc2∆atx1 yeast strain complemented with high-copy plasmids with wild-type Atox1 and WD-causing ATP7B mutants (as indicated on x-axis) in iron-limited conditions as indicated. Error bars are based on the calculation of weighted errors from six biological replicates. B The immunostaining of ATP7B mutants in yeast cells using ATP7B-specific antibodies. Nuclei were visualized by DAPI staining. Scale bars, 2.5 μm. Each image shows: (from left to right) aΔccc2Δatx1 yeast with wild-type ATP7B high-copy plasmid as control; b the G626A mutant; c the A604P mutant; d the R616W mutant; and e the L492S mutant
Localization of ATP7B in yeast cells
The diminished Cu transport may be due to direct effects on Cu transport, reduced protein expression, or protein mis-localization in the yeast. Western blot analysis was used to confirm that the expression levels of all ATP7B variants in yeast were similar to that of wild type ATP7B (Fig. S2). To assess localization of ATP7B variants in yeast cells, we used immunofluorescence. All ATP7B mutants except L492S showed cellular localization patterns similar to that of wild type ATP7B (Fig. 2b) and involved an evenly distribution throughout the yeast cell. However, in yeast expressing the L492S variant, the protein was mainly localized in a confined region in the yeast cells such that it appeared in a scattered pattern concentrated in the endoplasmic reticulum (ER) surrounding the nucleus (Fig. 2b). Thus, for this particular variant, the lack of Cu transport may be mainly due to improper biosynthesis, thereby trapping in the ER.
Molecular dynamics (MD) simulations of individual MBDs
To probe differences in protein dynamics caused by the mutations, we turned to MD simulations. To ensure a sufficient sampling time of the proteins, we focused on analysis of single-domain systems. MD simulations were executed for individual wild-type MBD5 and wild-type MBD6 as well as mutated MBD5 (MBD5L492S) and mutated MBD6 (MBD6R616W, MBD6G626A, and MBD6A604P). All simulations lasted for 500 ns and were performed in three replicas. To further enhance the sampling, the six single-domain systems were run at the temperature of 340 K, which has been rationalized by our previous work (Nilsson et al. 2013). To observe effects on structural dynamics due to the mutations, we mapped root mean square fluctuation (RMSF) values onto the corresponding average structures from the MD simulations (Figs. 3 and S3). A similar method has been successfully implemented in other proteins (Li and Nam 2017). Interestingly, the protein dynamics changes caused by the mutations were structurally distant form the mutation sites; the mutations did not significantly alter the local structure near the mutated residue. All four mutations altered the fluctuations of the Cu-binding loop, as well as of the loop between β2 and β3 that faces the Cu-binding loop, although the mutations were spatially distant from these loops. (The location of the Cu-binding residues are indicated by two black arrows in the first structure in Fig. 3.) MBD5L492S showed increased dynamics in the Cu-binding loop and the RMSF data showed altered dynamics throughout the polypeptide such that fluctuations increased in the first half of the domain sequence but decreased in the second half as compared to MBD5WT. These alterations (not dramatic but throughout the peptide chain) may explain why full-length ATP7B with this mutation did not become processed correctly in the yeast cells. The MBD6 variants showed lower RMSF fluctuations in selected parts of the polypeptide as compared to MBD6WT, although most had increased RMSF in the Cu-binding loop, especially MBD6R616W, as compared to MBD6WT.

Structural mapping of RMSF values for six different single-domain constructions. All MD snapshots from three replicas of each system are collected for calculating the RMSF values. The RMSF values are mapped on the protein backbones. The thin and blue ribbon gradually changes to thick and red ribbon with the increasing RMSF values. All six constructs are placed in a similar orientation. The two Cu-binding cysteines are indicated by black arrows on the wild-type WD5 structure. The mutated residues on mutant systems are pointed out by the combination of red circles and arrows. The plots of their RMSF values are presented in Fig. S3. (Color figure online)
As a complement to RMSF fluctuations, we analyzed secondary structure content throughout the simulations. This data showed that MBD5L492S exhibited somewhat less secondary structures, and large vairations in this parameter, compared to MBD5WT. For MBD6A604P and MBD6G626A the secondary structure content appeared larger than for WT MBD6, with the most dramatic difference for MBD6G626A, whereas MBD6R616W displayed reduced secondary structure compared to MBD6WT (Fig. 4). Notably, although MBD6WT showed reduced secondary structure compared to some variants, its Cu-binding loop was relatively stable throughout the simulation according to the RMSF data. Some dynamics in the MBD fold may be of importance for ATP7B functional interactions with other proteins. Notably, we found the Cu-dependent interaction of Atox1 with MBD4 to be driven by entropy (Niemiec et al. 2015; Rodriguez-Granillo et al. 2010). Increased dynamics in Cu-binding loops of MBD6A604P and MBD6R616W may explain their reduced Cu transport ability in the yeast assay. In case of the G626A mutation, the lack of dramatic effects on structural dynamics parallels the wild-type like Cu transport ability in yeast. However, the apparent subtle increase in structural stability (increased secondary structure and low RMSF) found for the MBD6G626A domain may result in modulation of other aspects of ATP7B function.

Analysis of secondary structures of six single-domain proteins. The black, red and blue lines indicate numbers of residues for constructing total secondary structures (α helix and β sheet), α helix and β sheet, respectively. Dashed green lines are used to separate MD replicas. (Color figure online)
Discussion
In earlier work, we employed MD simulations to analyze structural dynamics of individual MBDs of ATP7B, as well as two-domain constructs, with a major focus on the effects of Cu loading and domain–domain correlations (Rodriguez-Granillo et al. 2009, 2010). Here, we turned to WD-causing missense mutations in MBDs. Four variants were selected because these mutations’ specific positions in the MBD ferredoxin-like fold did not provide obvious reasons for dysfunction. First we tested the four ATP7B variants (L492S in MBD5 and A604P, R616W, and G626A in MBD6) in a straight-forward Cu transport assay in yeast where yeast growth in the exponential phase directly report on Cu transport (Ponnandai Shanmugavel et al. 2017). Cu transport is the most common way to probe WD mutations, and most studied WD mutations in the MBDs, e.g., G85V (Huster et al. 2012; Li et al. 2019), S406A (Huster et al. 2012), Y532H (Parisi et al. 2018; Hsi and Cox 2004), V456L (Huster et al. 2012), P410L (Lee et al. 2011), A629P (Payne and Gitlin 1998; Lutsenko et al. 2007), S637L (Donsante et al. 2007) and S653H (Kim et al. 2003), displayed reduced or lack of Cu transport in cell-, or vesicle-based, Cu transport assays. For our selected WD mutations, three of the variants showed reduced Cu transport in our yeast assay, but one variant (G626A) had wild-type Cu transport capacity.
To probe underlying structural dynamics effects in the variants, and search for (perhaps more subtle) alterations in the G626A variant, we performed MD simulations of individual MBDs with mutations introduced. The MD simulations demonstrated clearly that the effects of individual mutations extended throughout the domains and, in all cases, affected the dynamics of the Cu-binding loop, although this loop is distant from the site of mutation in each case. The structural dynamics in the L492S domain was most increased among the variants; in fact, the whole polypeptide was affected here. The high domain dynamics due to this mutation may thus explain why this variant (as a full length protein), did not fold properly in the yeast cells and became retained in the ER. The observed altered structural dynamics in the Cu-binding loops of A604P and R616W variants may explain these variants’ reduced Cu transport activity in the yeast assay. Increased Cu-loop dynamics may affect efficiency of Cu delivery to MBD6, from other MBDs or Atox1, as well as Cu discharge from MBD6 to the Cu entry site in the ATP7B membrane-spanning part. For the G626A mutation, the in silico data revealed little effects on structural dynamics (if anything, the fold became more ordered), which agrees with this variant being able to transport Cu as well as wild-type ATP7B in the yeast assay. The reason that the G626A mutation is linked to WD in patients cannot be established from our work but may be due to modulation of other, more subtle, aspects of ATP7B function.
There are no biophysical-molecular studies of WD-causing mutations in the MBDs, except for a recent study with introduced mutations in MBD4 (Kumar et al. 2017). However, there are limited data on functional properties of the here selected WD mutations when introduced in full-length ATP7B. In similarity to our observation in the yeast assay, in vitro vesicle-reconstitution studies reported low Cu transport activity for L492S and R616W variants but only somewhat decreased such activity for G626A ATP7B (A604P has not been investigated before our work) (Huster et al. 2012). In contrast to the Cu transport effects, ATP7B interactions with the regulatory protein COMMD1 appeared normal for L492S and R616W variants, but was found decreased for G616A and A604P ATP7B variants (de Bie et al. 2007). Notably, mutations in the ATP7B homolog, ATP7A, corresponding to the here studied A604P, R616W, and G626A mutations, have been linked to Menke’s disease (Gourdon et al. 2012), another Cu metabolism disease which involves mutations in ATP7A. Moreover, the I492T mutation in ATP7A (Tumer 2013), found at a position that corresponds to L492 in ATP7B, and F281N in ATP7A (Caicedo-Herrera et al. 2018) (at a similar position to L492 in the ferredoxin fold, but in MBD3) were also reported to cause Menke’s disease. In analogy to the Cu transport results for G626A in ATP7B, a similar mutation in ATP7A was reported to exhibit normal Cu transport to the Golgi, but the ATP7A variant showed trafficking problems at high Cu levels (Kim et al. 2003). However, the trafficking ability of G626A ATP7B has been reported to be wild-type like (Braiterman et al. 2014). Therefore, the mechanism of dysfunction due to G626A appears to involve subtle changes in interactions with regulatory proteins, such as COMMD1 (de Bie et al. 2007).
Taken together, the yeast assay provides a platform to screen putative disease-causing mutations in ATP7B for Cu transport ability. From this data (L492S showing no Cu transport, due to ER trapping, but G626A exhibiting wild-type Cu transport) one may predict that the L492S mutation will confer more severe WD than the G626A mutation, although clearly many additional factors are of importance. However, the yeast assay may miss to identify WD mutations (e.g., G626A) that promote more subtle dysfunction and/or affect other aspects of ATP7B function. To reveal molecular-mechanistic reasons for mutation-induced ATP7B dysfunction, MD simulations are helpful as it can probe mutation-induced local structural perturbations with atomic resolution. To ultimately predict disease progression in WD patients, much more basic knowledge of molecular mechanisms through which mutations cause ATP7B dysfunction is required.
References
Achila D et al (2006) Structure of human Wilson protein domains 5 and 6 and their interplay with domain 4 and the copper chaperone HAH1 in copper uptake. Proc Natl Acad Sci USA 103(15):5729–5734
Arioz C, Li Y, Wittung-Stafshede P (2017) The six metal binding domains in human copper transporter, ATP7B: molecular biophysics and disease-causing mutations. Biometals 30(6):823–840
Banci L et al (2008) Metal binding domains 3 and 4 of the Wilson disease protein: solution structure and interaction with the copper(I) chaperone HAH1. Biochemistry 47(28):7423–7429
Boal AK, Rosenzweig AC (2009) Structural biology of copper trafficking. Chem Rev 109(10):4760–4779
Braiterman LT et al (2014) Distinct phenotype of a Wilson disease mutation reveals a novel trafficking determinant in the copper transporter ATP7B. Proc Natl Acad Sci USA 111(14):E1364–E1373
Brooks BR et al (2009) CHARMM: the biomolecular simulation program. J Comput Chem 30(10):1545–1614
Caca K et al (2001) High prevalence of the H1069Q mutation in East German patients with Wilson disease: rapid detection of mutations by limited sequencing and phenotype-genotype analysis. J Hepatol 35(5):575–581
Caicedo-Herrera G et al (2018) Novel ATP7A gene mutation in a patient with Menkes disease. Appl Clin Genet 11:151–155
de Bie P et al (2007) Distinct Wilson’s disease mutations in ATP7B are associated with enhanced binding to COMMD1 and reduced stability of ATP7B. Gastroenterology 133(4):1316–1326
Dong Y et al (2016) Spectrum and classification of ATP7B variants in a large cohort of Chinese patients with Wilson’s disease guides genetic diagnosis. Theranostics 6(5):638–649
Donsante A et al (2007) Differences in ATP7A gene expression underlie intrafamilial variability in Menkes disease/occipital horn syndrome. J Med Genet 44(8):492–497
Forbes JR, Hsi G, Cox DW (1999) Role of the copper-binding domain in the copper transport function of ATP7B, the P-type ATPase defective in Wilson disease. J Biol Chem 274(18):12408–12413
Gietz RD, Woods RA (2006) Yeast transformation by the LiAc/SS carrier DNA/PEG method. Methods Mol Biol 313:107–120
Gonzalez-Guerrero M, Arguello JM (2008) Mechanism of Cu+-transporting ATPases: soluble Cu+ chaperones directly transfer Cu+ to transmembrane transport sites. Proc Natl Acad Sci USA 105(16):5992–5997
Gourdon P et al (2012) Structural models of the human copper P-type ATPases ATP7A and ATP7B. Biol Chem 393(4):205–216
Harada M et al (2001) A mutation of the Wilson disease protein, ATP7B, is degraded in the proteasomes and forms protein aggregates. Gastroenterology 120(4):967–974
Hsi G, Cox DW (2004) A comparison of the mutation spectra of Menkes disease and Wilson disease. Hum Genet 114(2):165–172
Huster D et al (2012) Diverse functional properties of Wilson disease ATP7B variants. Gastroenterology 142(4):947–956.e5
Jang JH et al (2017) Carrier frequency of Wilson’s disease in the Korean population: a DNA-based approach. J Hum Genet 62(9):815–818
Kim BE, Smith K, Petris MJ (2003) A copper treatable Menkes disease mutation associated with defective trafficking of a functional Menkes copper ATPase. J Med Genet 40(4):290–295
Kumar R et al (2017) Disease-causing point-mutations in metal-binding domains of Wilson disease protein decrease stability and increase structural dynamics. Biometals 30(1):27–35
Lee BH et al (2011) Distinct clinical courses according to presenting phenotypes and their correlations to ATP7B mutations in a large Wilson’s disease cohort. Liver Int 31(6):831–839
Li Y, Nam K (2017) Dynamic, structural and thermodynamic basis of insulin-like growth factor 1 kinase allostery mediated by activation loop phosphorylation. Chem Sci 8(5):3453–3464
Li X et al (2019) Complex ATP7B mutation patterns in Wilson disease and evaluation of a yeast model for functional analysis of variants. Hum Mutat 40(5):552–565
Ljubic H et al (2016) ATP7B gene mutations in croatian patients with Wilson disease. Genet Test Mol Biomark 20(3):112–117
Lo C, Bandmann O (2017) Epidemiology and introduction to the clinical presentation of Wilson disease. Handb Clin Neurol 142:7–17
Lutsenko S et al (2007) Function and regulation of human copper-transporting ATPases. Physiol Rev 87(3):1011–1046
Mohr I, Weiss KH (2019) Biochemical markers for the diagnosis and monitoring of Wilson disease. Clin Biochem Rev 40(2):59–77
Mondol T, Aden J, Wittung-Stafshede P (2016) Copper binding triggers compaction in N-terminal tail of human copper pump ATP7B. Biochem Biophys Res Commun 470(3):663–669
Morin I et al (2009) Dissecting the role of the N-terminal metal-binding domains in activating the yeast copper ATPase in vivo. FEBS J 276(16):4483–4495
Nagano K et al (1998) Intracellular distribution of the Wilson’s disease gene product (ATPase7B) after in vitro and in vivo exogenous expression in hepatocytes from the LEC rat, an animal model of Wilson’s disease. Hepatology 27(3):799–807
Niemiec MS, Weise CF, Wittung-Stafshede P (2012) In vitro thermodynamic dissection of human copper transfer from chaperone to target protein. PLoS ONE 7(5):e36102
Niemiec MS, Dingeldein APG, Wittung-Stafshede P (2015) Enthalpy-entropy compensation at play in human copper ion transfer. Sci Rep 5:10518
Nilsson L et al (2013) Small pH and salt variations radically alter the thermal stability of metal-binding domains in the copper transporter, Wilson disease protein. J Phys Chem B 117(42):13038–13050
Parisi S et al (2018) Characterization of the most frequent ATP7B mutation causing Wilson disease in hepatocytes from patient induced pluripotent stem cells. Sci Rep 8(1):6247
Park RH et al (1991) Wilson’s disease in Scotland. Gut 32(12):1541–1545
Payne AS, Gitlin JD (1998) Functional expression of the Menkes disease protein reveals common biochemical mechanisms among the copper-transporting P-type ATPases. J Biol Chem 273(6):3765–3770
Phillips JC et al (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26(16):1781–1802
Ponnandai Shanmugavel K, Petranovic D, Wittung-Stafshede P (2017) Probing functional roles of Wilson disease protein (ATP7B) copper-binding domains in yeast. Metallomics 9(7):981–988
Rodriguez-Castro KI, Hevia-Urrutia FJ, Sturniolo GC (2015) Wilson’s disease: a review of what we have learned. World J Hepatol 7(29):2859–2870
Rodriguez-Granillo A, Crespo A, Wittung-Stafshede P (2009) Conformational dynamics of metal-binding domains in Wilson disease protein: molecular insights into selective copper transfer. Biochemistry 48(25):5849–5863
Rodriguez-Granillo A et al (2010a) Copper-transfer mechanism from the human chaperone Atox1 to a metal-binding domain of Wilson disease protein. J Phys Chem B 114(10):3698–3706
Rodriguez-Granillo A, Crespo A, Wittung-Stafshede P (2010b) Interdomain interactions modulate collective dynamics of the metal-binding domains in the Wilson disease protein. J Phys Chem B 114(5):1836–1848
Shanmugavel KP, Wittung-Stafshede P (2019) Copper relay path through the N-terminus of Wilson disease protein, ATP7B. Metallomics. https://doi.org/10.1039/C9MT00147F
Tuan Pham LA et al (2017) Genetic analysis of 55 northern Vietnamese patients with Wilson disease: seven novel mutations in ATP7B. J Genet 96(6):933–939
Tumer Z (2013) An overview and update of ATP7A mutations leading to Menkes disease and occipital horn syndrome. Hum Mutat 34(3):417–429
Yu CH, Dolgova NV, Dmitriev OY (2017a) Dynamics of the metal binding domains and regulation of the human copper transporters ATP7B and ATP7A. IUBMB Life 69(4):226–235
Yu CH et al (2017b) The metal chaperone Atox1 regulates the activity of the human copper transporter ATP7B by modulating domain dynamics. J Biol Chem 292(44):18169–18177
Yu CH et al (2018) The structure of metal binding domain 1 of the copper transporter ATP7B reveals mechanism of a singular Wilson disease mutation. Sci Rep 8(1):581
Zong YN, Kong XD (2015) Analysis and application of ATP7B gene mutations in 35 patients with hepatolenticular degeneration. Genet Mol Res 14(4):18764–18770
Acknowledgements
Open access funding provided by Chalmers University of Technology. This work is supported by the Swedish Research Council, the Knut and Alice Wallenberg foundation and the International Postdoc Grant funded by the Swedish Research Council (to Y.L).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Shanmugavel, K.P., Kumar, R., Li, Y. et al. Wilson disease missense mutations in ATP7B affect metal-binding domain structural dynamics. Biometals 32, 875–885 (2019). https://doi.org/10.1007/s10534-019-00219-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-019-00219-y