
Abstract
Wilson Disease (WD) is a hereditary genetic disorder, which coincides with a dysfunctional copper (Cu) metabolism caused by mutations in ATP7B, a membrane-bound P1B-type ATPase responsible for Cu export from hepatic cells. The N-terminal part (~ 600 residues) of the multi-domain 1400-residue ATP7B constitutes six metal binding domains (MBDs), each of which can bind a copper ion, interact with other ATP7B domains as well as with different proteins. Although the ATP7B’s MBDs have been investigated in vitro and in vivo intensively, it remains unclear how these domains modulate overall structure, dynamics, stability and function of ATP7B. The presence of six MBDs is unique to mammalian ATP7B homologs, and many WD causing missense mutations are found in these domains. Here, we have summarized previously reported in vitro biophysical data on the MBDs of ATP7B and WD point mutations located in these domains. Besides the demonstration of where the research field stands today, this review showcasts the need for further biophysical investigation about the roles of MBDs in ATP7B function. Molecular mechanisms of ATP7B are important not only in the development of new WD treatment but also for other aspects of human physiology where Cu transport plays a role.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Copper (Cu) is an essential metal ion that plays key roles in metabolic processes acting as a reactive cofactor in proteins (e.g., connective tissue formation, tryptophan synthesis, iron bioavailability, respiration, free radical scavenging, pigmentation, and neurotransmitter synthesis) (Letelier et al. 2005; Matson Dzebo et al. 2016; Polishchuk et al. 2014; Prashanth et al. 2015). Because of its ability to switch between Cu (I) and Cu (II), this metal is often used by a number of copper-dependent enzymes in electron-transfer reactions (Festa and Thiele 2011; Pham et al. 2013; Yu et al. 2017a). The high redox potential of copper ions allow them, when in free form, to generate hydroxyl radicals (.OH) that can damage the structures of proteins, nucleic acids and lipids of cells (Festa and Thiele 2011). Therefore, free copper ions and/or their abnormal levels can be toxic for cells so that their distribution is tightly regulated via dedicated protein systems that facilitate its uptake, efflux and delivery to Cu-dependent enzymes (DiDonato et al. 2000; Kumar et al. 2017; Matson Dzebo et al. 2016; Mondol et al. 2016).
In humans, Cu enters the cell via the membrane-bound high-affinity copper uptake protein 1 (Ctr1) (Gupta and Lutsenko 2009; Wang et al. 2011). How Cu(II) in the blood is reduced to Cu(I) remains uncertain but the possible involvement of a membrane-bound copper reductase close to the Ctr1 was mentioned (Knöpfel and Solioz 2002; Lutsenko et al. 2007; Shatwell et al. 1996; Taylor et al. 2005). Nevertheless, after entering the cytoplasm copper is presumably in the reduced Cu(I) form because the cytoplasm is a highly-reducing environment due to its high glutathione (GSH) content (Kaplan and Maryon 2016; Lopez-Mirabal and Winther 2008). Therefore herein, the abbreviation “Cu” refers “Cu(I)” unless otherwise stated. In the cytoplasm, there are at least three distinct pathways of copper transport for metallation of Cu-dependent proteins/enzymes. One pathway involves the copper chaperone for superoxide dismutase (CCS) that carries Cu ions to Cu/Zn superoxide dismutase 1 (SOD1), the primary cytoplasmic scavenger of superoxide radical (.O2 −) (Matson Dzebo et al. 2016; Rakhit and Chakrabartty 2006). Another route is to the mitochondria: here, although there is much unknown, somehow the cytochrome c oxidase copper chaperone, Cox17, transfers Cu via the help of additional proteins (e.g., Sco1, Sco2, Cox11) to cytochrome c oxidase (COX), the final electron acceptor in the mitochondrial electron transport chain (Horn and Barrientos 2008). In the third and general cytoplasmic pathway for Cu, Atox1 (alias: HAH1), a 68-residue cytoplasmic Cu chaperone, transfers Cu to ATP7A (Menke’s disease protein) or ATP7B (Wilson disease protein), two homologous membrane proteins in the trans-Golgi network (TGN) (Banci et al. 2008; Barnes et al. 2009; Hamza et al. 1999). Following this, Cu ions are either used for the biosynthesis of various copper-dependent enzymes such as tyrosinase, lysyl oxidase and ceruloplasmin in the secretory pathway, or (if there is excess Cu) sequestered in cytoplasmic membrane vesicles for excretion from the cell to the bile (Cater et al. 2006; Dmitriev et al. 2006).
ATP7A and ATP7B are P1B-type ATPases, which use the energy released from ATP hydrolysis to transport Cu across Golgi membranes (Jayakanthan et al. 2017; Lutsenko et al. 2008; Yatsunyk and Rosenzweig 2007; Yu et al. 2017b). Although they share a close homology (67% amino acid similarity) (Yatsunyk and Rosenzweig 2007), they can be distinguished by unique properties (Hamza et al. 1999; Mercer et al. 2003). For example, ATP7A is primarily expressed in non-hepatic tissues such as intestines, brain, heart etc. whereas ATP7B is mostly found in liver and kidney, but also detected with lower levels in the lung, placenta and brain (Bunce et al. 2006; Cater et al. 2004; DiDonato et al. 2000). ATP7A is mostly responsible for Cu absorption from intestinal enterocytes to the blood and function-impairing mutations of this protein cause Menke’s disease (MD). As a result, intestinal cells accumulate excess amount of Cu, and thereby the blood-Cu level is decreased, leading to decreased Cu delivery to other tissues (Keller et al. 2012; Lutsenko et al. 2007). Patients with MD suffer from connective tissue abnormalities, lack of pigmentation, diminished functions of Cu-dependent enzymes and tortuosity of blood vessels due to the impaired delivery of Cu especially to the brain and the majority of these patients die at early childhood (Banci et al. 2009b). On the other hand, ATP7B is mostly responsible for Cu efflux from hepatocytes (Dmitriev et al. 2006; Keller et al. 2012) and impairment of ATP7B function due to mutations results in an autosomal recessive disorder known as Wilson disease (WD). The main biochemical phenotype for WD is a chronic copper toxicosis where hepatocytes fail to remove excess copper and is observed by clinical symptoms such as hepatic abnormalities, neurological defects, more commonly psychiatric/behavioral symptoms (Hedera 2017), and eventually liver failure which can cause death when untreated (Chesi et al. 2016; DiDonato et al. 2002; Yu et al. 2017c). For an adult, basic daily copper requirement is ~1–2 mg and under normal circumstances Cu overload is dissipated by ATP7B and maintained at levels less than 50 µg Cu/g dry liver. However, in WD patients hepatic Cu levels can reach up to 250–3000 µg Cu/g dry liver (Das and Ray 2006; Suzuki et al. 2002). To reduce it to normal levels, there are different strategies currently used as WD treatment involving removal of excess Cu by the administration of Cu chelators such as D-penicillamine (D-PCA; with very severe side effects such as immunological disturbances, skin defects, joint disorders and worsened neurological manifestations), Trientine (with lower side effects compared to D-penicillamine) and the novel chelating agents such as tetrathimolybdate (TTM; blocks Cu absorption) or choline tetrathiomolybdate (CTTM; a more stable salt formulation of tetrathiomolybdate) that are being currently evaluated (Chang et al. 2013; Schilsky 2014; Weiss 2016). After Cu detoxification, re-accumulation of Cu in tissues are prevented by a “maintenance therapy” throughout life in which patients use lowered dosages (25–33% of the initial dose) of above mentioned chelators/agents or zinc salts (prevents intestinal Cu absorption but increases metallothionein, an heavy-metal scavenger) but also maintain a low copper diet (Schilsky 2014). However, some of the agents described above have been reported to cause significant problems when they are used long-term (Roberts et al. 2008), therefore new drugs with less side effects are desired. Understanding how disease-causing mutations change ATP7B function at the molecular level is of great importance for developing new medication strategies for this disease.
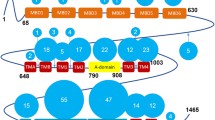
ATP7B has a basic P-type ATPase architecture (Banci et al. 2009b) with a cytosolic region composed of phosphorylation (P-), ATP-binding (N-) and actuator/dephosphorylation (A-) domains and a membrane part where eight transmembrane helices (transmembrane domains, TMDs) form an intramembranous Cu channel. A unique structural feature of this multi-domain protein, is the presence of a large, cytosolic N-terminal tail containing six 70-aa long independently folded Cu (I) binding domains (hereinafter metal binding domains, MBDs) (Fig. 1) (Banci et al. 2009b; LeShane et al. 2010; Mondol et al. 2016). MBDs are named from the N-terminal, with MBD1 being the first domain from the N-terminus and MBD6 the domain closest to the membrane-spanning part of ATP7B. The domains are connected by peptide linkers of various length (Fig. 3C). The complete N-terminus is about 630 residues long (Banci et al. 2009a) where roughly 200 residues belong to the inter-domain linkers. The longest linker is located between MBD4 and MBD5, consisting of 76 residues. The length of other linkers is relatively short: namely, 29 residues between MBD3-MBD4, 24 residues between MBD2-MBD3, 13 residues between MBD1-MBD2 and 6 residues between MBD5-MBD6 (Fig. 3C). The basic P-type ATPase architecture does not change from bacteria to humans but the number of MBDs greatly differentiate between phylogenetic families. Mammals, birds and reptiles have six MBDs; nematodes and insects contain four MBDs, whereas bacteria or lower eukaryotes (e.g. yeast) contain only one or two MBDs (Yu et al. 2017a). The increasing number of MBDs from bacteria to humans can be seen as an evolutionary advantage for human ATPases to fine-tune their activities. Such regulatory function can be fulfilled via interactions between MBDs and other domains of ATP7B (or other proteins) (Mondol et al. 2016; Sharma and Rosato 2009). Although some in vitro work have suggested the presence of domain–domain communication/s between MBDs and MBDs with the core of the ATP7B, the exact roles of these communications in vivo remain unknown. It is even not clear if MBDs are on the Cu-route towards the membrane channel or merely dead-end Cu reservoirs. Nonetheless, disease-causing mutations can be found in the ATP7B MBDs and thus these domains must critically impact normal Cu-transport activity. In contrast, MD mutations have not been reported to be positioned in any MBDs of ATP7A (Hamza et al. 1999), except for E628V and K633R, both of which are located in MBD6 of ATP7A (Skjorringe et al. 2017).

The structural organization of ATP7B. Cartoon illustrates the major functional domains and important motifs for Cu-ATPases. ATP7B and other P-type ATPases consist of three cytoplasmic domains; nucleotide binding domain (N-domain, blue), phosphorylation domain (P-domain, pink), actuator/dephosphorylation domain (A-domain, purple). A combination of N- and P-domains are usually referred as ATP-binding domain. ATP7B hydrolyzes ATP and the invariant aspartate (D) located at the DKTGT motif accepts γ-phosphate from ATP during the catalytic cycle forming a transient phosphorylated intermediate (E1P). SEHPL is a conserved motif among all ATPases transporting transition metals (P1-type ATPases or CPx-ATPases) and includes H1069 which is believed be involved in correct positioning of ATP prior to transfer of the γ-phosphate (Tsivkovskii et al. 2003). TGE motif in the A-domain is responsible for the removal of γ-phosphate from the DKTGT motif (dephosphorylation). The N-terminal contains six metal-binding domains (MBD1-MBD6, cyan) with conserved CxxC Cu-binding motifs. The transmembrane domain (TMD) encompasses eight transmembrane helices and position of residues that are predicted to be involved in Cu-coordination within the membrane are depicted as CPC, YN and MXXXS. The two leucine motifs (1459LL and 1487LL) located at the C-terminal have been shown to be essential for endocytosis and/or TGN relocalization from plasma membrane
ATP7B adheres to the working mechanism of all P-type ATPases, the Post-Albers catalytic cycle where a phosphorylation-dephosphorylation cascade occur with four principal conformations described as E1 (high affinity), E1P, E2 (low affinity) and E2P states (Gourdon et al. 2012; Skjorringe et al. 2017). In the catalytic cycle of ATP7B, ATP binding to N-domain and Cu binding to the CPC motif (in TMD6) (E1 state), are followed by the transient phosphorylation of the D1027 residue located at the P-domain upon the transfer of the γ-phosphate from ATP. This transfer (E1P state) enables Cu to be occluded from the intramembrane region to the Golgi lumen and is followed by a conformational change within ATP7B structure which allows the Cu to be translocated across the membrane (E2P state). This is followed by dephosphorylation of the invariant D1027 residue by the A-domain (E2 state) (Banci et al. 2009b). These steps are altogether coupled to conformational changes within ATP7B and it is proposed that alterations of domain–domain interactions, notably involving the MBDs, are of high relevance for ATPase activity (Banci et al. 2009a; Huang et al. 2014; Tsivkovskii et al. 2001). ATP7B normally resides in trans-Golgi network (TGN) of hepatocytes under low Cu conditions (Polishchuk et al. 2014) but as Cu levels increase, it exists TGN and re-localizes into cytoplasmic membrane vesicles to sequester excess cytosolic Cu. Later, these vesicles move close to the plasma membrane where excess Cu is extruded from the cell via vesicle-mediated exocytosis (Wu et al. 2015). As Cu levels are depleted, ATP7B returns from vesicles to the TGN to continue its role in Cu-loading of proteins (Huang et al. 2014).
To display what is known today about ATP7B MBDs from a biophysical viewpoint, here we have summarized previously reported results for various constructs of the six MBDs of ATP7B (Supplementary Table S1) and also gathered the limited biophysical information of known WD point mutations that are located in the six human MBDs (Table 1).
Cu coordination in ATP7B MBDs
Several in vitro and in silico studies have investigated Cu coordination of MBDs and its effects on the protein structure using individual MBD domains or multi-domain MBD constructs (Supplementary Table S1). Based on previous work, the overall structure of each domain is similar to each other and to the MBDs of ATP7A and to that of Atox1 (Achila et al. 2006; Banci et al. 2008; Fatemi et al. 2010; Walker et al. 2004). All MBDs have a ferredoxin-like fold (Achila et al. 2006; Banci et al. 2008) where the Cu-binding site is situated in a solvent-exposed loop between β1-strand and α1-helix (Braiterman et al. 2015; LeShane et al. 2010). Here, each MBD includes a strictly conserved Cu-binding motif GMX1CX2X3CV, known also as the Cu site, which can bind a single Cu(I) ion via di-cysteine coordination of the two sulfur residues in cysteines (C1 and C2) (DiDonato et al. 1997, 2000, 2002; Hussain et al. 2009; Larin et al. 1999; Lutsenko et al. 1997; Niemiec et al. 2014; Rodriguez-Granillo et al. 2009; Walker et al. 2004). This coordination geometry does not change in any of the MBDs and was shown with X-ray absorption spectroscopy (XAS) to be very similar to that for Cu in Atox1 (Ralle et al. 1998). Both in vitro and in silico work with different constructs of MBDs (e.g. MBD1-6 as single domains or as entire MBD1-6) (DiDonato et al. 2002; Rodriguez-Granillo et al. 2009; Walker et al. 2004) showed that increasing Cu concentration does not affect Cu coordination. However, an XAS/EXAFS analysis of MBD1-6 construct revealed that Cu sites in each MBD can be distorted from the ideal linear geometry of a two-coordinate Cu(I) center to a geometry with a S–Cu–S angle between 120° and 180° maintaining a Cu–S distance of 2.17–2.19 Å (DiDonato et al. 2000; Ralle et al. 2004; Walker et al. 2004). A similar Cu(I) coordination environment with a Cu–S distance of 2.16 Å was also shown to exist for MBDs of ATP7A (Ralle et al. 1998) and Atox1 (Ralle et al. 2003; Walker et al. 2004) via EXAFS measurements.
At first glance, all Cu-binding motifs of MBDs appear identical (Banci et al. 2009a; Walker et al. 2004). However, the sequence alignment of all MBDs (Fig. 2) indicates some positions within this motif may differ from one MBD to another with the highest conservation around the Cu-binding motif (GMX1CX2X3CV). Here, in most MBDs except MBD3, X1 position is occupied by threonine (T), whilst in MBD3, it is replaced by a histidine (H). Another substitution around the Cu motif is position X2 where a polar/uncharged glutamine (Q) in both MBD1 and MBD2 is changed to a lysine (K) in MBD3 and an alanine (A) in MBD4-6. Besides the Cu-binding motif, there is a second cluster of conserved residues which are located around phenylalanine (F66) (Fig. 2, F66 marked with asterisk). In silico, F66 was predicted to be in the α2-β4 loop in close proximity to Cu-binding motif (Rodriguez-Granillo et al. 2009). Here, a D63MGFEA68 motif is preserved for MBD1, MBD2 and MBD4. In MBD3, the motif is instead A63LPPGN68, and in MBD6 there are D63E, M64I and E67H substitutions. In MBD5, there is only one substitution (M64L) in this motif. The functional importance of these substitutions in individual MBDs is unclear but could be related to unique properties of individual domains (Bartee et al. 2009; Cater et al. 2004; Fatemi et al. 2010) and perhaps might help to explain why not all MBDs are functionally equivalent (Banci et al. 2009a; Cater et al. 2004; Iida et al. 1998; Rodriguez-Granillo et al. 2009; Wernimont et al. 2004).

Multiple sequence alignment of individual ATP7B MBDs (MBD1-6) and Atox1. Residues that are highly conserved are highlighted with a dark purple color and color tone decays in parallel with the degree of conservation for that specific residue. Sequences between 3 and 70 for all MBDs and 1–68 for Atox1 were used. Sequence alignment was performed using Jalview version 2.10.2b1 (http://www.jalview.org/)
In silico studies showed that the Cu-loop of most individual MBDs is stabilized by the hydrogen bonds between C1, the highly conserved threonine (T = X1) and serine (S = X3). The stabilization of the Cu-loop is strengthened by the methionine (M) residue preceding X1, which is fulfilled through the hydrophobic interaction between the M residue and the conserved phenylalanine (F66). C2 is buried in the core of the domains, and interacts with F66 or X3 (= S), M64 (Fig. 2, marked with asterisk) or with C1 to maintain a stable Cu-loop. C2 is buried deeper in MBD3 than in other domains, but does not interact with X3 or M64, instead of interacting with valine (V18) in helix α1 and V36/L38 in a β-strand. In contrast to other MBDs, MBD4 lacks most of the described interactions around the Cu loop due to the increased dynamics between α1-helix and last part of the Cu loop, which is absent in the other MBDs. The Cu-site in MBD4 is only stabilized through interactions of M with L40 and G43 that are located in the β2-β3 loop. C1 here interacts only with L40 but not with X1 (= T) or X3 (= S), and C2 of MBD4 is buried to the core and makes contacts with several residues including X1 (= T), X3 (= S), V20 in α1-helix, V38 in β2-strand and L40. Upon Cu-binding, C1 and C2 of MBD2 and MBD6 form hydrogen bonds with X1 (= T) and X3 (= S) and M became completely buried within the structure but like in the apo forms, continued to interact with residues in the β2–β3 loop, F66 and C2. The positioning of C2 in MBD6 becomes more buried than it is in MBD2 upon Cu binding, because it points toward the loop backbone. In MBD4, upon Cu-binding the interactions of M residue greatly varies but ultimately this residue points to the Cu-loop, contributing to the increased fluctuations of the Cu-loop. Moreover, Cu-bound C2 stops interacting with the F66 side chain and hence F66 forms contacts with the hydrophobic core. Although the Cu loop becomes more flexible in MBD4 upon Cu-binding, Cu-coordination was observed to reduce overall backbone fluctuations for all domains including MBD4 (Rodriguez-Granillo et al. 2009), which stabilizes the ferredoxin-fold of each domain (Portmann and Solioz 2005; Tadini-Buoninsegni et al. 2010).
Long-range copper-induced effects in MBDs
Despite differences described above, all MBDs can bind Cu(I) with a similar binding affinity (Lutsenko et al. 1997; Yatsunyk and Rosenzweig 2007). However, isothermal titration calorimetry (ITC) experiments indicated that both MBD3–4 and MBD5–6 bind Cu-more tightly than MBD1–2 when they are studied as two-domain constructs but this distinction disappears when the domains are either accommodated in MBD1-6 (Wernimont et al. 2004; Yatsunyk and Rosenzweig 2007) or monitored separately in a two-domain construct (Banci et al. 2008). In silico studies with two-domain constructs revealed that the motional freedom of the Cu-binding loop undergoes a different behavior when covalently linked to a consecutive domain (Rodriguez-Granillo et al. 2010b). Although it has been proposed that progressive Cu binding induces conformational rearrangements along the overall N-terminal (DiDonato et al. 2000; DiDonato et al. 1997), NMR, CD and EXAFS studies performed on MBD1–6 construct (Banci et al. 2009a; Banci et al. 2009b; Banci et al. 2008) showed that Cu binding does not affect the overall fold of individual domains except for some changes localized only to the Cu-binding loop. These changes were proposed to be transmitted to other parts of the MBD1–6 construct through the peptide backbone (Banci et al. 2008; DiDonato et al. 2000). This suggestion agrees well with NMR and proteolysis results showing that Cu binding to MBD1–4 makes four domains to be organized in a more compact manner without triggering any major structural changes within each domain (Bartee et al. 2009; Mondol et al. 2016). However, NMR studies with MBD4-6 and MBD5-6 revealed that Cu-induced changes were only limited to the residues surrounding the Cu-binding motif and the rest of the protein structure remained largely unaffected (Achila et al. 2006; Fatemi et al. 2010). A recent in vitro study with MBD1-6 reported that MBD1-3 was compacted without Cu and adopted an extended conformation after Cu loading (Yu et al. 2017b), indicating a possible impact of Cu binding on the structural arrangements of both linkers and MBDs. Clearly, there should be more analyses to perform because constructs with different number of MBDs may behave differently.
Thermal unfolding experiments with MBD1–4 indicated that upon Cu-binding thermal midpoint (Tm) increased from 51 to 59 °C. This occurrence may be caused by the increased interactions between individual MBDs (Mondol et al. 2016). Similarly, Cu-binding to MBD5–6 construct increased Tm from 68 to 78 °C, which was reasoned that the binding of Cu to one domain increased the stability of both domains (Nilsson et al. 2013). Earlier an in silico study suggested that the interface region between MBD5 and MBD6 contains charged residues where R53 in MBD6 facing the protein surface interacts with F7 and E45 that are located in β1/β3 strands of MBD5. These interactions contributed to the stabilization of the entire construct (Rodriguez-Granillo et al. 2009). The NMR structure of MBD56 (PDB code: 2EW9) also shows that conserved K119 of MBD6 interacts with negatively charged residues (E73, E83 and D74) that are located either in the linker region or in strand β5 of MBD6. Due to these ionic interaction network, altered salt and pH conditions were found to dramatically affect MBD5-6 thermal stability (Nilsson et al. 2013). The deprotonation/protonation changes of histidine (H100 and H148) residues were proposed to affect the overall network of interactions on the protein’s surface (Nilsson et al. 2013). In addition, conformational changes within connecting linkers might modulate structural stability of the overall N-terminus (Bartee et al. 2009; DiDonato et al. 2000; Huster and Lutsenko 2003). These flexible linkers can also provide docking sites for the trafficking machinery (Hasan et al. 2012; Lim et al. 2006) and other interacting partners such as Atox1 and COMMD1 (Fatemi et al. 2010). The linker between MBD3 and MBD4 was shown with NMR, single-molecule fluorescence resonance energy transfer (smFRET) and proteolysis experiments to undergo conformational changes, such that it became more dynamic when the MBDs were loaded with Cu (Bartee et al. 2009; Fatemi et al. 2010; Keller et al. 2012). Taken all together, it is plausible that Cu coordination induces altered spatial organization of the six MBDs locally or globally, thereby modulating the overall protein activity.
MBD interactions with other domains and proteins
High-resolution structures of homologs to ATP7B have been reported, including class 1B Legionella pneumophila CopA (LpCopA) (Gourdon et al. 2011), class IIA sarcoplasmic reticulum Ca2+-ATPase (SERCA1a) (Olesen et al. 2004), class IIC Na+, K+-ATPase (Morth et al. 2007; Ogawa et al. 2009; Shinoda et al. 2009) and class IIIA H+-ATPase (Abrahams et al. 1994; Stock et al. 1999), but in none of them the position of an MBD was resolved. Although Gourdon et al. have proposed a full-length model of ATP7B using homology modeling (Gourdon et al. 2012) where MBDs were arranged as ‘stack of logs’, the true orientation of the MBDs is still unknown. To visualize the organization of the six MBDs at the atomic level, we developed a full-length model of the six MBDs by integrating current structural and biophysical data (Dmitriev et al. 2016; Yu et al. 2017a). The detailed information for building the model is described in Fig. 3. The initial model (Fig. 3A) was then sampled by MD simulations in implicit solvent to optimize the added linkers between different domains. In the resulting model (Fig. 3B), MBD1–3 and MBD5–6 act as two independent structural units and the orientation of MBD1-3 agrees with the data from recent small-angle X-ray scattering (SAXS) experiments (Yu et al. 2017b). By contrast, MBD4 undergoes few contacts with other domains in the model, in line with its proposed role in merely connecting other structural units (Yu et al. 2017b). Multi-scale modeling that integrates all-atom and coarse-gained models may provide a strategy to simulate a full-length ATP7B model (including MBDs, membrane-spanning parts, lipids etc.) in the future.

An all-atom full-length model of MBD1-6. A The initial model. B The optimized model. Six subdomains are colored differently, and their unstructured linkers are colored in cyan. NMR structures in Table 2 were used. The initial domain arrangement was determined based on the sequence length of linkers and electrostatic complementarity of the protein surfaces between different domains. 3D coordinates of the missing linkers were generated by SWISS-MODEL (Biasini et al. 2014) server using the arranged model as a template, leading to a full-length model of MBD1-6 without the inclusion of the first 56 residues. The initial model was then optimized by MD simulations using NAMD (version 2.12) (Phillips et al. 2005) for 5 nanoseconds (ns). The solvent was presented in the generalized Born/solvent-accessible surface area implicit solvent model (Tanner et al. 2011), and the protein system was described in CHARMM36 force field (Huang and MacKerell 2013). C The schematic representation of N-terminal MBDs of ATP7B in tandem. The length of each MBD and linkers (as the position of amino acids) in between is indicated according to the PDB structures of the respective MBDs (Table 2). A linker is defined as the segment between folded 70-aa MBDs except only for MBD6 which is 68-aa long. TMD represents the transmembrane part of ATP7B and the linker between MBD6 and transmembrane helix 1 (TMD1) is 20-aa long (Lorinczi et al. 2008)
An interaction of MBDs with another ATP7B domain was first reported upon co-purification of MBDs with the N-domain. The interaction was found to depend on Cu such that it was detected only in the absence of Cu in the MBDs (Tsivkovskii et al. 2001). This interaction was proposed to keep ATP7B inactive at low Cu-conditions (Bartee et al. 2009; Tsivkovskii et al. 2001). Upon Cu-binding to MBDs, the structural reorganization resulted in dissociation of the MBD-N-domain complex and thereby allowed ATP to bind to the N-domain (Bartee et al. 2009). However, NMR studies with MBD1–4 and MBD1–6 constructs could not detect any stable interaction with the N-domain in solution although very high concentrations were used. Mondol et al. therefore suggested that the N-domain-MBD complex formed only transiently, or constituted a minor species in the co-purification experiments (Mondol et al. 2016). The homology model of ATP7B, based on the LpCopA, also indicated that MBD6, with an overall positive surface charge, may interact with the negatively charged P-domain (Gourdon et al. 2012) and N-domain (Dmitriev et al. 2006). As the latest piece of information, a synthetic peptide comprising residues 33–63 of ATP7B (i.e., peptide stretch just before MBD1) was found by NMR to interact at the interface between N- and A-domains despite the usage of a high concentration of the peptide (Yu et al. 2017b). More investigations are needed to understand how the N-terminal MBDs interact with the rest of the ATPase cytoplasmic domains, and the resulting consequences for Cu transport function.
Atox1 interactions with MBDs
Prokaryotic ATPases from different organisms can contain up to four MBDs and many of them sequester its metal directly from cytoplasm where they do not strictly require a cytoplasmic chaperone (Sharma and Rosato 2009). In humans, however direct protein–protein interactions between Atox1 and MBDs are essential for the delivery of Cu to the MBDs (Hamza et al. 1999; Hussain et al. 2009; Larin et al. 1999; Walker et al. 2002, 2004; Yu et al. 2017b). After the initial Cu-transfer from the chaperone, Cu is most likely channeled through MBDs according to the proposed “bucket-brigade” model where Cu is moved along MBDs (Bunce et al. 2006; Walker et al. 2004) and later delivered to MBD6, which is believed to forward Cu ions to the intramembrane Cu-sites (CIACPC motif in TMD6, Fig. 1). An inter-MBD Cu-transfer reaction was shown in vitro by NMR to take place from MBD4 to MBD6 in a construct containing MBD4-6 (Achila et al. 2006). ITC data revealed that affinities of MBDs for Cu was very similar among six domains and to that of Atox1 (Benitez et al. 2011) suggesting that Cu-exchange between Atox1 and MBDs, and among MBDs, may be under kinetic rather than thermodynamic control (Wernimont et al. 2004; Yatsunyk and Rosenzweig 2007).
Atox1 can deliver Cu (I) to all MBDs in vitro (Banci et al. 2008; Walker et al. 2002; Yatsunyk and Rosenzweig 2007) but not all MBDs are equivalent in terms of receiving Cu from Atox1 (Achila et al. 2006; Fatemi et al. 2010). Titration of apo-MBD2 (Walker et al. 2004), apo-MBD4 and apo-MBD5–6 (Achila et al. 2006) with Atox1-Cu complexes revealed that Atox1 delivers copper specifically to MBD2 and MBD4 forming an adduct with the chaperone whereas MBD56 is partially metallated without complex formation in accordance with the findings of a two-hybrid assay (Banci et al. 2009a; Larin et al. 1999). In fact, only MBD1, MBD2 and MBD4 were demonstrated to form Cu(I)-bridged adducts with Atox1, whereas no adduct formation takes place for MBD3, MBD5 and MBD6 but there is still Atox1-mediated Cu transport (Banci et al. 2008; Bunce et al. 2006). The electrostatic potential surface of Atox1 is dominated by the positive potential due to the presence of arginine (R) and lysine (L) residues in the helix α1 and helix α2. However, the surface charge distribution varies in different MBD domains; MBD1 (the least negative MBD), MBD2, MBD4 and MBD5 contain more negatively charged patches while MBD3 and MBD6 have more positively charged surfaces. MBD4 is the most negative one among the six domains, suggesting that this domain contributes more than others to the interaction between the positively charged Atox1 and negatively-charged MBDs (Rodriguez-Granillo et al. 2009). In accordance with this, NMR studies with MBD2 and MBD4 indicated that both MBD2 and MBD4 interact similarly with Atox1 (through similar residues) where positively charged residues in Atox1; R21, K25, K56 and K57 showed significant chemical shifts in the presence of these MBDs (Achila et al. 2006) indicating a possible ionic interaction network at the interface. A mutational study also showed that K60 of Atox1 is a critical position for complex formation with MBDs so that K60A/K60Y Atox1 mutations disrupted MBD4 binding (Hussain et al. 2009). Taken together, it could be stated that electrostatic interactions between these positively charged residues in Atox1 and negatively charged residues at the surface of MBDs complement each other, facilitating Cu-dependent interaction and thereby Cu-transfer (Bunce et al. 2006; Hamza et al. 1999).
MD calculations showed that Atox1 binds to MBD4 stronger than to any other MBDs (Rodriguez-Granillo et al. 2009; van Dongen et al. 2004). In the Atox1-MBD4 complex, Cu bridges the proteins by coordinating C residues of both proteins (Banci et al. 2009a; Benitez et al. 2011; Larin et al. 1999; Lutsenko et al. 1997). In silico, it has been shown that 2-coordinated intermediates are not likely to occur during Atox1-Cu-MBD4 complex formation and a stable adduct is formed with a 3-coordinated intermediate geometry that is; Cu is coordinated via two cysteines from one protein and one cysteine from the other. Such adduct is considered as a more stable geometry than 2- or 4-coordinated counterparts (Rodriguez-Granillo et al. 2010a), which is supported by the experimental findings with MBD4 (Niemiec et al. 2015) and MBD1–6 (Ralle et al. 2004). Using a combination of SEC and ITC experiments performed on MBD4 and Atox1, Niemiec et al. showed that hetero-protein complex formation occur via two steps; reactants to hetero-protein intermediate (step 1) and intermediate complex to products (step 2) (Niemiec et al. 2012) where the first step involves large favorable enthalpy and entropy changes (Niemiec et al. 2015). In contrast to the bulk solution results, smFRET experiments showed that apo-forms of Atox1 and MBD4 interact with each other, but Cu-bridging was shown to further stabilize the protein complex (Benitez et al. 2011).
WD-causing mutations in MBDs of ATP7B
WD is caused by mutations which can impede every step of the catalytic cycle of ATP7B and the final impact on the protein can range from mild to severe depending on which residues are affected (Huster et al. 2012). WD disease-causing mutations can be missense, frameshift, non-sense or splice-site mutations (Harada et al. 2001), however a large of number of reported WD mutations are missense (Chen et al. 2015; Huster et al. 2012). Most mutations observed in patients tend to be clustered in conserved regions of the ATPase core especially in the transmembrane region (Forbes and Cox 1998; Huster et al. 2012; Schushan et al. 2012) but some are also observed at other parts of the ATPase structure (e.g. A-domain, MBDs). The three most common mutations are as follows; R778L (located in TMD4 close to the A-domain) is the most common mutation worldwide (58% of all WD cases) and observed in patients of southeastern Asian descent (mostly Korean and Chinese patients), the second most common mutation H1069Q (found in N-domain; 35–45% of all mutations) is prevalent among European descent (Europe and North America) and is followed by R778W mutation which is prevalent among Indian population (Hedera 2017; Inesi et al. 2014; Jang et al. 2017).
To date, more than 800 mutations are reported with observed clinical symptoms (Human Gene Mutation Database, HGMD; http://www.hgmd.org) but the wide-spectrum of phenotypic variations of these mutations often makes it hard to make an accurate and early clinical diagnose for WD (Braiterman et al. 2014; Dong et al. 2016; Jang et al. 2017). Due to a wide variety of symptoms and age of onset, knowledge of how mutations in ATP7B link to symptoms in patients is still inconclusive. A number of studies, both biological/biochemical and biophysical, characterized WD-causing mutations which showed that these mutations may have various effects on ATP7B function (Braiterman et al. 2014; Chen et al. 2015). The most commonly observed effect is “protein misfolding” where ATP7B is retained in endoplasmic reticulum (ER) with a marked decrease in protein stability and thus results in the loss of Cu-transport in cells (Concilli et al. 2016; Payne et al. 1998; Tsivkovskii et al. 2003). Moreover, other effects such as impaired protein–protein interactions, decreased/increased phosphorylation state, altered binding affinities for ATP or Cu, altered conformational changes or cellular localization/vesicle trafficking behavior (Chen et al. 2015; Huster et al. 2012) have also been reported. We here gathered the known WD missense mutations localized in the six MBDs (Table 1). It is notable that WD-causing mutations in every MBD has been reported, but MBD5 and MBD6 contain two thirds of reported mutations. This is likely due to their location close to the Cu entry site in the membrane part of ATP7B and hints to key roles of this region in ATP7B function. The majority of the mutated positions are part of α-helices (e.g., R198G, I390V, E541K, L549P, T587M and R616Q etc.), linker regions (e.g., M67V, R136G, G515V and G591S etc.), whereas a few reside in β-strands (e.g., V519M and A595T etc.) (Fig. 4). Most of the WD mutations in Table 1 lack in vitro or in vivo information for how they affect ATP7B function, but below follows what has been reported.

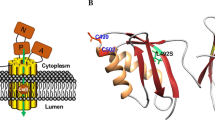
The positioning of WD-causing mutations (Table 1) in each MBD. The disease-causing mutations were visualized in magenta using UCSF Chimera molecular-modeling software. For all MBDs Cu-binding cysteines (C1 and C2) are shown with ball-stick representation in cyan. Mutations N41S in MBD1 and S406A, V456L, A476T in MBD5 could not be represented here due to the absence of these positions in reported PDB structures
G85V (in MBD1) and L492S (in MBD5) ATP7B variants were reported to result in complete loss of catalytic Cu transport activity (Huster et al. 2012; van den Berghe et al. 2009), whereas S406A, Y532H and G626A ATP7B variants were shown to maintain a normal Cu-transport activity in cell assays. G85, L492 and Y532 positions are distant to the Cu-site, and the mutations may thus affect domain–domain interactions or intrinsic structural stability. On the other hand, G626 is situated near the CxxC motif in MBD6 and MD simulations showed that G626A alters the hydrogen-bonding network thereby affecting Cu-binding ability (Hsi et al. 2008). We previously reported that G386V and G386D mutants (equivalent to G85 V and G591D but incorporated in MBD4) displayed dramatically reduced thermal stability compared to wild-type MBD4. Complementary MD simulations showed that the mutated residues enhance overall domain fluctuations (Kumar et al. 2017) which may alter how MBDs interact with Atox1, other MBDs, or other interacting partners. In accordance with this, ATP7B with the G85V mutant was reported to have a decreased interaction with Atox1 in cell-based assays. This was also found for G591D and L492S variants of full-length ATP7B when using GST-pulldown analysis (Hamza et al. 1999; Pilankatta et al. 2011). All three mutations showed increased interactions with MURR1-containing domain 1 (COMMD1), a scaffold protein responsible for protein ubiquitination and proteosomal degradation (de Bie et al. 2007; Hamza et al. 1999; Vonk et al. 2014). G85V ATP7B was observed in cell studies to mislocalize to the ER along with reduced protein expression (van den Berghe et al. 2009). In contrast, the G591D (Hamza et al. 1999; Pilankatta et al. 2011; Vanderwerf et al. 2001) and L492S (Hamza et al. 1999) variants were shown to traffic normally, but L492S was reported to be defective in Cu transport (Huster et al. 2012) whereas G591D resulted in impaired Cu-mediated ATPase activity (Vanderwerf et al. 2001). Characterization of additional WD mutations in individual domains and in full-length ATP7B using in vitro and in vivo approaches may contribute to further knowledge of underlying mechanisms.
Outlook
In Atox1-ATP7B pathway, Cu ions are believed to be transferred from Atox1 to some or all MBDs, then forwarded to the Cu-site in the transmembrane channel and finally delivered to Cu-dependent proteins passing the lumen of the Golgi. To better understand how the MBDs modulate ATP7B function, a high-resolution X-ray structure of full-length ATP7B would be extremely helpful but appears to be difficult to obtain as there are many different arrangements of the different domains in ATP7B. Instead, cryo-EM may be a promising approach for information on the spatial organization of the MBDs in ATP7B as a function of conditions. In the bigger picture, Cu-transfer mechanisms is of great importance to identify since Cu-transport dysregulation has been reported for several cancers (e.g., lymphoma, reticulum cell sarcoma, bronchogenic and laryngeal squamous cell carcinomas, cervical, breast, stomach and lung cancers) (Denoyer et al. 2015) and in some neurodegenerative diseases (Alzheimer’s disease, amyotrophic lateral sclerosis, Huntington’s disease, Parkinson’s disease, and prion diseases etc.) (Chen et al. 2016). In fact, in some cancers ATP7B expression has been connected to tumor-cell differentiation and alteration of outcome for platinum-based chemotherapy drugs (Dmitriev 2011; Kuo et al. 2007; Li et al. 2014; Martinez-Balibrea et al. 2009; Yang et al. 2015). For instance, cisplatin, a potent anti-cancer agent used against solid tumors of various cancers has been shown to bind to both Atox1 and ATP7B, perhaps processes governing cell resistance against this drug (Dmitriev 2011; Palm-Espling et al. 2013; Palm et al. 2011). Despite a lot of excellent biophysical work, further studies on ATP7B mechanisms, and the roles of the MBDs, are desired. This will aid not only better understanding of normal Cu transport and development of WD, but also the role of Cu in cancer and neurodegeneration processes.
References
Abdelghaffar TY, Elsayed SM, Elsobky E, Bochow B, Buttner J, Schmidt H (2008) Mutational analysis of ATP7B gene in Egyptian children with Wilson disease: 12 novel mutations. J Hum Genet 53(8):681–687
Abrahams JP, Leslie AG, Lutter R, Walker JE (1994) Structure at 2.8 a resolution of F1-ATPase from bovine heart mitochondria. Nature 370(6491):621–628
Achila D, Banci L, Bertini I, Bunce J, Ciofi-Baffoni S, Huffman DL (2006) Structure of human Wilson protein domains 5 and 6 and their interplay with domain 4 and the copper chaperone HAH1 in copper uptake. Proc Natl Acad Sci USA 103(15):5729–5734
Banci L, Bertini I, Cantini F, Rosenzweig AC, Yatsunyk LA (2008) Metal binding domains 3 and 4 of the Wilson disease protein: solution structure and interaction with the copper(I) chaperone HAH1. Biochemistry 47(28):7423–7429
Banci L, Bertini I, Cantini F, Massagni C, Migliardi M, Rosato A (2009a) An NMR study of the interaction of the N-terminal cytoplasmic tail of the Wilson disease protein with copper(I)-HAH1. J Biol Chem 284(14):9354–9360
Banci L, Bertini I, Cantini F, Migliardi M, Natile G, Nushi F, Rosato A (2009b) Solution structures of the actuator domain of ATP7A and ATP7B, the Menkes and Wilson disease proteins. Biochemistry 48(33):7849–7855
Barnes N, Bartee MY, Braiterman L, Gupta A, Ustiyan V, Zuzel V, Kaplan JH, Hubbard AL, Lutsenko S (2009) Cell-specific trafficking suggests a new role for renal ATP7B in the intracellular copper storage. Traffic 10(6):767–779
Bartee MY, Ralle M, Lutsenko S (2009) The loop connecting metal-binding domains 3 and 4 of ATP7B is a target of a kinase-mediated phosphorylation. Biochemistry 48(24):5573–5581
Benitez JJ, Keller AM, Huffman DL, Yatsunyk LA, Rosenzweig AC, Chen P (2011) Relating dynamic protein interactions of metallochaperones with metal transfer at the single-molecule level. Faraday Discuss 148:71–82 discussion 97–108
Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L et al (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42(Web Server issue):W252–W258
Braiterman L, Nyasae L, Guo Y, Bustos R, Lutsenko S, Hubbard A (2009) Apical targeting and Golgi retention signals reside within a 9-amino acid sequence in the copper-ATPase, ATP7B. Am J Physiol Gastrointest Liver Physiol 296(2):G433–G444
Braiterman LT, Murthy A, Jayakanthan S, Nyasae L, Tzeng E, Gromadzka G, Woolf TB, Lutsenko S, Hubbard AL (2014) Distinct phenotype of a Wilson disease mutation reveals a novel trafficking determinant in the copper transporter ATP7B. Proc Natl Acad Sci U S A 111(14):E1364–E1373
Braiterman LT, Gupta A, Chaerkady R, Cole RN, Hubbard AL (2015) Communication between the N and C Termini Is required for copper-stimulated Ser/Thr Phosphorylation of Cu(I)-ATPase (ATP7B). J Biol Chem 290(14):8803–8819
Bugbee D, Davies L, Kenney S, Cox WD (2010) Wilson Disease Mutation Database. University of Alberta, Edmonton
Bunce J, Achila D, Hetrick E, Lesley L, Huffman DL (2006) Copper transfer studies between the N-terminal copper binding domains one and four of human Wilson protein. Biochim Biophys Acta 1760(6):907–912
Cater MA, Forbes J, La Fontaine S, Cox D, Mercer JF (2004) Intracellular trafficking of the human Wilson protein: the role of the six N-terminal metal-binding sites. Biochem J 380(Pt 3):805–813
Cater MA, La Fontaine S, Shield K, Deal Y, Mercer JF (2006) ATP7B mediates vesicular sequestration of copper: insight into biliary copper excretion. Gastroenterology 130(2):493–506
Chang H, Xu A, Chen Z, Zhang Y, Tian F, Li T (2013) Long-term effects of a combination of D-penicillamine and zinc salts in the treatment of Wilson’s disease in children. Exp Ther Med 5(4):1129–1132
Chen C, Shen B, Xiao JJ, Wu R, Duff Canning SJ, Wang XP (2015) Currently clinical views on genetics of Wilson’s disease. Chin Med J (Engl) 128(13):1826–1830
Chen P, Miah MR, Aschner M. (2016) Metals and Neurodegeneration. F1000Res 5
Chesi G, Hegde RN, Iacobacci S, Concilli M, Parashuraman S, Festa BP, Polishchuk EV, Di Tullio G, Carissimo A, Montefusco S et al (2016) Identification of p38 MAPK and JNK as new targets for correction of Wilson disease-causing ATP7B mutants. Hepatology 63(6):1842–1859
Concilli M, Iacobacci S, Chesi G, Carissimo A, Polishchuk R (2016) A systems biology approach reveals new endoplasmic reticulum-associated targets for the correction of the ATP7B mutant causing Wilson disease. Metallomics 8(9):920–930
Das SK, Ray K (2006) Wilson’s disease: an update. Nat Clin Pract Neurol 2(9):482–493
de Bie P, van de Sluis B, Burstein E, van de Berghe PV, Muller P, Berger R, Gitlin JD, Wijmenga C, Klomp LW (2007) Distinct Wilson’s disease mutations in ATP7B are associated with enhanced binding to COMMD1 and reduced stability of ATP7B. Gastroenterology 133(4):1316–1326
Denoyer D, Masaldan S, La Fontaine S, Cater MA (2015) Targeting copper in cancer therapy: ‘Copper that cancer’. Metallomics 7(11):1459–1476
DiDonato M, Narindrasorasak S, Forbes JR, Cox DW, Sarkar B (1997) Expression, purification, and metal binding properties of the N-terminal domain from the wilson disease putative copper-transporting ATPase (ATP7B). J Biol Chem 272(52):33279–33282
DiDonato M, Hsu HF, Narindrasorasak S, Que L Jr, Sarkar B (2000) Copper-induced conformational changes in the N-terminal domain of the Wilson disease copper-transporting ATPase. Biochemistry 39(7):1890–1896
DiDonato M, Zhang J, Que L Jr, Sarkar B (2002) Zinc binding to the NH2-terminal domain of the Wilson disease copper-transporting ATPase: implications for in vivo metal ion-mediated regulation of ATPase activity. J Biol Chem 277(16):13409–13414
Dmitriev OY (2011) Mechanism of tumor resistance to cisplatin mediated by the copper transporter ATP7B. Biochem Cell Biol 89(2):138–147
Dmitriev O, Tsivkovskii R, Abildgaard F, Morgan CT, Markley JL, Lutsenko S (2006) Solution structure of the N-domain of Wilson disease protein: distinct nucleotide-binding environment and effects of disease mutations. Proc Natl Acad Sci U S A 103(14):5302–5307
Dmitriev OY, Lutsenko S, Muyldermans S (2016) Nanobodies as Probes for protein dynamics in vitro and in cells. J Biol Chem 291:3767–3775
Dolgova NV, Nokhrin S, Yu CH, George GN, Dmitriev OY (2013) Copper chaperone Atox1 interacts with the metal-binding domain of Wilson’s disease protein in cisplatin detoxification. Biochem J 454(1):147–156
Dong Y, Ni W, Chen WJ, Wan B, Zhao GX, Shi ZQ, Zhang Y, Wang N, Yu L, Xu JF et al (2016) Spectrum and classification of ATP7B variants in a large cohort of chinese patients with wilson’s disease guides genetic diagnosis. Theranostics 6(5):638–649
Fatemi N, Korzhnev DM, Velyvis A, Sarkar B, Forman-Kay JD (2010) NMR characterization of copper-binding domains 4–6 of ATP7B. Biochemistry 49(39):8468–8477
Festa RA, Thiele DJ (2011) Copper: an essential metal in biology. Curr Biol 21(21):R877–R883
Forbes JR, Cox DW (1998) Functional characterization of missense mutations in ATP7B: wilson disease mutation or normal variant? Am J Hum Genet 63(6):1663–1674
Gourdon P, Liu XY, Skjorringe T, Morth JP, Moller LB, Pedersen BP, Nissen P (2011) Crystal structure of a copper-transporting PIB-type ATPase. Nature 475(7354):59–64
Gourdon P, Sitsel O, Lykkegaard Karlsen J, Birk Moller L, Nissen P (2012) Structural models of the human copper P-type ATPases ATP7A and ATP7B. Biol Chem 393(4):205–216
Gupta A, Lutsenko S (2009) Human copper transporters: mechanism, role in human diseases and therapeutic potential. Future Med Chem 1(6):1125–1142
Hamza I, Schaefer M, Klomp LW, Gitlin JD (1999) Interaction of the copper chaperone HAH1 with the Wilson disease protein is essential for copper homeostasis. Proc Natl Acad Sci USA 96(23):13363–13368
Harada M, Sakisaka S, Terada K, Kimura R, Kawaguchi T, Koga H, Kim M, Taniguchi E, Hanada S, Suganuma T et al (2001) A mutation of the Wilson disease protein, ATP7B, is degraded in the proteasomes and forms protein aggregates. Gastroenterology 120(4):967–974
Hasan NM, Gupta A, Polishchuk E, Yu CH, Polishchuk R, Dmitriev OY, Lutsenko S (2012) Molecular events initiating exit of a copper-transporting ATPase ATP7B from the trans-Golgi network. J Biol Chem 287(43):36041–36050
Hedera P (2017) Update on the clinical management of Wilson’s disease. Appl Clin Genet 10:9–19
Hinz AVH (2014) Biophysical Characterization of the First Four Metal-Binding Domains of Human Wilson Disease Protein. Dissertations. 280.: Western Michigan University
Horn D, Barrientos A (2008) Mitochondrial copper metabolism and delivery to cytochrome c oxidase. IUBMB Life 60(7):421–429
Hsi G, Cullen LM, Macintyre G, Chen MM, Glerum DM, Cox DW (2008) Sequence variation in the ATP-binding domain of the Wilson disease transporter, ATP7B, affects copper transport in a yeast model system. Hum Mutat 29(4):491–501
Huang J, MacKerell AD Jr (2013) CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem 34(25):2135–2145
Huang YP, Nokhrin S, Hassanzadeh-Ghassabeh G, Yu CH, Yang HJ, Barry AN, Tonelli M, Markley JL, Muyldermans S, Dmitriev OY et al (2014) Interactions between metal-binding domains modulate intracellular targeting of Cu(I)-ATPase ATP7B, as revealed by nanobody binding. J Biol Chem 289(47):32682–32693
Hussain F, Rodriguez-Granillo A, Wittung-Stafshede P (2009) Lysine-60 in copper chaperone Atox1 plays an essential role in adduct formation with a target Wilson disease domain. J Am Chem Soc 131(45):16371–16373
Huster D, Lutsenko S (2003) The distinct roles of the N-terminal copper-binding sites in regulation of catalytic activity of the Wilson’s disease protein. J Biol Chem 278(34):32212–32218
Huster D, Kuhne A, Bhattacharjee A, Raines L, Jantsch V, Noe J, Schirrmeister W, Sommerer I, Sabri O, Berr F et al (2012) Diverse functional properties of Wilson disease ATP7B variants. Gastroenterology 142(4):947–956
Iida M, Terada K, Sambongi Y, Wakabayashi T, Miura N, Koyama K, Futai M, Sugiyama T (1998) Analysis of functional domains of Wilson disease protein (ATP7B) in Saccharomyces cerevisiae. FEBS Lett 428(3):281–285
Inesi G, Pilankatta R, Tadini-Buoninsegni F (2014) Biochemical characterization of P-type copper ATPases. Biochem J 463(2):167–176
Jang JH, Lee T, Bang S, Kim YE, Cho EH (2017) Carrier frequency of Wilson’s disease in the Korean population: a DNA-based approach. J Hum Genet 62(9):815–818
Jayakanthan S, Braiterman LT, Hasan NM, Unger VM, Lutsenko S (2017) Human copper transporter Atp7b (Wilson disease protein) forms stable dimers in vitro and in cells. J Biol Chem. doi:10.1074/jbc.M117.807263
Kaplan JH, Maryon EB (2016) How mammalian cells acquire copper: an essential but potentially toxic metal. Biophys J 110:7–13
Keller AM, Benitez JJ, Klarin D, Zhong L, Goldfogel M, Yang F, Chen TY, Chen P (2012) Dynamic multibody protein interactions suggest versatile pathways for copper trafficking. J Am Chem Soc 134(21):8934–8943
Knöpfel M, Solioz M (2002) Characterization of a cytochrome b558 Ferric/Cupric reductase from rabbit duodenal brush border membranes. Biochem Biophys Res Commun 291:220–225
Kroll CA, Ferber MJ, Dawson BD, Jacobson RM, Mensink KA, Lorey F, Sherwin J, Cunningham G, Rinaldo P, Matern D et al (2006) Retrospective determination of ceruloplasmin in newborn screening blood spots of patients with Wilson disease. Mol Genet Metab 89(1–2):134–138
Kumar R, Arioz C, Li Y, Bosaeus N, Rocha S, Wittung-Stafshede P (2017) Disease-causing point-mutations in metal-binding domains of Wilson disease protein decrease stability and increase structural dynamics. Biometals 30(1):27–35
Kuo MT, Chen HH, Song IS, Savaraj N, Ishikawa T (2007) The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev 26(1):71–83
Larin D, Mekios C, Das K, Ross B, Yang AS, Gilliam TC (1999) Characterization of the interaction between the Wilson and Menkes disease proteins and the cytoplasmic copper chaperone, HAH1p. J Biol Chem 274(40):28497–28504
LeShane ES, Shinde U, Walker JM, Barry AN, Blackburn NJ, Ralle M, Lutsenko S (2010) Interactions between copper-binding sites determine the redox status and conformation of the regulatory N-terminal domain of ATP7B. J Biol Chem 285(9):6327–6336
Letelier ME, Lepe AM, Faundez M, Salazar J, Marin R, Aracena P, Speisky H (2005) Possible mechanisms underlying copper-induced damage in biological membranes leading to cellular toxicity. Chem Biol Interact 151(2):71–82
Li XP, Yin JY, Wang Y, He H, Li X, Gong WJ, Chen J, Qian CY, Zheng Y, Li F et al (2014) The ATP7B genetic polymorphisms predict clinical outcome to platinum-based chemotherapy in lung cancer patients. Tumour Biol 35(8):8259–8265
Lim CM, Cater MA, Mercer JF, La Fontaine S (2006) Copper-dependent interaction of glutaredoxin with the N termini of the copper-ATPases (ATP7A and ATP7B) defective in Menkes and Wilson diseases. Biochem Biophys Res Commun 348(2):428–436
Lin CW, Er TK, Tsai FJ, Liu TC, Shin PY, Chang JG (2010) Development of a high-resolution melting method for the screening of Wilson disease-related ATP7B gene mutations. Clin Chim Acta 411(17–18):1223–1231
Lopez-Mirabal HR, Winther JR (2008) Redox characteristics of the eukaryotic cytosol. Biochim Biophys Acta 1783(4):629–640
Lorinczi E, Tsivkovskii R, Haase W, Bamberg E, Lutsenko S, Friedrich T (2008) Delivery of the Cu-transporting ATPase ATP7B to the plasma membrane in Xenopus oocytes. Biochim Biophys Acta 1778(4):896–906
Loudianos G, Kostic V, Solinas P, Lovicu M, Dessi V, Svetel M, Major T, Cao A (2003) Characterization of the molecular defect in the ATP7B gene in Wilson disease patients from Yugoslavia. Genet Test 7(2):107–112
Lutsenko S, Petrukhin K, Cooper MJ, Gilliam CT, Kaplan JH (1997) N-terminal domains of human copper-transporting adenosine triphosphatases (the Wilson’s and Menkes disease proteins) bind copper selectively in vivo and in vitro with stoichiometry of one copper per metal-binding repeat. J Biol Chem 272(30):18939–18944
Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY (2007) Function and regulation of human copper-transporting ATPases. Physiol Rev 87(3):1011–1046
Lutsenko S, Gupta A, Burkhead JL, Zuzel V (2008) Cellular multitasking: the dual role of human Cu-ATPases in cofactor delivery and intracellular copper balance. Arch Biochem Biophys 476(1):22–32
Mak CM, Lam CW, Lai ST, Hui Y, Tam S (2008) Rapid diagnosis of Wilson disease by a 28-mutation panel: real-time amplification refractory mutation system in diagnosing acute Wilsonian liver failure. Clin Chim Acta 398(1–2):39–42
Martinez-Balibrea E, Martinez-Cardus A, Musulen E, Gines A, Manzano JL, Aranda E, Plasencia C, Neamati N, Abad A (2009) Increased levels of copper efflux transporter ATP7B are associated with poor outcome in colorectal cancer patients receiving oxaliplatin-based chemotherapy. Int J Cancer 124(12):2905–2910
Matson Dzebo M, Arioz C, Wittung-Stafshede P (2016) Extended functional repertoire for human copper chaperones. Biomol Concepts 7(1):29–39
Mercer JF, Barnes N, Stevenson J, Strausak D, Llanos RM (2003) Copper-induced trafficking of the cU-ATPases: a key mechanism for copper homeostasis. Biometals 16(1):175–184
Mondol T, Aden J, Wittung-Stafshede P (2016) Copper binding triggers compaction in N-terminal tail of human copper pump ATP7B. Biochem Biophys Res Commun 470(3):663–669
Morth JP, Pedersen BP, Toustrup-Jensen MS, Sorensen TL, Petersen J, Andersen JP, Vilsen B, Nissen P (2007) Crystal structure of the sodium-potassium pump. Nature 450(7172):1043–1049
Mukherjee S, Dutta S, Majumdar S, Biswas T, Jaiswal P, Sengupta M, Bhattacharya A, Gangopadhyay PK, Bavdekar A, Das SK et al (2014) Genetic defects in Indian Wilson disease patients and genotype-phenotype correlation. Parkinsonism Relat Disord 20(1):75–81
Nagasaka H, Miida T, Inui A, Inoue I, Tsukahara H, Komatsu H, Hiejima E, Fujisawa T, Yorifuji T, Hiranao K et al (2012) Fatty liver and anti-oxidant enzyme activities along with peroxisome proliferator-activated receptors gamma and alpha expressions in the liver of Wilson’s disease. Mol Genet Metab 107(3):542–547
Niemiec MS, Weise CF, Wittung-Stafshede P (2012) In vitro thermodynamic dissection of human copper transfer from chaperone to target protein. PLoS ONE 7(5):e36102
Niemiec MS, Dingeldein AP, Wittung-Stafshede P (2014) T versus D in the MTCXXC motif of copper transport proteins plays a role in directional metal transport. J Biol Inorg Chem 19(6):1037–1047
Niemiec MS, Dingeldein APG, Wittung-Stafshede P (2015) Enthalpy-entropy compensation at play in human copper ion transfer. Sci Rep-Uk 5
Nilsson L, Aden J, Niemiec MS, Nam K, Wittung-Stafshede P (2013) Small pH and salt variations radically alter the thermal stability of metal-binding domains in the copper transporter, Wilson disease protein. J Phys Chem B 117(42):13038–13050
Ogawa H, Shinoda T, Cornelius F, Toyoshima C (2009) Crystal structure of the sodium-potassium pump (Na + , K + -ATPase) with bound potassium and ouabain. Proc Natl Acad Sci USA 106(33):13742–13747
Olesen C, Sorensen TL, Nielsen RC, Moller JV, Nissen P (2004) Dephosphorylation of the calcium pump coupled to counterion occlusion. Science 306(5705):2251–2255
Palm ME, Weise CF, Lundin C, Wingsle G, Nygren Y, Bjorn E, Naredi P, Wolf-Watz M, Wittung-Stafshede P (2011) Cisplatin binds human copper chaperone Atox1 and promotes unfolding in vitro. Proc Natl Acad Sci USA 108(17):6951–6956
Palm-Espling ME, Andersson CD, Bjorn E, Linusson A, Wittung-Stafshede P (2013) Determinants for simultaneous binding of copper and platinum to human chaperone atox1: hitchhiking not Hijacking. PLoS ONE 8(7):e70473
Payne AS, Kelly EJ, Gitlin JD (1998) Functional expression of the Wilson disease protein reveals mislocalization and impaired copper-dependent trafficking of the common H1069Q mutation. Proc Natl Acad Sci USA 95(18):10854–10859
Pham AN, Xing GW, Miller CJ, Waite TD (2013) Fenton-like copper redox chemistry revisited: hydrogen peroxide and superoxide mediation of copper-catalyzed oxidant production. J Catal 301:54–64
Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26(16):1781–1802
Pilankatta R, Lewis D, Inesi G (2011) Involvement of protein kinase D in expression and trafficking of ATP7B (copper ATPase). J Biol Chem 286(9):7389–7396
Polishchuk EV, Concilli M, Iacobacci S, Chesi G, Pastore N, Piccolo P, Paladino S, Baldantoni D, van IJzendoorn SC, Chan J et al (2014) Wilson disease protein ATP7B utilizes lysosomal exocytosis to maintain copper homeostasis. Dev Cell 29(6):686–700
Portmann R, Solioz M (2005) Purification and functional reconstitution of the human Wilson copper ATPase, ATP7B. FEBS Lett 579(17):3589–3595
Prashanth L, Kattapagari KK, Chitturi R, Baddam VRR, Prasad LK (2015) A review on role of essential trace elements in health and disease. J Dr NTR Univ Health Sci 4(2):75–85
Rakhit R, Chakrabartty A (2006) Structure, folding, and misfolding of Cu, Zn superoxide dismutase in amyotrophic lateral sclerosis. Biochim Biophys Acta 1762(11–12):1025–1037
Ralle M, Cooper MJ, Lutsenko S, Blackburn NJ (1998) The Menkes disease protein binds copper via novel 2-coordinate Cu(I)-cysteinates in the N-terminal domain. J Am Chem Soc 120(51):13525–13526
Ralle M, Lutsenko S, Blackburn NJ (2003) X-ray absorption spectroscopy of the copper chaperone HAH1 reveals a linear two-coordinate Cu(I) center capable of adduct formation with exogenous thiols and phosphines. J Biol Chem 278(25):23163–23170
Ralle M, Lutsenko S, Blackburn NJ (2004) Copper transfer to the N-terminal domain of the Wilson disease protein (ATP7B): x-ray absorption spectroscopy of reconstituted and chaperone-loaded metal binding domains and their interaction with exogenous ligands. J Inorg Biochem 98(5):765–774
Roberts EA, Schilsky ML, American Association for Study of Liver D (2008) Diagnosis and treatment of Wilson disease: an update. Hepatology 47(6):2089–2111
Rodriguez-Granillo A, Crespo A, Wittung-Stafshede P (2009) Conformational dynamics of metal-binding domains in Wilson disease protein: molecular insights into selective copper transfer. Biochemistry 48(25):5849–5863
Rodriguez-Granillo A, Crespo A, Estrin DA, Wittung-Stafshede P (2010a) Copper-transfer mechanism from the human chaperone Atox1 to a metal-binding domain of Wilson disease protein. J Phys Chem B 114(10):3698–3706
Rodriguez-Granillo A, Crespo A, Wittung-Stafshede P (2010b) Interdomain interactions modulate collective dynamics of the metal-binding domains in the Wilson disease protein. J Phys Chem B 114(5):1836–1848
Schilsky ML (2014) Wilson disease: clinical manifestations, diagnosis, and treatment. Clinical Liver Disease 3(5):104–107
Schushan M, Bhattacharjee A, Ben-Tal N, Lutsenko S (2012) A structural model of the copper ATPase ATP7B to facilitate analysis of Wilson disease-causing mutations and studies of the transport mechanism. Metallomics 4(7):669–678
Scvortova E (2013) Functional analysis and genotype-phenotype correlations in Wilson Disease. Analele Stiintifice ale Universitatii “Alexandru Ioan Cuza” din Iasi Sec II a Genetica si Biologie Moleculara 14 (3):35–42
Sharma S, Rosato A (2009) Role of the N-terminal Tail of metal-transporting P-1B-type ATPases from genome-wide analysis and molecular dynamics simulations. J Chem Inf Model 49(1):76–83
Shatwell KP, Dancis A, Cross AR, Klausner RD, Segal AW (1996) The FRE1 ferric reductase of Saccharomyces cerevisiae is a cytochrome b similar to that of NADPH oxidase. J Biol Chem 271(24):14240–14244
Shinoda T, Ogawa H, Cornelius F, Toyoshima C (2009) Crystal structure of the sodium-potassium pump at 2.4 A resolution. Nature 459(7245):446–450
Skjorringe T, Amstrup Pedersen P, Salling Thorborg S, Nissen P, Gourdon P, Birk Moller L (2017) Characterization of ATP7A missense mutants suggests a correlation between intracellular trafficking and severity of Menkes disease. Sci Rep 7(1):757
Stock D, Leslie AG, Walker JE (1999) Molecular architecture of the rotary motor in ATP synthase. Science 286(5445):1700–1705
Suzuki KT, Someya A, Komada Y, Ogra Y (2002) Roles of metallothionein in copper homeostasis: responses to Cu-deficient diets in mice. J Inorg Biochem 88(2):173–182
Tadini-Buoninsegni F, Bartolommei G, Moncelli MR, Pilankatta R, Lewis D, Inesi G (2010) ATP dependent charge movement in ATP7B Cu+-ATPase is demonstrated by pre-steady state electrical measurements. FEBS Lett 584(22):4619–4622
Tanner DE, Chan KY, Phillips JC, Schulten K (2011) Parallel generalized born implicit solvent calculations with NAMD. J Chem Theory Comput 7(11):3635–3642
Taylor AB, Stoj CS, Ziegler L, Kosman DJ, Hart PJ (2005) The copper-iron connection in biology: structure of the metallo-oxidase Fet3p. Proc Natl Acad Sci USA 102(43):15459–15464
Todorov T, Savov A, Jelev H, Panteleeva E, Konstantinova D, Krustev Z, Mihaylova V, Tournev I, Tankova L, Tzolova N et al (2005) Spectrum of mutations in the Wilson disease gene (ATP7B) in the Bulgarian population. Clin Genet 68(5):474–476
Tsivkovskii R, MacArthur BC, Lutsenko S (2001) The Lys 1010-Lys1325 fragment of the Wilson’s disease protein binds nucleotides and interacts with the N-terminal domain of this protein in a copper-dependent manner. J Biol Chem 276(3):2234–2242
Tsivkovskii R, Efremov RG, Lutsenko S (2003) The role of the invariant His-1069 in folding and function of the Wilson’s disease protein, the human copper-transporting ATPase ATP7B. J Biol Chem 278(15):13302–13308
van den Berghe PV, Stapelbroek JM, Krieger E, de Bie P, van de Graaf SF, de Groot RE, van Beurden E, Spijker E, Houwen RH, Berger R et al (2009) Reduced expression of ATP7B affected by Wilson disease-causing mutations is rescued by pharmacological folding chaperones 4-phenylbutyrate and curcumin. Hepatology 50(6):1783–1795
van Dongen EM, Klomp LW, Merkx M (2004) Copper-dependent protein-protein interactions studied by yeast two-hybrid analysis. Biochem Biophys Res Commun 323(3):789–795
Vanderwerf SM, Cooper MJ, Stetsenko IV, Lutsenko S (2001) Copper specifically regulates intracellular phosphorylation of the Wilson’s disease protein, a human copper-transporting ATPase. J Biol Chem 276(39):36289–36294
Vonk WI, Kakkar V, Bartuzi P, Jaarsma D, Berger R, Hofker MH, Klomp LW, Wijmenga C, Kampinga HH, van de Sluis B (2014) The Copper Metabolism MURR1 domain protein 1 (COMMD1) modulates the aggregation of misfolded protein species in a client-specific manner. PLoS ONE 9(4):e92408
Walker JM, Tsivkovskii R, Lutsenko S (2002) Metallochaperone Atox1 transfers copper to the NH2-terminal domain of the Wilson’s disease protein and regulates its catalytic activity. J Biol Chem 277(31):27953–27959
Walker JM, Huster D, Ralle M, Morgan CT, Blackburn NJ, Lutsenko S (2004) The N-terminal metal-binding site 2 of the Wilson’s Disease Protein plays a key role in the transfer of copper from Atox1. J Biol Chem 279(15):15376–15384
Wang Y, Hodgkinson V, Zhu S, Weisman GA, Petris MJ (2011) Advances in the understanding of mammalian copper transporters. Adv Nutr 2(2):129–137
Weiss KH (2016) Wilson Disease Synonym: Hepatolenticular Degeneration. Gene reviews
Wernimont AK, Yatsunyk LA, Rosenzweig AC (2004) Binding of copper(I) by the Wilson disease protein and its copper chaperone. J Biol Chem 279(13):12269–12276
Wu F, Wang J, Pu C, Qiao L, Jiang C (2015) Wilson’s disease: a comprehensive review of the molecular mechanisms. Int J Mol Sci 16(3):6419–6431
Yang T, Chen M, Chen T, Thakur A (2015) Expression of the copper transporters hCtr1, ATP7A and ATP7B is associated with the response to chemotherapy and survival time in patients with resected non-small cell lung cancer. Oncol Lett 10(4):2584–2590
Yatsunyk LA, Rosenzweig AC (2007) Cu(I) binding and transfer by the N terminus of the Wilson disease protein. J Biol Chem 282(12):8622–8631
Yu C, Lee W, Dmitriev O (2016) NMR structure of metal-binding domain 1 of ATP7B. Protein Data Bank
Yu CH, Dolgova NV, Dmitriev OY (2017a) Dynamics of the metal binding domains and regulation of the human copper transporters ATP7B and ATP7A. IUBMB Life 69(4):226–235
Yu CH, Yang N, Bothe J, Tonelli M, Nokhrin S, Dolgova NV, Braiterman LT, Lutsenko S, Dmitriev OY (2017b) The metal chaperone Atox1 regulates the activity of the human copper transporter ATP7B by modulating domain dynamics. The Journal of biological chemistry. doi:10.1074/jbc.M117.811752
Yu H, Xie JJ, Chen YC, Dong QY, Dong Y, Ni W, Wu ZY (2017c) Clinical features and outcome in patients with osseomuscular type of Wilson’s disease. BMC Neurol 17(1):34
Acknowledgements
This work was supported by Knut and Alice Wallenberg foundation, the Swedish Research Council, the Chalmers Foundation and the Chemistry Department at Umeå University. All MD simulations were conducted using the resources provided by the Swedish National Infrastructure for Computing (SNIC) at the High Performance Computing Center North (HPC2N).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ariöz, C., Li, Y. & Wittung-Stafshede, P. The six metal binding domains in human copper transporter, ATP7B: molecular biophysics and disease-causing mutations. Biometals 30, 823–840 (2017). https://doi.org/10.1007/s10534-017-0058-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-017-0058-2